Synthesis and Characterization of Carbon/Nitrogen/Iron Based Nanoparticles by Laser Pyrolysis as Non-Noble Metal Electrocatalysts for Oxygen Reduction

 , ,
, ,
Abstract

1. Introduction
2. Materials and Methods
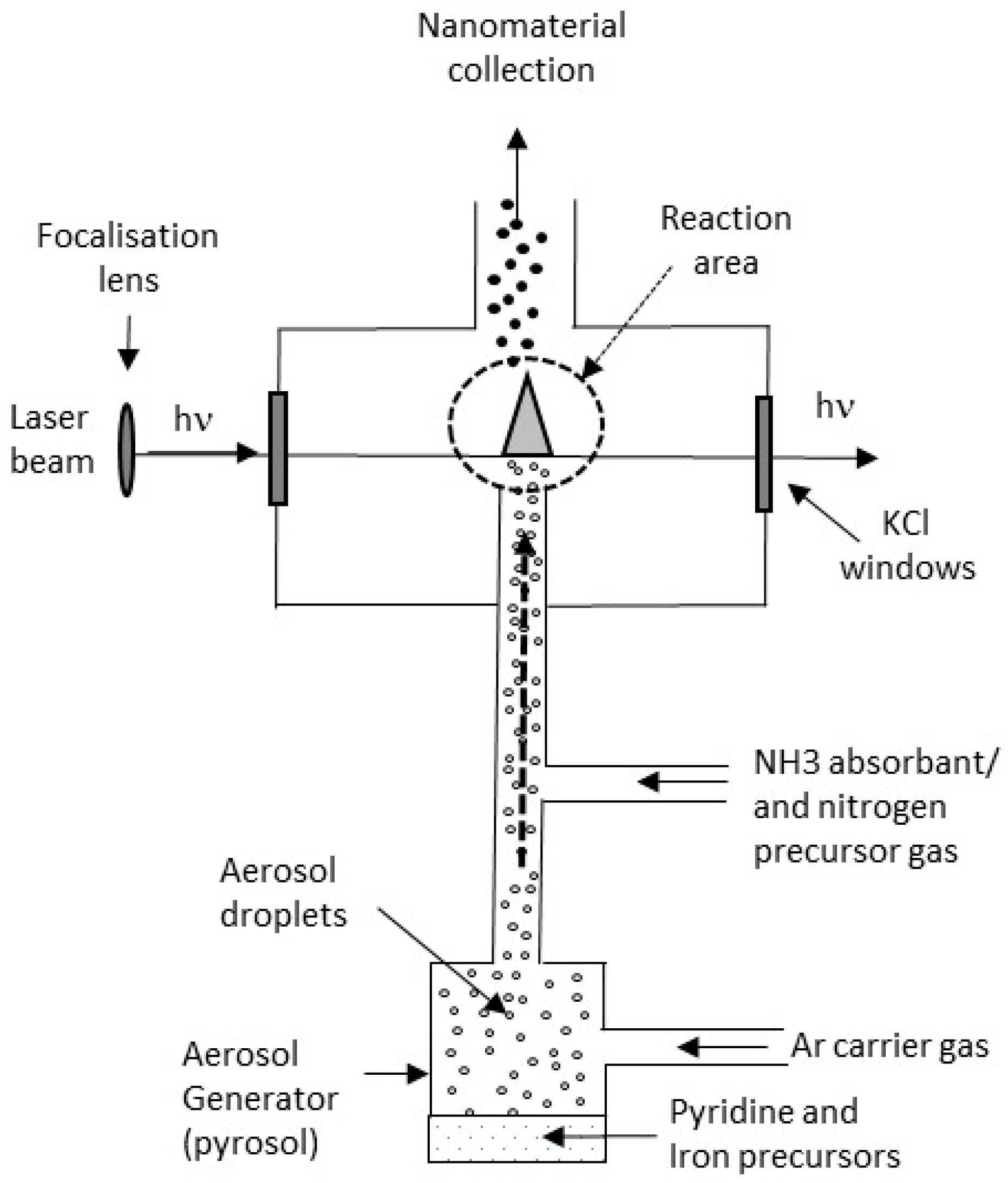
2.1. Synthesis
2.2. Characterization
2.3. Electrode Preparation and Electrochemical Measurement
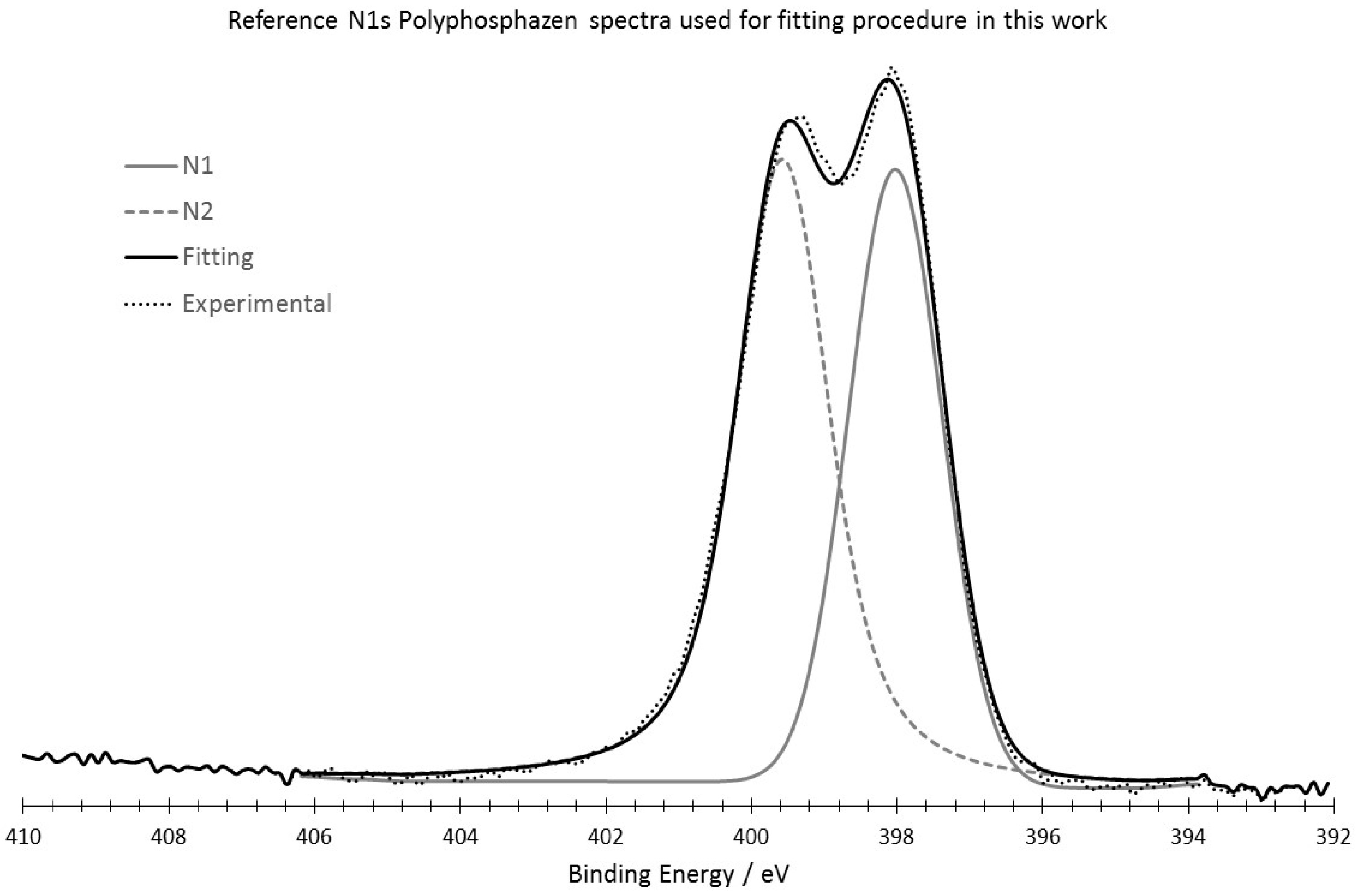
2.4. XPS Analysis
3. Results
