
Relieving the Phenotyping Bottleneck for Grape Bunch Architecture in Grapevine Breeding Research: Implementation of a 3D-Based Phenotyping Approach for Quantitative Trait Locus Mapping

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Sampling
2.3. Three-Dimensional Data Acquisition and Analysis
2.4. Two-Dimensional and Manual Data Acquisition
2.5. Applied Genetic Maps
2.6. QTL Analysis
2.7. Statistical Analysis
2.8. Approximation of Physical QTL Positions
3. Results
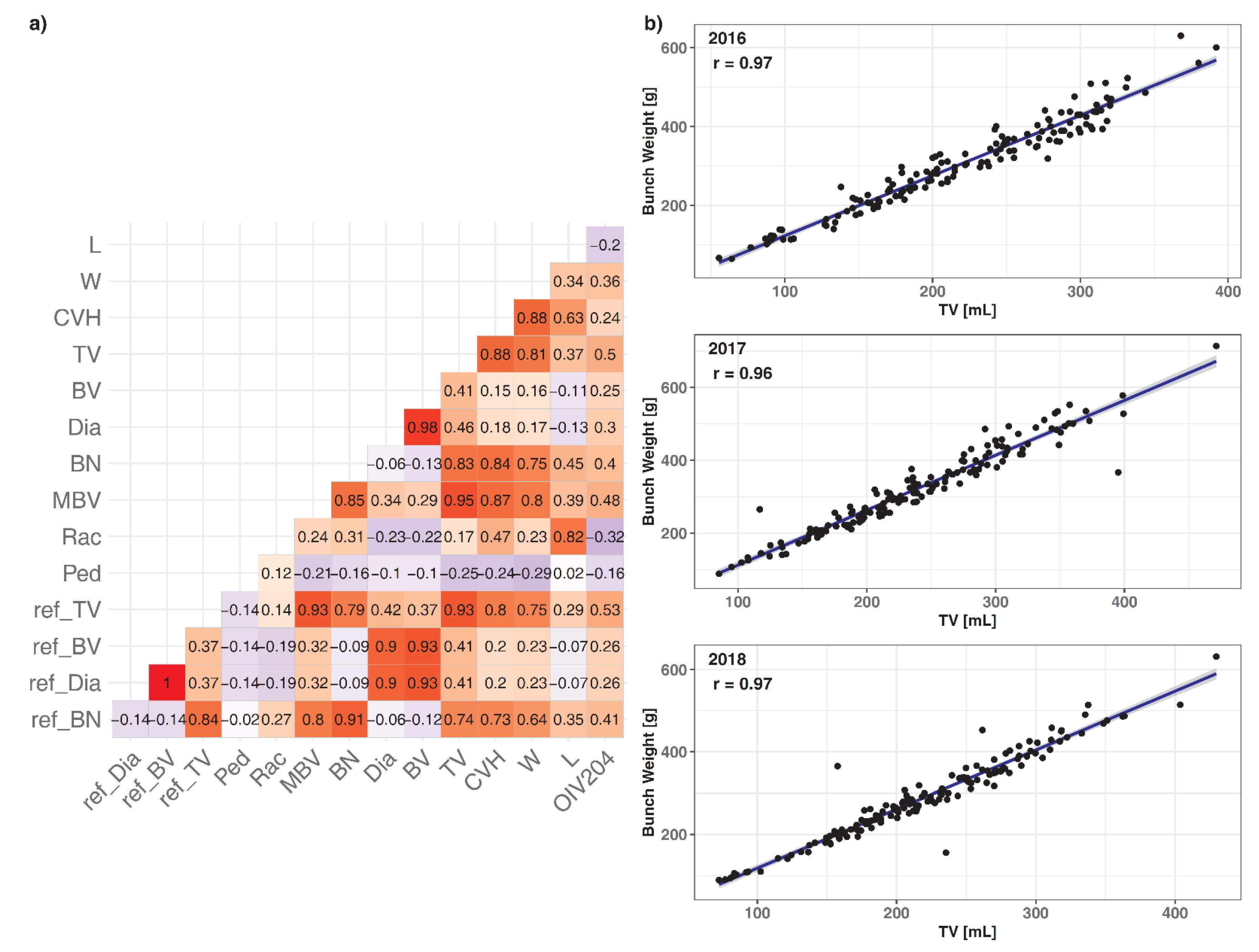
3.1. Direct Comparison between 3D, 2D, and Manually Measured Data of 150 Genotypes
3.2. Comparison of Detected QTLs in CxV_150 Based on 3D and Corresponding 2D Data
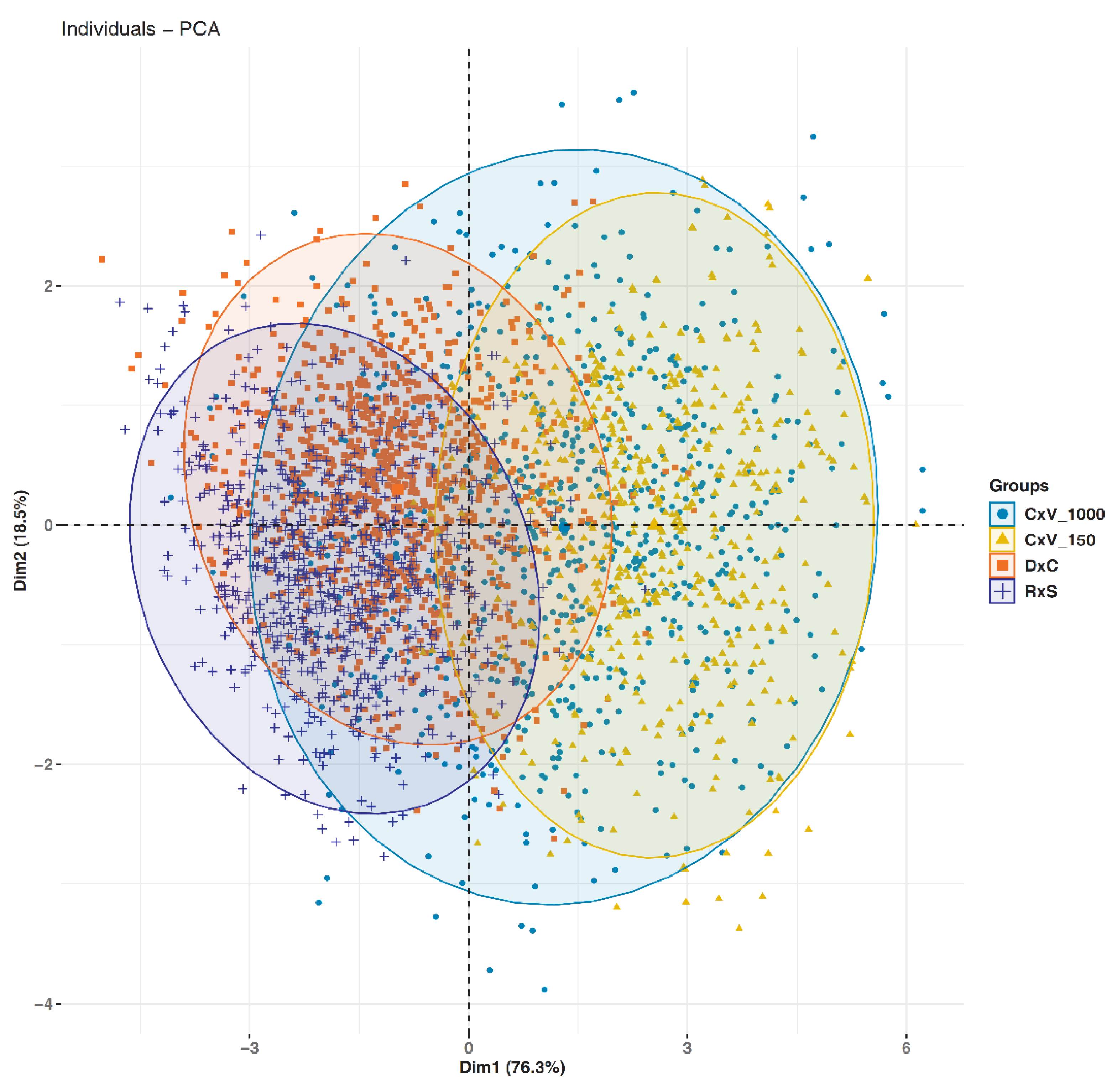
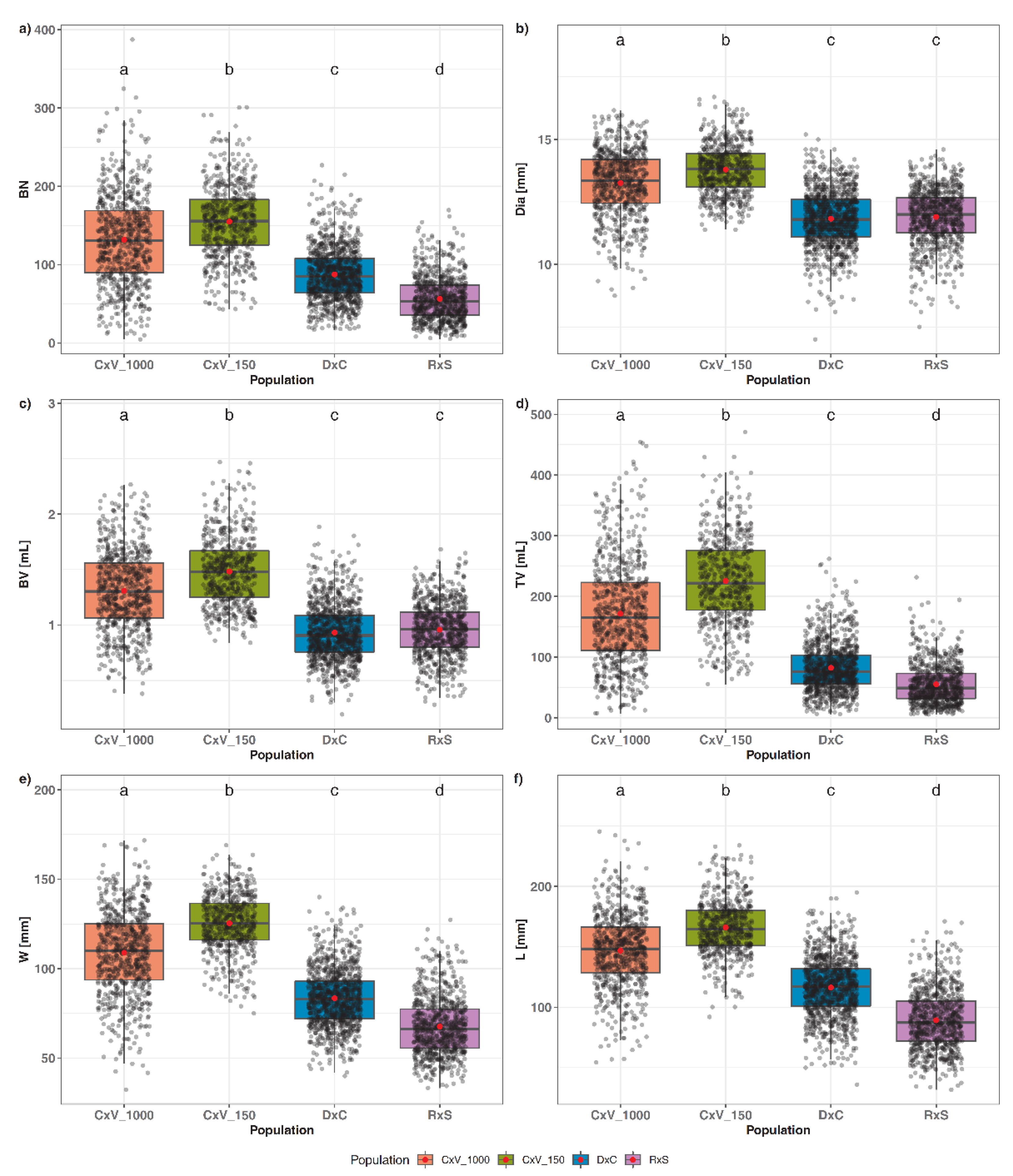
3.3. High-Throughput 3D Phenotyping of Different Populations
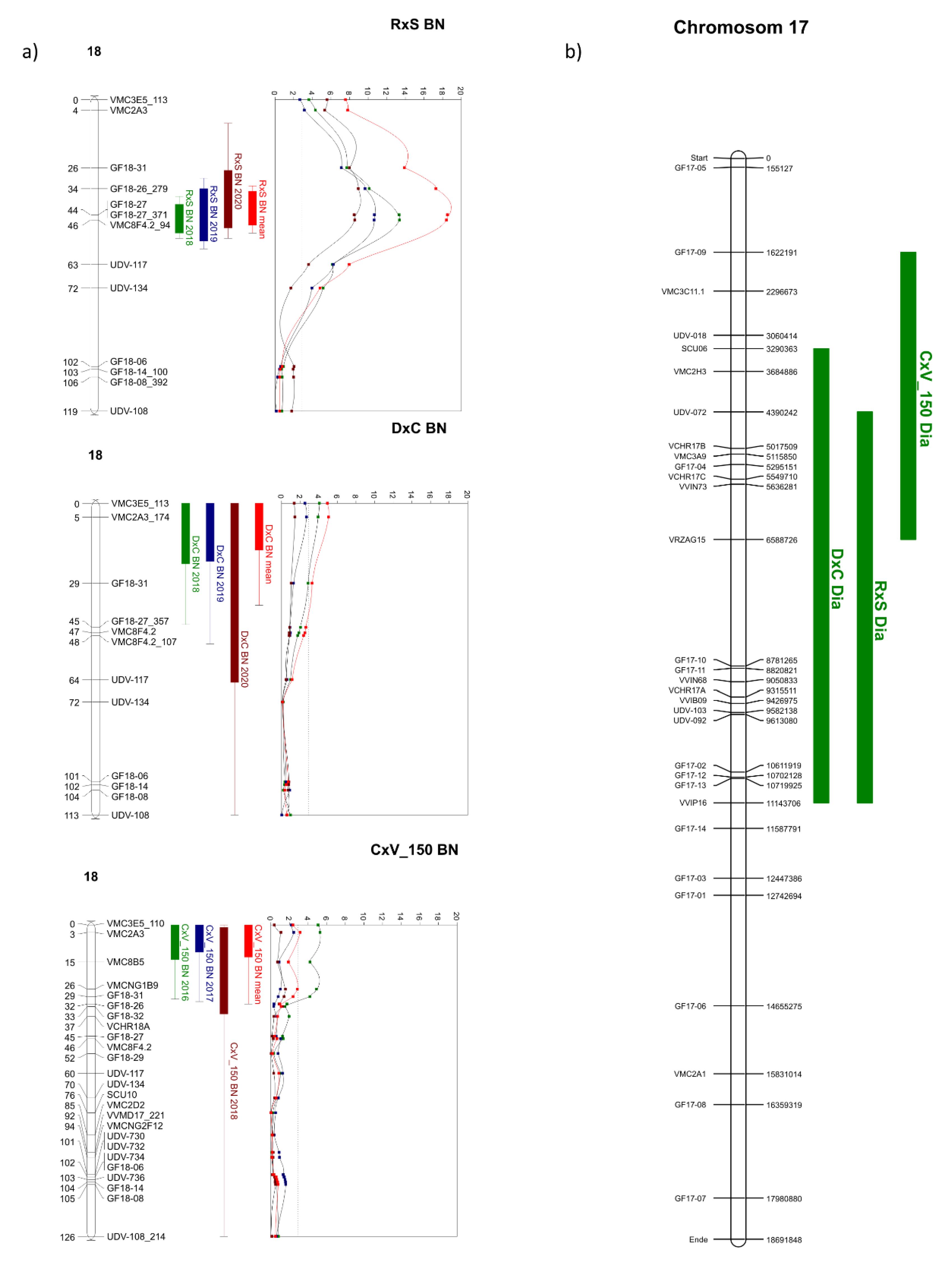
3.4. QTL Analysis for the Investigated Populations
4. Discussion
4.1. Three-Dimensional-Based Approach Enables Phenotypic Studies with High Throughput and Precision
4.2. Three-Dimensional vs. Two-Dimensional QTL Comparison
4.3. Sample Size of Investigated Populations
4.4. Seasonal Effect
4.5. Comparison of QTL Positions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Migicovsky, Z.; Sawler, J.; Gardner, K.M.; Aradhya, M.K.; Prins, B.H.; Schwaninger, H.R.; Bustamante, C.D.; Buckler, E.S.; Zhong, G.Y.; Brown, P.J.; et al. Patterns of Genomic and Phenomic Diversity in Wine and Table Grapes. Hortic. Res. 2017, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Herzog, K.; Wind, R.; Töpfer, R. Impedance of the Grape Berry Cuticle as a Novel Phenotypic Trait to Estimate Resistance to Botrytis Cinerea. Sensors 2015, 15, 12498–12512. [Google Scholar] [CrossRef] [PubMed]
- Tello, J.; Torres-Pérez, R.; Flutre, T.; Grimplet, J.; Ibáñez, J. VviUCC1 Nucleotide Diversity, Linkage Disequilibrium and Association with Rachis Architecture Traits in Grapevine. Genes 2020, 11, 598. [Google Scholar] [CrossRef]
- Molitor, D.; Behr, M.; Hoffmann, L.; Evers, D. Impact of Grape Cluster Division on Cluster Morphology and Bunch Rot Epidemic. Am. J. Enol. Vitic. 2012, 63, 508–514. [Google Scholar] [CrossRef]