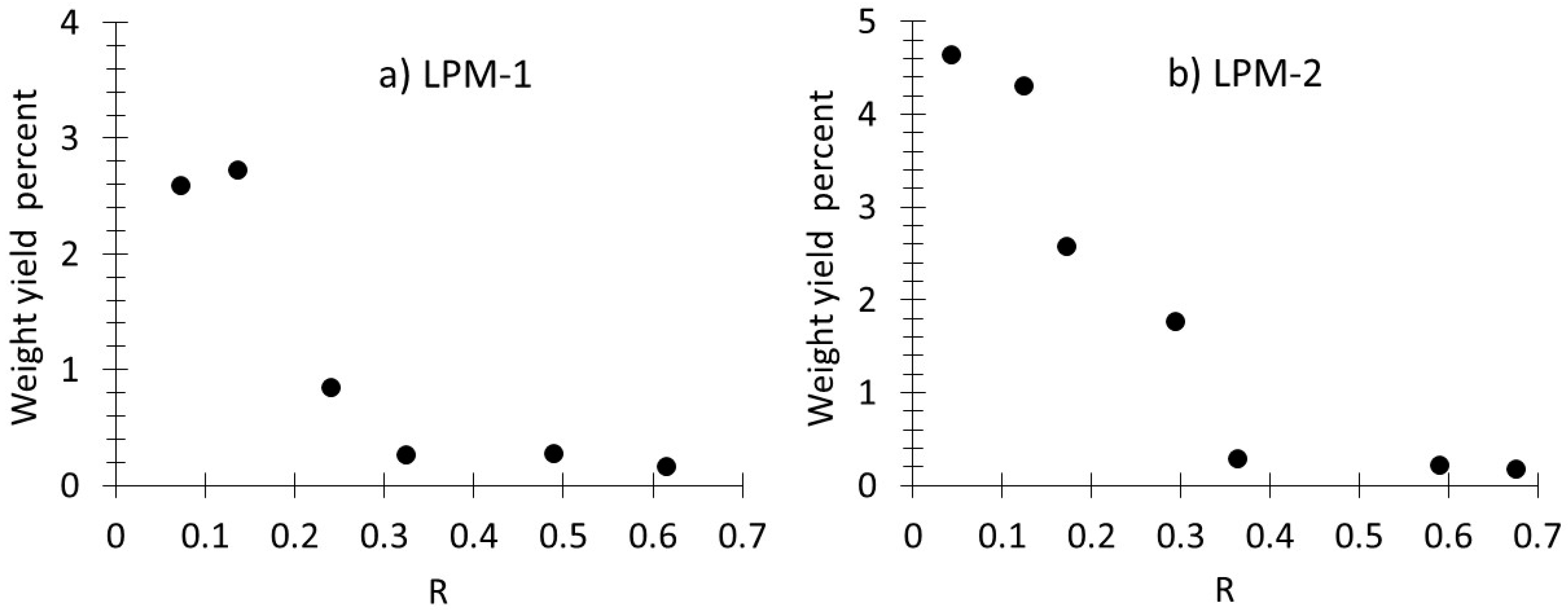
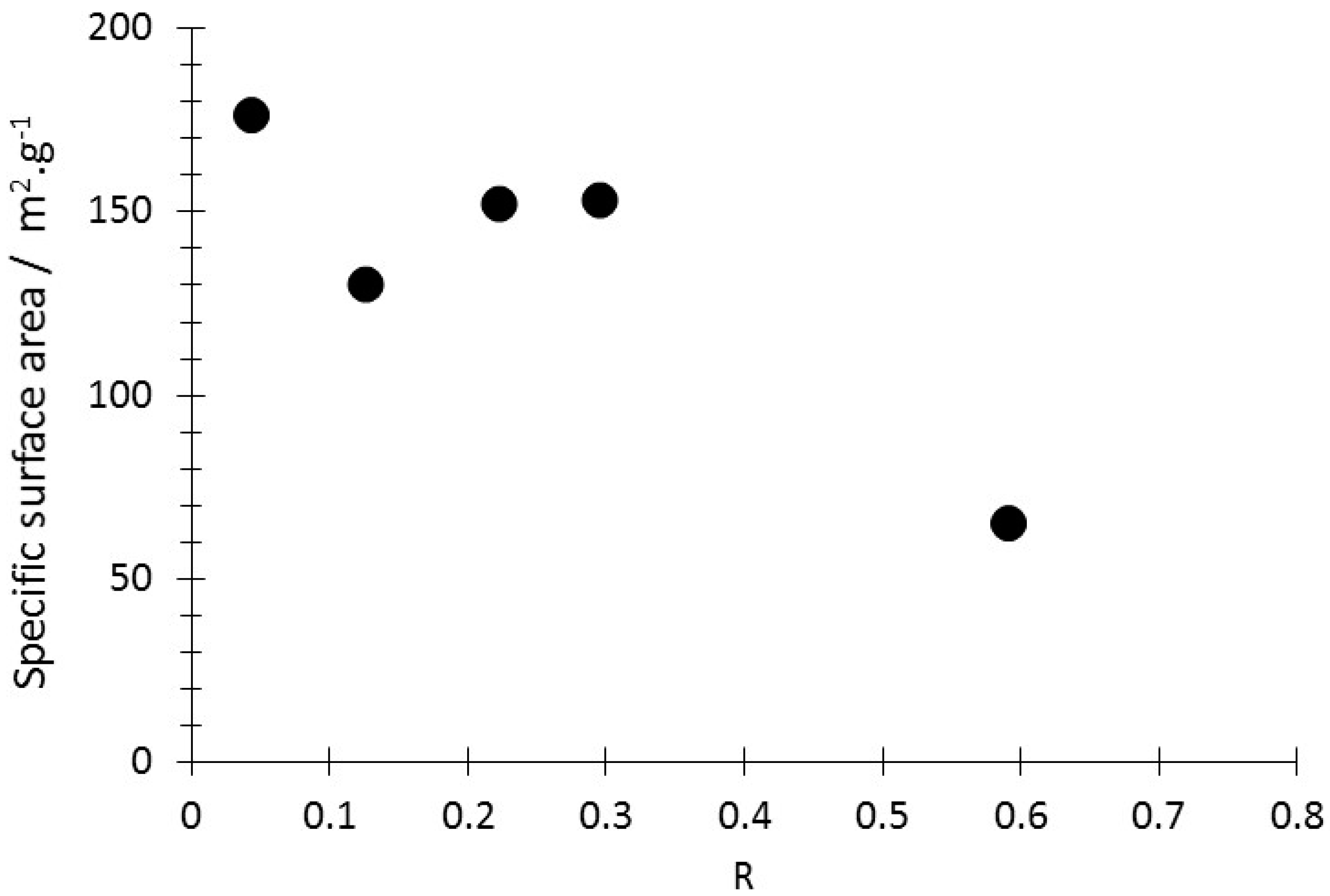
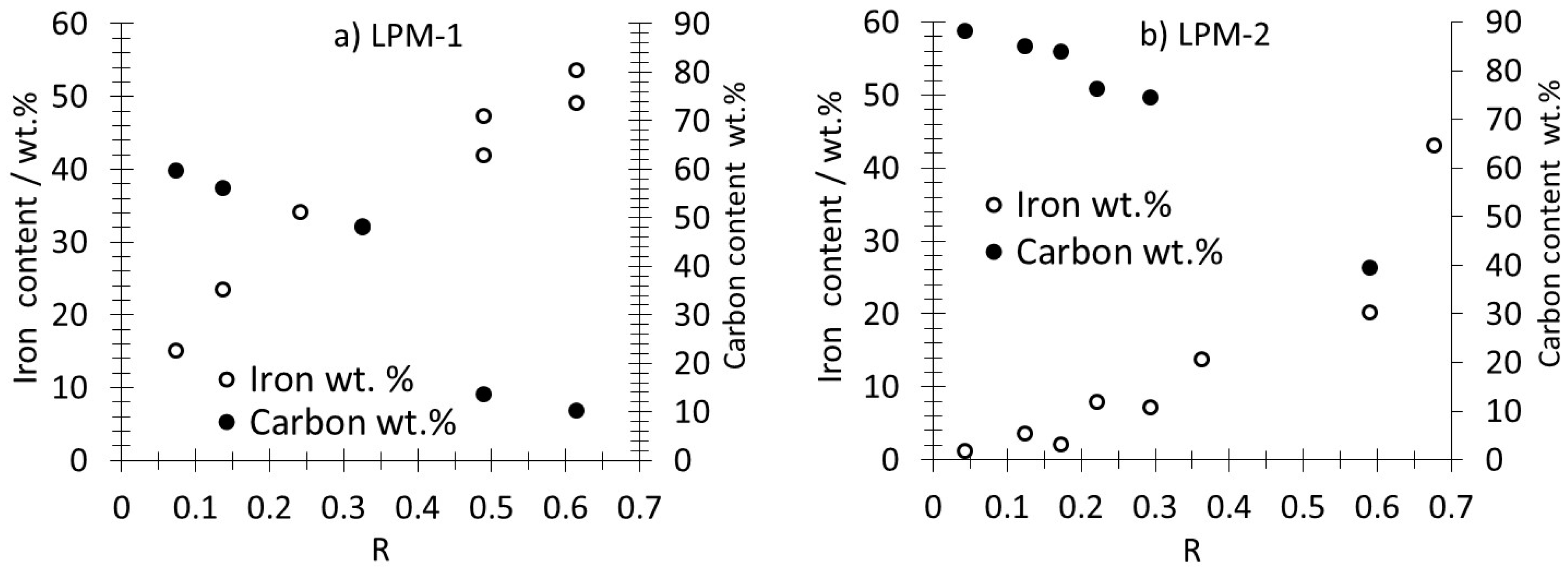
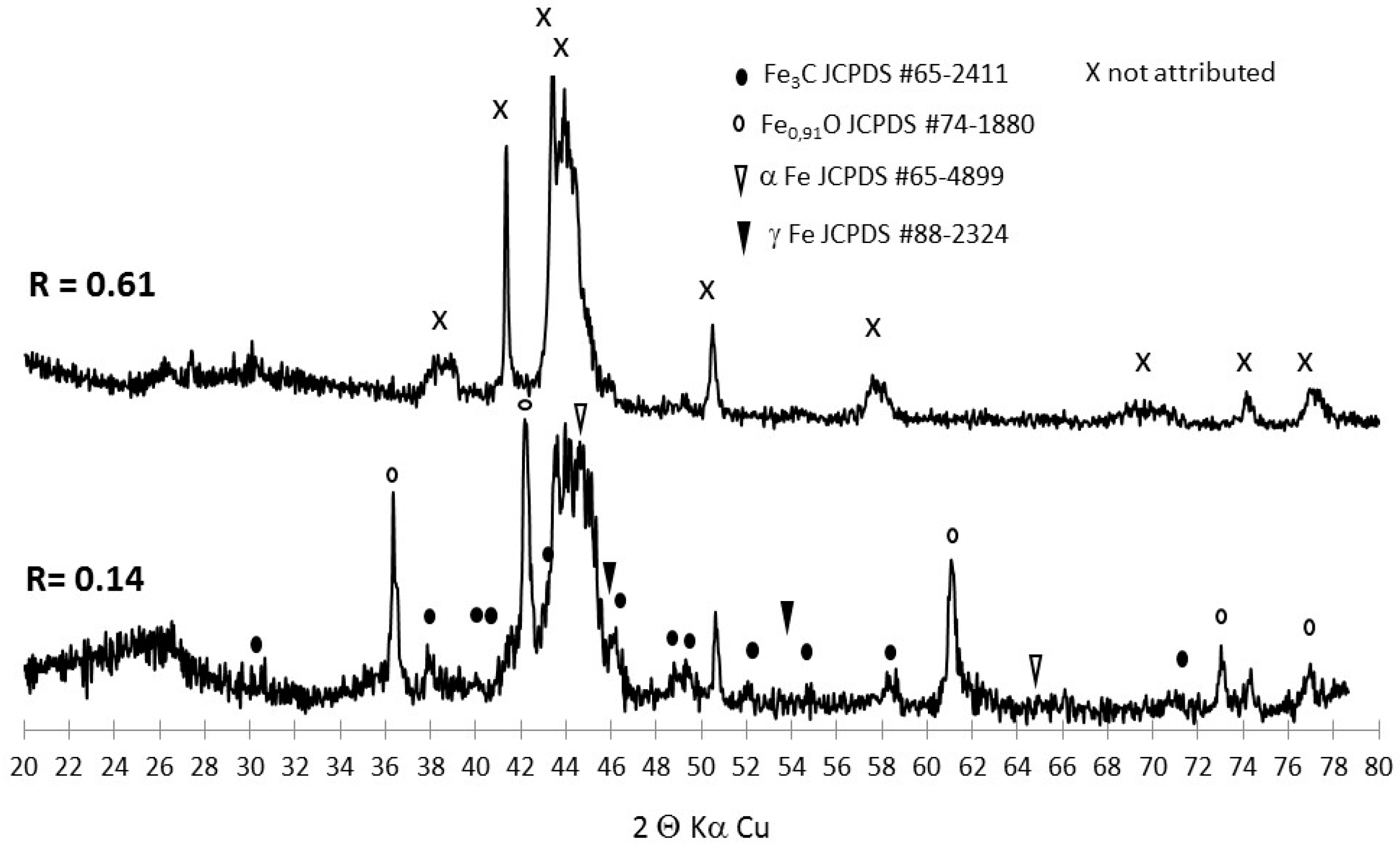
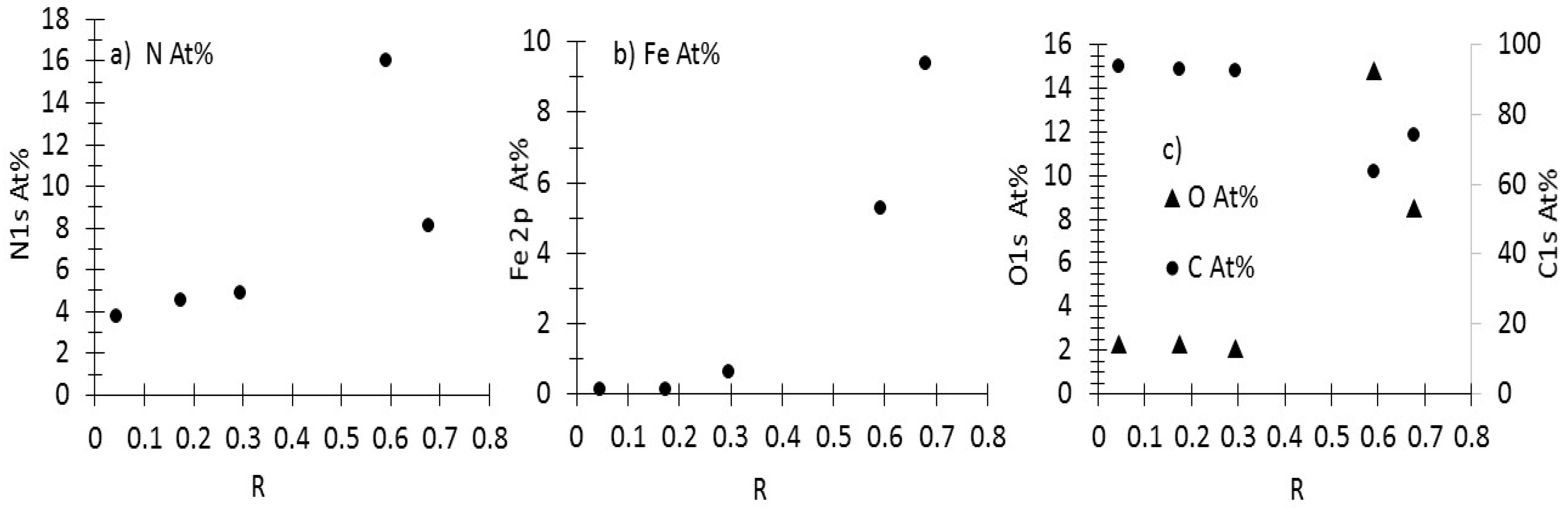