- Herzog, K.; Schwander, F.; Kassemeyer, H.-H.; Bieler, E.; Dürrenberger, M.; Trapp, O.; Töpfer, R. Towards Sensor-Based Phenotyping of Physical Barriers of Grapes to Improve Resilience to Botrytis Bunch Rot. Front. Plant Sci. 2022, 12, 3299. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.; Van Kan, J.A.L.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 Fungal Pathogens in Molecular Plant Pathology. Molecular plant pathology 2012, 13, 414–430. [Google Scholar] [CrossRef]
- Tello, J.; Ibáñez, J. What Do We Know about Grapevine Bunch Compactness? A State-of-the-Art Review. Aust. J. Grape Wine Res. 2018, 24, 6–23. [Google Scholar] [CrossRef]
- Pertot, I.; Caffi, T.; Rossi, V.; Mugnai, L.; Hoffmann, C.; Grando, M.S.; Gary, C.; Lafond, D.; Duso, C.; Thiery, D.; et al. A Critical Review of Plant Protection Tools for Reducing Pesticide Use on Grapevine and New Perspectives for the Implementation of IPM in Viticulture. Crop Prot. 2017, 97, 70–84. [Google Scholar] [CrossRef]
- Richter, R.; Rossmann, S.; Gabriel, D.; Töpfer, R.; Theres, K.; Zyprian, E. Differential Expression of Transcription Factor- and Further Growth-Related Genes Correlates with Contrasting Cluster Architecture in Vitis Vinifera ‘Pinot Noir’ and Vitis Spp. Genotypes. Theor. Appl. Genet. 2020, 1, 3. [Google Scholar] [CrossRef]
- Töpfer, R.; Hausmann, L.; Harst, M.; Maul, E.; Zyprian, E. New Horizons for Grapevine Breeding. Methods Temp. Fruit Breed. 2011, 5, 79–100. [Google Scholar]
- Gabler, F.M.; Smilanick, J.L.; Mansour, M.; Ramming, D.W.; Mackey, B.E. Correlations of Morphological, Anatomical, and Chemical Features of Grape Berries with Resistance to Botrytis Cinerea. Phytopathology 2003, 93, 1263–1273. [Google Scholar] [CrossRef]
- Tello, J.; Ibáñez, J. Evaluation of Indexes for the Quantitative and Objective Estimation of Grapevine Bunch Compactness. Vitis J. Grapevine Res. 2014, 53, 9–16. [Google Scholar]
- Rist, F.; Herzog, K.; Mack, J.; Richter, R.; Steinhage, V.; Töpfer, R. High-Precision Phenotyping of Grape Bunch Architecture Using Fast 3D Sensor and Automation. Sensors 2018, 18, 763. [Google Scholar] [CrossRef] [PubMed]
- Organization Internationale de la Vigne et du Vin (OIV). Available online: https://www.oiv.int/en/technical-standards-and-documents/description-of-grape-varieties/oiv-descriptor-list-for-grape-varieties-and-vitis-species-2nd-edition (accessed on 26 September 2022).