3.1. Production, Morphology and Composition Data of the Materials as a Function of R
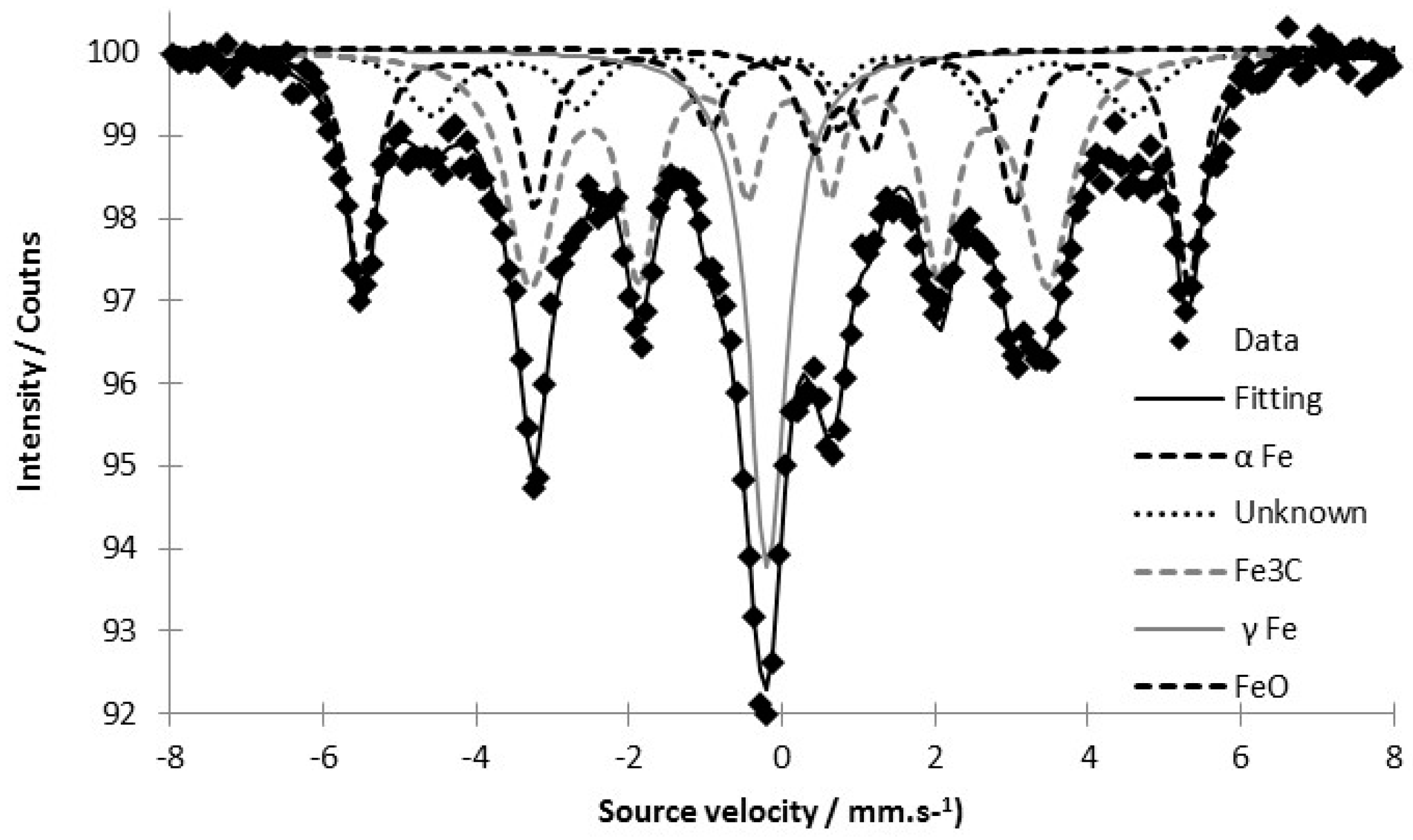
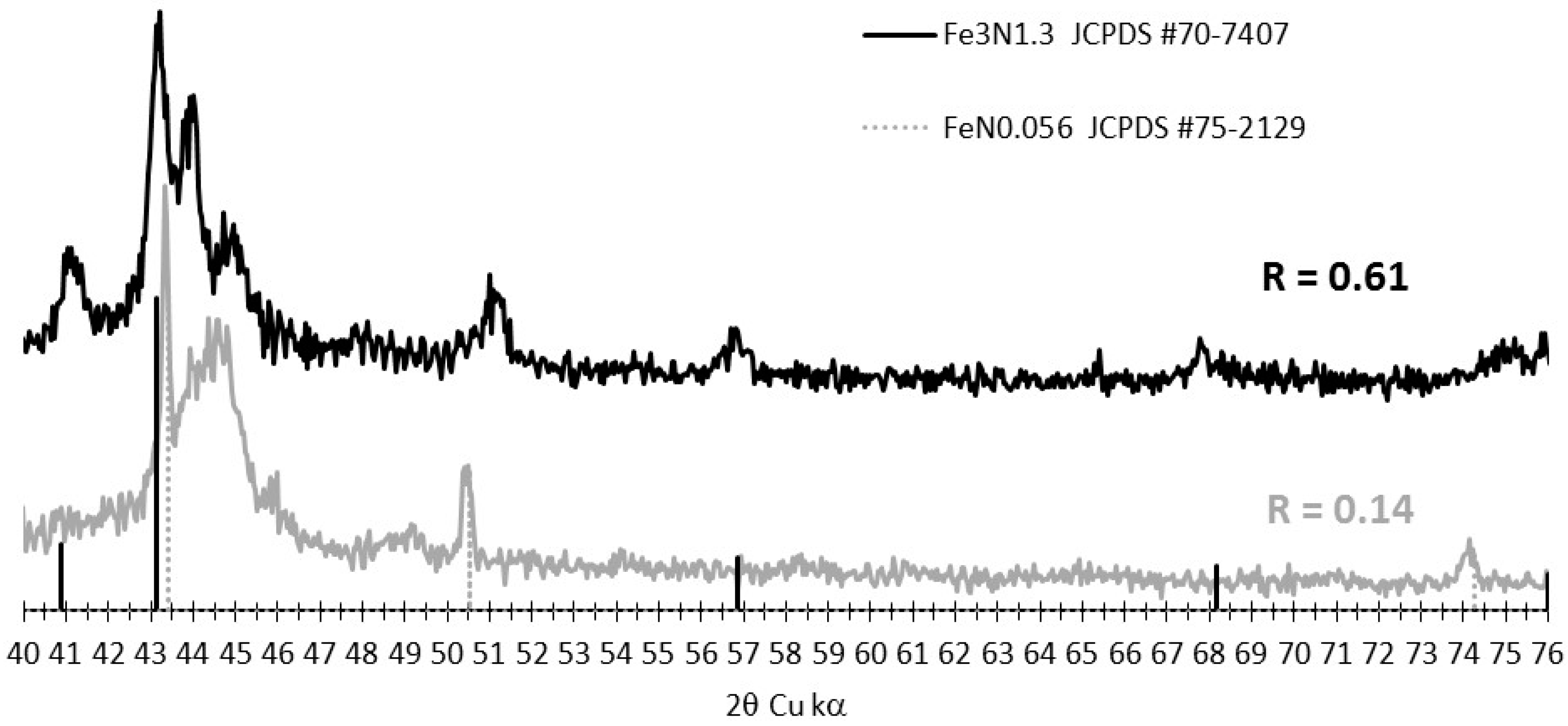
3.2. Tracking the Nature of the Iron Phases
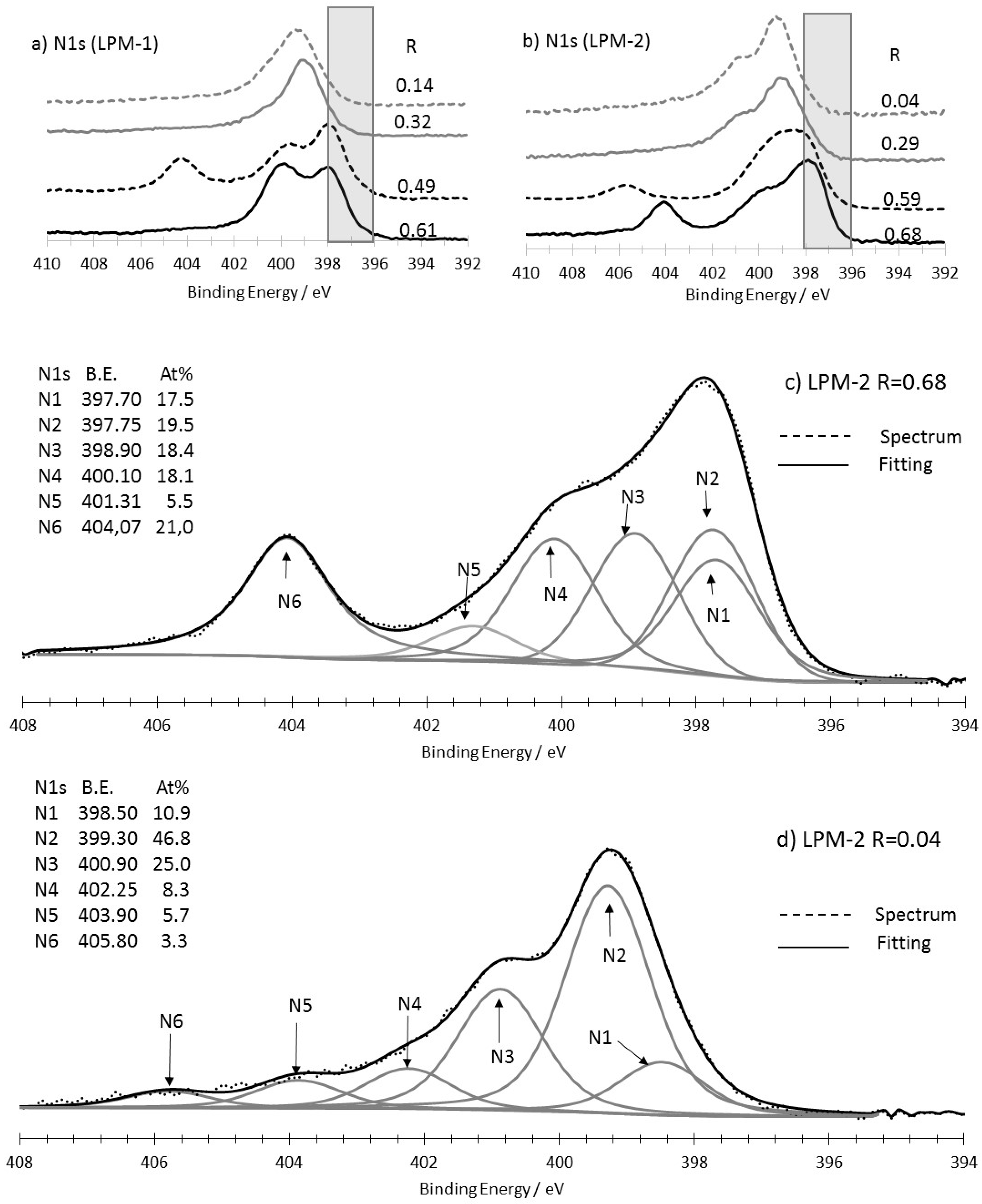