- Palacios, F.; Diago, M.P.; Tardaguila, J. A Non-Invasive Method Based on Computer Vision for Grapevine Cluster Compactness Assessment Using a Mobile Sensing Platform under Field Conditions. Sensors 2019, 19, 3799. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ding, H.; Yuan, L.M.; Cai, J.R.; Chen, X.; Lin, Y. New Approach of Simultaneous, Multi-Perspective Imaging for Quantitative Assessment of the Compactness of Grape Bunches. Aust. J. Grape Wine Res. 2018, 24, 413–420. [Google Scholar] [CrossRef]
- Rist, F.; Gabriel, D.; Mack, J.; Steinhage, V.; Töpfer, R.; Herzog, K. Combination of an Automated 3D Field Phenotyping Workflow and Predictive Modelling for High-Throughput and Non-Invasive Phenotyping of Grape Bunches. Remote Sens. 2019, 11, 2953. [Google Scholar] [CrossRef]
- Furbank, R.T.; Tester, M. Phenomics-Technologies to Relieve the Phenotyping Bottleneck. Trends Plant Sci. 2011, 16, 635–644. [Google Scholar] [CrossRef]
- Kurtser, P.; Ringdahl, O.; Rotstein, N.; Berenstein, R.; Edan, Y. In-Field Grape Cluster Size Assessment for Vine Yield Estimation Using a Mobile Robot and a Consumer Level RGB-D Camera. IEEE Robot. Autom. Lett. 2020, 5, 2031–2038. [Google Scholar] [CrossRef]
- Underhill, A.; Hirsch, C.; Clark, M. Image-Based Phenotyping Identifies Quantitative Trait Loci for Cluster Compactness in Grape. J. Am. Soc. Hortic. Sci. 2020, 145, 363–373. [Google Scholar] [CrossRef]
- Vezzulli, S.; Doligez, A.; Bellin, D. Molecular Mapping of Grapevine Genes. In The Grape Genome; Springer: Cham, Switzerland, 2019; pp. 103–136. [Google Scholar]
- Asins, M.J.; Bernet, G.P.; Villalta, I.; Carbonell, E.A. QTL Analysis in Plant Breeding. In Molecular Techniques in Crop Improvement, 2nd ed.; Springer: Dordrecht, The Netherlands, 2009; pp. 3–21. ISBN 9789048129669. [Google Scholar]
- Richter, R.; Gabriel, D.; Rist, F.; Töpfer, R.; Zyprian, E. Identification of Co-Located QTLs and Genomic Regions Affecting Grapevine Cluster Architecture. Theor. Appl. Genet. 2018, 132, 1159–1177. [Google Scholar] [CrossRef]
- Shavrukov, Y.N.; Dry, I.B.; Thomas, M.R. Inflorescence and Bunch Architecture Development in Vitis Vinifera, L. Aust. J. Grape Wine Res. 2004, 10, 116–124. [Google Scholar] [CrossRef]
- Marguerit, E.; Boury, C.; Manicki, A.; Donnart, M.; Butterlin, G.; Némorin, A.; Wiedemann-Merdinoglu, S.; Merdinoglu, D.; Ollat, N.; Decroocq, S. Genetic Dissection of Sex Determinism, Inflorescence Morphology and Downy Mildew Resistance in Grapevine. Theor. Appl. Genet. 2009, 118, 1261–1278. [Google Scholar] [CrossRef]
- Correa, J.; Mamani, M.; Muñoz-Espinoza, C.; Laborie, D.; Muñoz, C.; Pinto, M.; Hinrichsen, P. Heritability and Identification of QTLs and Underlying Candidate Genes Associated with the Architecture of the Grapevine Cluster (Vitis Vinifera L.). Theor. Appl. Genet. 2014, 127, 1143–1162. [Google Scholar] [CrossRef] [PubMed]
- Tello, J.; Aguirrezábal, R.; Hernáiz, S.; Larreina, B.; Montemayor, M.I.; Vaquero, E.; Ibáñez, J. Multicultivar and Multivariate Study of the Natural Variation for Grapevine Bunch Compactness. Aust. J. Grape Wine Res. 2015, 21, 277–289. [Google Scholar] [CrossRef]
- Fanizza, G.; Lamaj, F.; Costantini, L.; Chaabane, R.; Grando, M.S. QTL Analysis for Fruit Yield Components in Table Grapes (Vitis Vinifera). Theor. Appl. Genet. 2005, 111, 658–664. [Google Scholar] [CrossRef]
- Sun, L.; Li, S.; Jiang, J.; Tang, X.; Fan, X.; Zhang, Y.; Liu, J.; Liu, C. New Quantitative Trait Locus (QTLs) and Candidate Genes Associated with the Grape Berry Color Trait Identified Based on a High-Density Genetic Map. BMC Plant Biol. 2020, 20, 302. [Google Scholar] [CrossRef]
- Zyprian, E.; Ochßner, I.; Schwander, F.; Šimon, S.; Hausmann, L.; Bonow-Rex, M.; Moreno-Sanz, P.; Grando, M.S.; Wiedemann-Merdinoglu, S.; Merdinoglu, D.; et al. Quantitative Trait Loci Affecting Pathogen Resistance and Ripening of Grapevines. Mol. Genet. Genom. 2016, 291, 1573–1594. [Google Scholar] [CrossRef]
- COOMBE, B.G. Growth Stages of the Grapevine: Adoption of a System for Identifying Grapevine Growth Stages. Aust. J. Grape Wine Res. 1995, 1, 104–110. [Google Scholar] [CrossRef]
- Mack, J.; Schindler, F.; Rist, F.; Herzog, K.; Töpfer, R.; Steinhage, V. Semantic Labeling and Reconstruction of Grape Bunches from 3D Range Data Using a New RGB-D Feature Descriptor. Comput. Electron. Agric. 2018, 155, 96–102. [Google Scholar] [CrossRef]
- Kicherer, A.; Roscher, R.; Herzog, K.; Šimon, S.; Förstner, W.; Töpfer, R. BAT (Berry Analysis Tool): A high-throughput image interpretation tool to acquire the number, diameter, and volume of grapevine berries. VITIS J. Grapevine Res. 2015, 52, 129–135. [Google Scholar]
- Ferreira, J.H.S.; Marais, P.G. Effect of Rootstock Cultivar, Pruning Method and Crop Load on Botrytis Cinerea Rot of Vitis Vinifera Cv. Chenin Blanc Grapes. S. Afr. J. Enol. Vitic. 2017, 8, 41–44. [Google Scholar] [CrossRef][Green Version]
- Jaillon, O.; Aury, J.M.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The Grapevine Genome Sequence Suggests Ancestral Hexaploidization in Major Angiosperm Phyla. Nature 2007, 449, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Van Ooijen, J.W. MapQTL® 6, Software for the Mapping of Quantita Tive Trait Loci. In Experimental Populations of Diploid Species; Kyazma, B.V., Ed.; Kyazma: Wageningen, The Netherlands, 2009. [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing; 2011; ISBN 3900051070. [Google Scholar]
- Length, R. emmeans: Estimated Marginal Means, aka Least-Squares Means. R Package Version 1.3.4. 2019. Available online: https://CRAN.R-project.org/package=emmeans (accessed on 26 September 2022).
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009; ISBN 978-0-387-98140-6. [Google Scholar]
- Kassambara, A. ggpubr: ‘ggplot2’ Based Publication Ready Plots. R Package Version 0.2. 2018. Available online: https://CRAN.R-project.org/package=ggpubr (accessed on 26 September 2022).
- Wei, T.; Simko, V. R Package “Corrplot”: Visualization of a Correlation Matrix (Version 0.84). 2017. Available online: https://github.com/taiyun/corrplot (accessed on 26 September 2022).
- Kassambara, A. factoextra: Extract and Visualize the Results of Multivariate Data Analyses. 2017. Available online: https://cran.r-project.org/web/packages/factoextra/index.html (accessed on 26 September 2022).
- Canaguier, A.; Grimplet, J.; Di Gaspero, G.; Scalabrin, S.; Duchêne, E.; Choisne, N.; Mohellibi, N.; Guichard, C.; Rombauts, S.; Le Clainche, I.; et al. A New Version of the Grapevine Reference Genome Assembly (12X.v2) and of Its Annotation (VCost.V3). Genom. Data 2017, 14, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Paulus, S. Measuring Crops in 3D: Using Geometry for Plant Phenotyping. Plant Methods 2019, 15, 103. [Google Scholar] [CrossRef]
- Paproki, A.; Sirault, X.; Berry, S.; Furbank, R.; Fripp, J. A Novel Mesh Processing Based Technique for 3D Plant Analysis. BMC Plant Biol. 2012, 12, 63. [Google Scholar] [CrossRef]
- Paulus, S.; Dupuis, J.; Mahlein, A.K.; Kuhlmann, H. Surface Feature Based Classification of Plant Organs from 3D Laserscanned Point Clouds for Plant Phenotyping. BMC Bioinform. 2013, 14, 238. [Google Scholar] [CrossRef]
- Wahabzada, M.; Paulus, S.; Kersting, K.; Mahlein, A.-K. Automated Interpretation of 3D Laserscanned Point Clouds for Plant Organ Segmentation. BMC Bioinform. 2015, 16, 248. [Google Scholar] [CrossRef]
- He, J.Q.; Harrison, R.J.; Li, B. A Novel 3D Imaging System for Strawberry Phenotyping. Plant Methods 2017, 13, 93. [Google Scholar] [CrossRef]
- Tello, J.; Cubero, S.; Blasco, J.; Tardaguila, J.; Aleixos, N.; Ibáñez, J. Application of 2D and 3D Image Technologies to Characterise Morphological Attributes of Grapevine Clusters. J. Sci. Food Agric. 2016, 96, 4575–4583. [Google Scholar] [CrossRef]