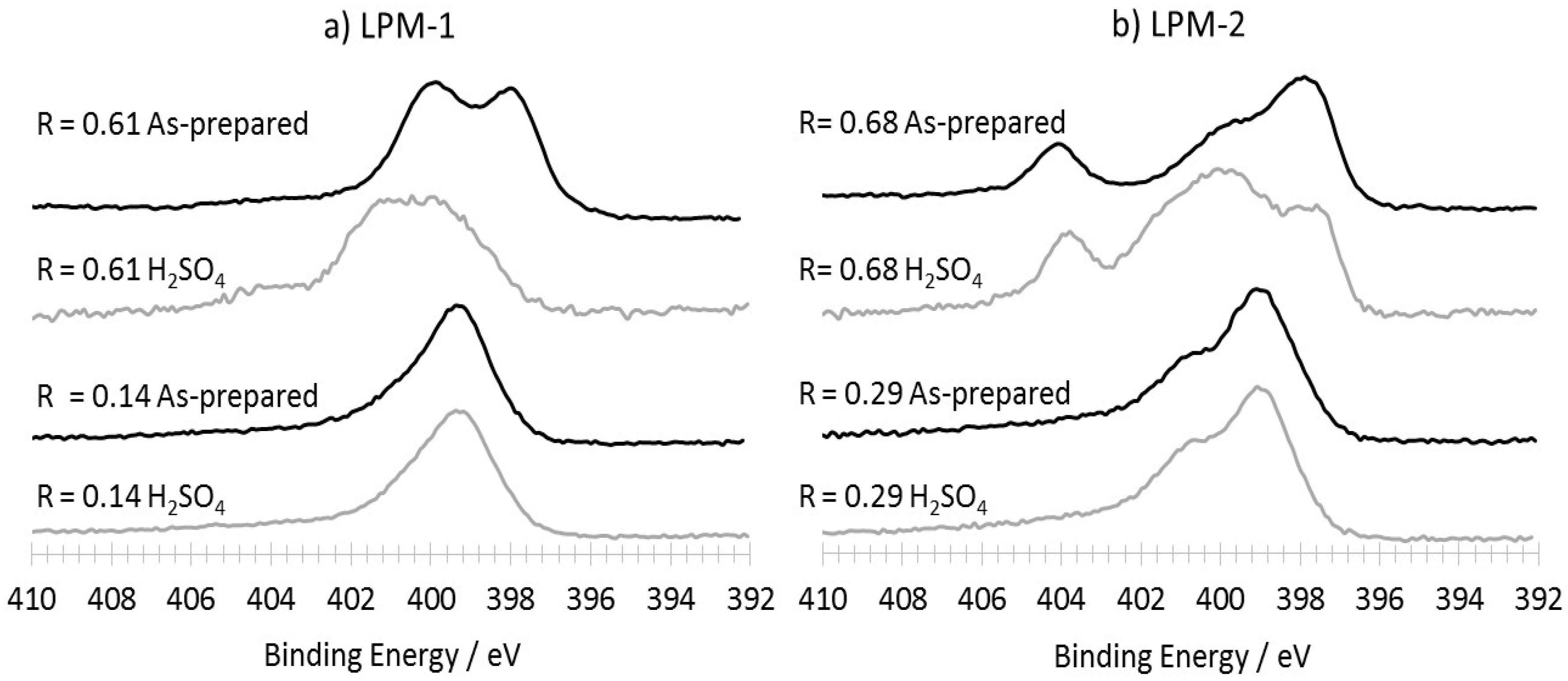
3.3. XPS Analysis
3.3.1. As-Prepared Materials
3.3.2. Characterization of the Materials Obtained from LPM-1 after Acidic Treatment
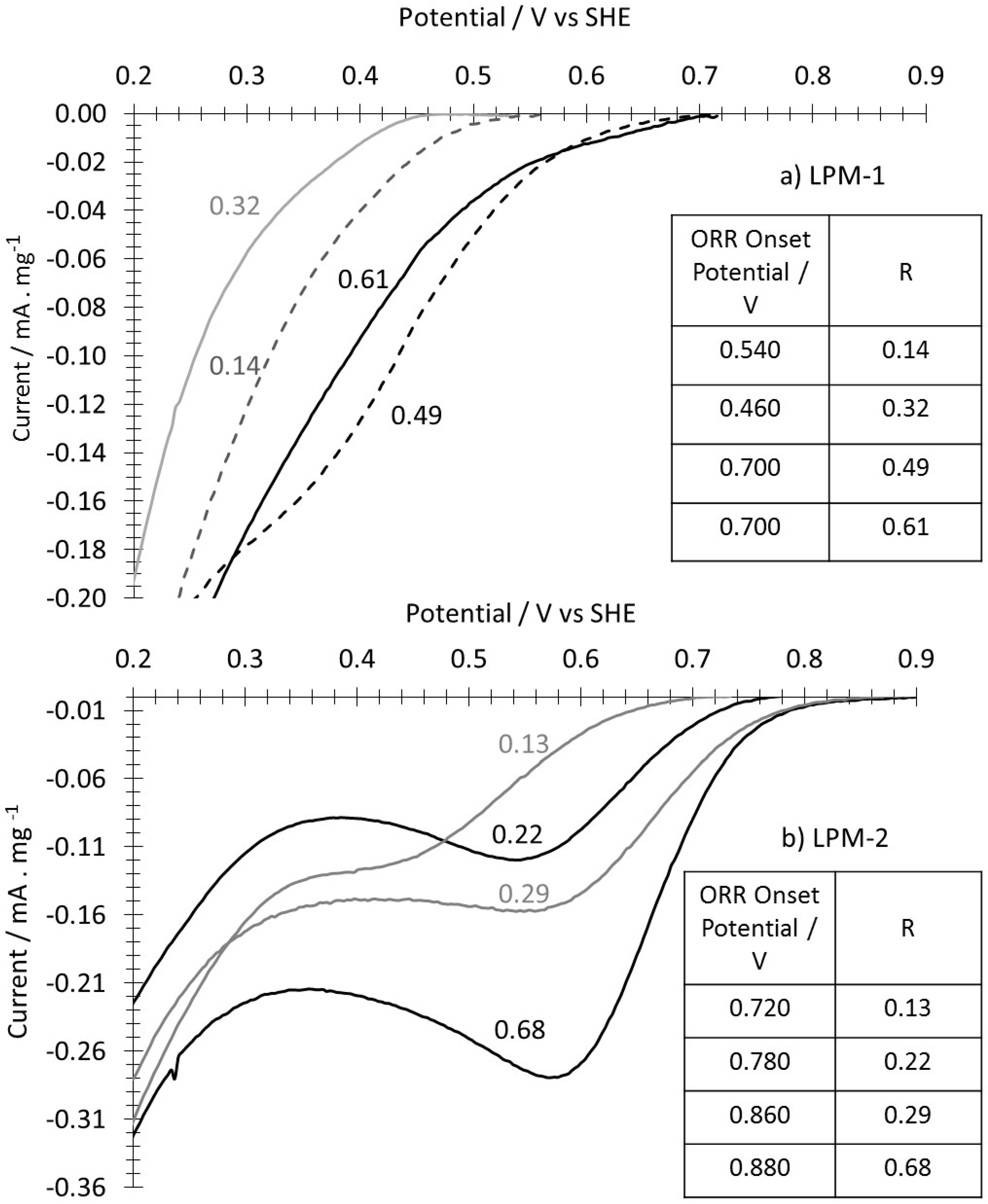
4. Preliminary Oxygen Reduction Study
5. Discussion
- -
- Heating decomposes precursors to monomers which upon saturation grow by molecular coagulation or nucleation, to critical/primary particles.