- Hacking, C.; Poona, N.; Manzan, N.; Poblete-Echeverría, C. Investigating 2-D and 3-D Proximal Remote Sensing Techniques for Vineyard Yield Estimation. Sensors 2019, 19, 3652. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.; Kicherer, A.; Wieland, M.; Klingbeil, L.; Töpfer, R.; Kuhlmann, H. Towards Automated Large-Scale 3D Phenotyping of Vineyards under Field Conditions. Sensors 2016, 16, 2136. [Google Scholar] [CrossRef] [PubMed]
- Pieruschka, R.; Schurr, U. Plant Phenotyping: Past, Present, and Future. Plant Phenomics 2019, 2019, 7507131. [Google Scholar] [CrossRef]
- Entling, W.; Anslinger, S.; Jarausch, B.; Michl, G.; Hoffmann, C. Berry Skin Resistance Explains Oviposition Preferences of Drosophila Suzukii at the Level of Grape Cultivars and Single Berries. J. Pest Sci. 2018, 92, 477–484. [Google Scholar] [CrossRef]
- Riffle, V.; Palmer, N.; Federico Casassa, L.; Peterson, J.C.D. The Effect of Grapevine Age (Vitis Vinifera L. Cv. Zinfandel) on Phenology and Gas Exchange Parameters over Consecutive Growing Seasons. Plants 2021, 10, 311. [Google Scholar] [CrossRef]
- Darvasi, A.; Weinreb, A.; Minke, V.; Weller, J.I.; Soller, M. Detecting Marker-Qtl Linkage and Estimating Qtl Gene Effect and Map Location Using a Saturated Genetic Map. Genetics 1993, 134, 943. [Google Scholar] [CrossRef]
- Xu, Y.; Li, P.; Yang, Z.; Xu, C. Genetic Mapping of Quantitative Trait Loci in Crops. Crop J. 2017, 5, 175–184. [Google Scholar] [CrossRef]
- Van Leeuwen, C.; Roby, J.P.; De Rességuier, L. Soil-Related Terroir Factors: A Review. Oeno ONE 2018, 52, 173–188. [Google Scholar] [CrossRef]
- Grimplet, J.; Ibáñez, S.; Baroja, E.; Tello, J.; Ibáñez, J. Phenotypic, Hormonal, and Genomic Variation among Vitis Vinifera Clones with Different Cluster Compactness and Reproductive Performance. Front. Plant Sci. 2019, 9, 1917. [Google Scholar] [CrossRef]
- Li-Mallet, A.; Rabot, A.; Geny, L. Factors Controlling Inflorescence Primordia Formation of Grapevine: Their Role in Latent Bud Fruitfulness? A Review. Botany 2016, 94, 147–163. [Google Scholar] [CrossRef]
- Tello, J.; Torres-Pérez, R.; Grimplet, J.; Ibáñez, J. Association Analysis of Grapevine Bunch Traits Using a Comprehensive Approach. Theor. Appl. Genet. 2016, 129, 227–242. [Google Scholar] [CrossRef]
- Lebon, G.; Wojnarowiez, G.; Holzapfel, B.; Fontaine, F.; Vaillant-Gaveau, N.; Clément, C. Sugars and Flowering in the Grapevine (Vitis Vinifera L.). J. Exp. Bot. 2008, 59, 2565–2578. [Google Scholar] [CrossRef] [PubMed]
- (PDF) The Flowering Process of Vitis Vinifera: A Review. Available online: https://www.researchgate.net/publication/233865529_The_Flowering_Process_of_Vitis_vinifera_A_Review (accessed on 14 May 2021).
- Tello, J.; Torres-Pérez, R.; Grimplet, J.; Carbonell-Bejerano, P.; Martínez-Zapater, J.M.; Ibáñez, J. Polymorphisms and Minihaplotypes in the VvNAC26 Gene Associate with Berry Size Variation in Grapevine. BMC Plant Biol. 2015, 15, 1–19. [Google Scholar] [CrossRef]
- Doligez, A.; Bertrand, Y.; Farnos, M.; Grolier, M.; Romieu, C.; Esnault, F.; Dias, S.; Berger, G.; François, P.; Pons, T.; et al. New Stable QTLs for Berry Weight Do Not Colocalize with QTLs for Seed Traits in Cultivated Grapevine (Vitis Vinifera L.). BMC Plant Biol. 2013, 13, 217. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.L.; Zhao, H.L.; Li, Q.; Zhang, G.H.; Jiang, J.F.; Liu, C.H.; Yu, Y.H. Genome-Wide Association Study of Berry-Related Traits in Grape [Vitis Vinifera L.] Based on Genotyping-by-Sequencing Markers. Hortic. Res. 2019, 6, 1–13. [Google Scholar] [CrossRef]
- Rossmann, S.; Richter, R.; Sun, H.; Schneeberger, K.; Töpfer, R.; Zyprian, E.; Theres, K. Mutations in the MiR396 Binding Site of the Growth-Regulating Factor Gene VvGRF4 Modulate Inflorescence Architecture in Grapevine. Plant J. 2020, 101, 1234–1248. [Google Scholar] [CrossRef] [PubMed]
- Delfino, P.; Zenoni, S.; Imanifard, Z.; Tornielli, G.B.; Bellin, D. Selection of Candidate Genes Controlling Veraison Time in Grapevine through Integration of Meta-QTL and Transcriptomic Data. BMC Genom. 2019, 20, 657. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3D | 2D | ||||||
|---|---|---|---|---|---|---|---|
| Chromosome | LODmax Position [cM] | LODmax | Phenotypic Variation [%] | LODmax Position [cM] | LODmax | Phenotypic Variation [%] | |
| BN | 4 | - | - | - | 2.3 | 3.01 | 9.1 |
| 8 | 19.5 | 3.8 | 11.3 | 19.5 | 3.8 | 11.2 | |
| 10 | 69.3 | 3.6 | 10.7 | 69.3 | 4.8 | 14 | |
| 17 | 50.3 | 3.7 | 11 | 37 | 4.3 | 12.7 | |
| 18 | 13.6 | 5.9 | 16.8 | 14.6 | 5.6 | 16.2 | |
| L | 1 | 35.3 | 3.3 | 9.8 | - | - | - |
| 2 | - | - | - | 19.7 | 2.8 | 8.3 | |
| 4 | - | - | - | 34.6 | 4 | 11.8 | |
| 8 | 38.1 | 3.5 | 10.4 | 22.2 | 3.5 | 10.4 | |
| 9 | 26.4 | 4.4 | 12.8 | 21.5 | 4.1 | 11.9 | |
| 15 | - | - | - | 4 | 3.7 | 10.9 | |
| Dia | 6 | 30.2 | 3 | 8.9 | 30.2 | 3 | 9.1 |
| 12 | 59.5 | 4.3 | 12.7 | 59.5 | 4.8 | 14.1 | |
| 17 | 7.3 | 3.4 | 10.2 | 17.4 | 5.1 | 14.9 | |
| 18 | 21.5 | 3 | 9 | 21.5 | 3.7 | 11.1 | |
| BV | 6 | 30.2 | 2.9 | 8.7 | 30.2 | 2.7 | 8.1 |
| 8 | 0 | 3.1 | 9.3 | - | - | - | |
| 12 | 58.8 | 4.6 | 13.4 | 59.5 | 4.6 | 13.5 | |
| 17 | 7.3 | 4.3 | 12.6 | 17.4 | 5 | 14.6 | |
| 18 | 21.5 | 3.6 | 10.7 | 21.5 | 3.6 | 10.7 | |
| TV | 7 | 79.4 | 3 | 8.9 | 79.4 | 3.2 | 9.6 |
| 10 | 67.3 | 3.2 | 9.4 | 66.3 | 4.4 | 13.2 | |
| 17 | 49.5 | 4.1 | 12 | 37 | 5.1 | 15.1 | |
| 18 | 14.6 | 5.6 | 16.1 | 14.6 | 5.9 | 17.1 | |
| LODmax | |||||
|---|---|---|---|---|---|
| Chromosome | Trait | R × S | D × C | C × V_150 | C × V_1000 |
| 1 | L | 2.92 | 4.57 | ||
| BN | 3.82 | 7.26 | |||
| 2 | BV | 4.38 | 4.88 | ||
| Dia | 4.35 | 9.65 | 4.32 | ||
| W | 4.16 | 2.76 | |||
| 3 | Dia | 3.44 | 4.51 | ||
| BV | 3.4 | 4.65 | |||
| 4 | BN | 6.09 | 4.15 | ||
| CVH | 3.83 | 3 | |||
| 6 | Dia | 3.77 | 5.28 | ||
| BV | 3.46 | 5.16 | |||
| 7 | W | 5.49 | 3.64 | ||
| 8 | L | 4.49 | 4.97 | 4.84 | |
| BV | 3.47 | 3.33 | 3.8 | ||
| BN | 3.24 | 3.05 | 4.15 | ||
| CVH | 3.97 | 3.95 | |||
| TV | 3.15 | 3.61 | 3.12 | ||
| 9 | CVH | 3.79 | 2.9 | ||
| 11 | Dia | 6.1 | 3.26 | ||
| 14 | W | 4.92 | 4.13 | ||
| 17 | CVH | 3.44 | 2.63 | 11.55 | |
| Dia | 4 | 13.18 | 6.39 | 12.7 | |
| TV | 3.07 | 14.47 | |||
| BV | 5.11 | 7.13 | 14.88 | ||
| W | 3.62 | 4.24 | 12.24 | ||
| 18 | BN | 18.93 | |||
| TV | 15.1 | 4.68 | |||
| W | 15.67 | 5.75 | |||
| L | 20.06 | 4.56 | 3.12 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rist, F.; Schwander, F.; Richter, R.; Mack, J.; Schwandner, A.; Hausmann, L.; Steinhage, V.; Töpfer, R.; Herzog, K. Relieving the Phenotyping Bottleneck for Grape Bunch Architecture in Grapevine Breeding Research: Implementation of a 3D-Based Phenotyping Approach for Quantitative Trait Locus Mapping. Horticulturae 2022, 8, 907. https://doi.org/10.3390/horticulturae8100907
Rist F, Schwander F, Richter R, Mack J, Schwandner A, Hausmann L, Steinhage V, Töpfer R, Herzog K. Relieving the Phenotyping Bottleneck for Grape Bunch Architecture in Grapevine Breeding Research: Implementation of a 3D-Based Phenotyping Approach for Quantitative Trait Locus Mapping. Horticulturae. 2022; 8(10):907. https://doi.org/10.3390/horticulturae8100907
Chicago/Turabian StyleRist, Florian, Florian Schwander, Robert Richter, Jennifer Mack, Anna Schwandner, Ludger Hausmann, Volker Steinhage, Reinhard Töpfer, and Katja Herzog. 2022. "Relieving the Phenotyping Bottleneck for Grape Bunch Architecture in Grapevine Breeding Research: Implementation of a 3D-Based Phenotyping Approach for Quantitative Trait Locus Mapping" Horticulturae 8, no. 10: 907. https://doi.org/10.3390/horticulturae8100907
APA StyleRist, F., Schwander, F., Richter, R., Mack, J., Schwandner, A., Hausmann, L., Steinhage, V., Töpfer, R., & Herzog, K. (2022). Relieving the Phenotyping Bottleneck for Grape Bunch Architecture in Grapevine Breeding Research: Implementation of a 3D-Based Phenotyping Approach for Quantitative Trait Locus Mapping. Horticulturae, 8(10), 907. https://doi.org/10.3390/horticulturae8100907