- -
- Surface reaction of monomers on primary particles can contribute to their growth, while evaporation/sublimation contribute to particle size decrease and forming monomers again.
- -
- Further coagulation and/or coalescence process between primary particles may also contribute to nanoparticles growth.
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Iron-Based Phase | Hyperfine Field—H(T) | Isomer Shift mm·s−1 | Area % |
|---|---|---|---|
| α Fe | 33.59 | 0.02 | 24.3 |
| γ Fe | - | −0.056 | 16.5 |
| Fe3C | 20.90 | 0.196 | 42.8 |
| FeO | - | 0.897 | 5.0 |
| Unknown phase | 28.33 | 0.11 | 11.4 |
Appendix B

Appendix C

References
- Jasinski, R. A New Fuel Cell Cathode Catalyst. Nature 1964, 201, 1212–1213. [Google Scholar] [CrossRef]
- Jahnke, H.; Schönborn, M.; Zimmermann, G. Organic dyestuffs as catalysts for fuel cells. Top. Curr. Chem. 1976, 61, 133–181. [Google Scholar] [PubMed]
- Gupta, S.; Tryk, D.; Bae, I.; Aldred, W.; Yeager, E. Heat-treated polyacrylonitrile-based catalysts for oxygen electroreduction. J. Appl. Electrochem. 1989, 19, 19–27. [Google Scholar] [CrossRef]
- Lefèvre, M.; Proietti, E.; Jaouen, F.; Dodelet, J.-P. Iron-Based Catalysts with Improved Oxygen Reduction Activity in Polymer Electrolyte Fuel Cells. Science 2009, 324, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Chang, Q.; Dodelet, J.-P.; Chenitz, R. Recent advances in electrocatalysts for oxygen reduction. Chem. Rev. 2016, 116, 3594–3657. [Google Scholar] [CrossRef] [PubMed]
- Masa, J.; Xia, W.; Mulher, M.; Schuhmann, W. On the role of Metals in the Nitrogen-doped carbon electrocatalyst for oxygen reduction. Angew. Chem. Int. Ed. 2015, 54, 10102–10120. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Zelenay, P. Nanostructured Nonprecious Metal Catalysts for Oxygen Reduction Reaction. Acc. Chem. Res. 2013, 46, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- Dombrovskis, J.K.; Palmqvist, A.E.C. Recent Progress in Synthesis, Characterization and Evaluation of Non-Precious Metal Catalysts for the Oxygen Reduction Reaction. Fuel Cells 2016, 16, 4–22. [Google Scholar] [CrossRef]
- Liu, J.; Li, E.; Ruan, M.; Song, P.; Xu, W. Recent Progress on Fe/N/C Electrocatalysts for the Oxygen Reduction Reaction in Fuel Cells. Catalysts 2015, 5, 1167–1192. [Google Scholar] [CrossRef]
- Russel, D.K. Infrared laser powered homogeneous pyrolysis. Chem. Soc. Rev. 1990, 19, 407–437. [Google Scholar] [CrossRef]
- David, B.; Scheneeweiss, O.; Pizurova, N.; Klementova, M.; Bezdicka, P.; Alexandrescu, R.; Dimitrache, F.; Morjan, I. Fe3C nanopowder synthesized by laser pyrolysis and its annealing behaviour. Surf. Interface Anal. 2006, 38, 482–485. [Google Scholar] [CrossRef]
- Leconte, Y.; Maskrot, H.; Combemale, L.; Herlin-Boime, N.; Reynaud, C. Application of the laser pyrolysis to the synthesis of SiC, TiC and ZrC pre-ceramics nanopowders. J. Anal. Appl. Pyrolysis 2007, 79, 465–470. [Google Scholar] [CrossRef]
- Grimes, C.A.; Qian, D.; Dickey, E.C.; Allen, J.L.; Eklund, P.C. Laser Pyrolysis fabrication of ferromagnetic γ’-Fe4N and Fe3C nanoparticles. J. Appl. Phys. 2000, 87, 5642–5644. [Google Scholar] [CrossRef] [PubMed]
- Leconte, Y.; Veintemillas-Verdaguer, S.; Morales, M.P.; Costo, R.; Rodríguez, I.; Bonville, P.; Bouchet-Fabre, B.; Herlin-Boime, N. Continuous production of water dispersible carbon–iron nanocomposites by laser pyrolysis: Application as MRI contrasts. J. Colloid Interface Sci. 2007, 313, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Simon, P.; Pignon, B.; Miao, B.; Coste-Leconte, S.; Leconte, Y.; Marguet, S.; Jegou, P.; Bouchet-Fabre, B.; Reynaud, C.; Herlin-Boime, N. N-Doped Titanium Monoxide Nanoparticles with TiO Rock-Salt Structure, Low Energy Band Gap and Visible Light Activity. Chem. Mater. 2010, 22, 3704–3711. [Google Scholar] [CrossRef]
- Bomati-Miguel, O.; Tartaj, P.; Morales, M.P.; Bonville, P.; Golla-Schindler, U.; Zhao, X.Q.; Veintemillas-Verdaguer, S. Core–Shell Iron–Iron Oxide Nanoparticles Synthesized by Laser-Induced Pyrolysis. Small 2006, 2, 1476–1483. [Google Scholar] [CrossRef] [PubMed]
- Alexandrescu, R.; Morjan, I.; Tomescu, A.; Simion, C.; Scarisoreanu, M.; Birjega, R.; Fleaca, C.; Gavrila, L.; Soare, I.; Dumitrache, F. Direct production of a novel iron-based nanocomposite from the laser pyrolysis of Fe(CO)5/MMA mixtures: Structural and sensing properties. J. Nanomater. 2010, 2010, 1–12. [Google Scholar] [CrossRef]
- Sourice, J.; Bordes, A.; Boulineau, A.; Alper, J.P.; Franger, S.; Quinsac, A.; Habert, A.; De Vito, Y.E.; Porcher, W.; Reynaud, C.; et al. Core–shell amorphous silicon–carbon nanoparticles for high performance anodes in lithium ion batteries. J. Power Sources 2016, 328, 527–535. [Google Scholar] [CrossRef]
- Wakizaka, Y.; Shishikura, T. Process for Production and Use of Carbonitride Mixture Particles or Oxycarbonitride Mixture Particles. U.S. Patent 2011/0183234, 28 July 2011. [Google Scholar]
- Cheng, X.; Than, X.-T.; Pinault, M.; Mayne, M.; Reynaud, C.; Vigneron, J.; Etcheberry, A.; Perez, H. Determination of selectivity and specific area related to oxygen reduction reaction as a function of catalyst loading on non-noble metal based electrocatalyst porous electrodes: An example on nitrogen doped carbon nanotube. Electrochim. Acta 2014, 135, 293–300. [Google Scholar] [CrossRef]
- Baret, B.; Aubert, P.-H.; Mayne-L’Hermite, M.; Pinault, M.; Reynaud, C.; Etcheberry, A.; Perez, H. Nanocomposite electrodes based on pre-synthesized organically capped platinum nanoparticles and carbon nanotubes part I: Tuneable low platinum loadings, specific H upd feature and evidence for oxygen reduction. Electrochim. Acta 2009, 54, 5421–5430. [Google Scholar] [CrossRef]
- March, G.; Volatron, F.; Lachaud, F.; Cheng, X.; Baret, B.; Pinault, M.; Etcheberry, A.; Perez, H. Nanocomposite electrodes based on pre-synthesized organically capped platinum nanoparticles and carbon nanotubes. Part II: Determination of diffusion area for oxygen reduction reflects platinum accessibility. Electrochim. Acta 2011, 56, 5151–5157. [Google Scholar] [CrossRef]
- Cheng, X.; Volatron, F.; Pardieu, E.; Borta, A.; Carrot, G.; Reynaud, C.; Mayne, M.; Pinault, M.; Etcheberry, A.; Perez, H. Nanocomposite electrodes based on pre-synthesized organically grafted platinum nanoparticles and carbon nanotubes III. Determination of oxygen reduction reaction selectivity and specific area of porous electrode related to the oxygen reduction reaction ranging from 2 m2·gPt−1 to 310 m2·gPt−1. Electrochim. Acta 2013, 89, 1–12. [Google Scholar] [CrossRef]
- Oya, A.; Marsh, H. Phenomena of catalytic graphitization. J. Mater. Sci. 1982, 17, 309–322. [Google Scholar] [CrossRef]
- Hargreaives, J.S.J. Heterogeneous catalysis with metal nitrides. Coord. Chem. Rev. 2013, 257, 2015–2031. [Google Scholar] [CrossRef]
- Han, Y.; Wang, H.; Zang, M.; Su, M.; Li, W.; Tao, K. Low-temperature approach to synthesize iron nitride from amorphous Iron. Inorg. Chem. 2008, 47, 1261–1263. [Google Scholar] [CrossRef] [PubMed]
- Dieckmann, W.; Panzner, G.; Grabke, H.J. The bonding state of nitrogen segregated on Fe (100) and on iron nitrides Fe4N and Fe2N. Surf. Sci. 1989, 218, 507–518. [Google Scholar] [CrossRef]
- Lin, Y.C.; Hong, J.-Y.; Yen, C.-H.; Tong, S.-Y.; Tung, M.-J.; Shiu, H.-W.; Lin, M.-T. X-ray photelectron spectroscopic investigation on Fe geometrical sites of iron nitride thin films. Jpn. J. Appl. Phys. 2015, 54, 033002. [Google Scholar] [CrossRef]
- Torres, J.; Perry, C.C.; Bransfield, S.J.; Fairbrother, D.H. Low temperature oxidation of nitride iron surfaces. J. Phys. Chem. B 2003, 107, 5558–5567. [Google Scholar] [CrossRef]
- Susi, T.; Pichler, T.; Ayala, P. X-ray photoelectron spectroscopy of graphitic carbon nanomaterials doped with heteroatoms. Beilstein J. Nanotechnol. 2015, 6, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Casolo, S.; Suzuki, T.; Shikano, T.; Sakurai, M.; Harada, Y.; Saito, M.; Oshima, M.; Trioni, M.I.; Tantardini, G.F.; et al. Atomic-scale characterization of nitrogen-doped graphite: Effects of dopant nitrogen on the local electronic structure of the surrounding carbon atoms. Phys. Rev. B 2012, 86, 035436. [Google Scholar] [CrossRef]
- Pels, J.R.; Kapteijn, F.; Moulijn, J.A.; Zhu, Q.; Thomas, K.M. Evolution of nitrogen functionalities in carbonaceous materials during pyrolysis. Carbon 1995, 33, 1641–1653. [Google Scholar] [CrossRef]
- Kapteijn, F.; Moulijn, J.A.; Matzner, S.; Boehm, H.-P. The development of nitrogen functionality in model chars during gasification in CO2 and O2. Carbon 1999, 37, 1143–1150. [Google Scholar] [CrossRef]
- Marton, D.; Boyd, K.J.; Al-Bayati, A.H.; Todorov, S.S.; Rabalais, J.W. Carbon Nitride Deposited Using Energetic Species: A Two-Phase System. Phys. Rev. Lett. 1994, 73, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.C.; Park, J. Distribution and Structure of N Atoms in Multiwalled Carbon Nanotubes Using Variable-Energy X-ray Photoelectron Spectroscopy. J. Phys. Chem. B 2005, 109, 4333–4340. [Google Scholar] [CrossRef] [PubMed]
- Gammon, W.J.; Kraft, O.; Reilly, A.C.; Holloway, B.C. Experimental comparison of N(1s) X-ray photoelectron spectroscopy binding energies of hard and elastic amorphous carbon nitride films with reference organic compounds. Carbon 2003, 41, 1917–1923. [Google Scholar] [CrossRef]
- Artyushkova, K.; Serov, A.; Rojas-Carbonell, S.; Atanassov, P. Chemistry of Multitudinous Active Sites for Oxygen Reduction Reaction in Transition Metal−Nitrogen−Carbon. J. Phys. Chem. C 2015, 119, 25917–25928. [Google Scholar] [CrossRef]
- Casanovas, J.; Ricart, J.M.; Rubio, J.; Illas, F.; Jimenez-Mateos, J.M. Origin of the Large N1s Binding Energy in X-ray Photoelectron Spectra of Calcined Carbonaceous Materials. J. Am. Chem. Soc. 1996, 118, 8071–8076. [Google Scholar] [CrossRef]
- Jimenez Mateos, J.M.; Fierro, J.L.G. X-ray Photoelectron Spectroscopic Study of Petroleum Fuel Cokes. Surf. Int. Anal. 1996, 2, 223–236. [Google Scholar] [CrossRef]
- Buesser, B.; Pratsinis, S.E. Design of Nanomaterial Synthesis by Aerosol Processes. Annu. Rev. Chem. Biomol. Eng. 2012, 3, 103–127. [Google Scholar] [CrossRef] [PubMed]
- Buesser, B.; Gröhn, A.J. Multiscale aspect of modeling gas phase nanoparticle synthesis. Chem. Eng. Technol. 2012, 35, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Kelesidis, G.A.; Goudeli, E.; Pratsinis, S.E. Flame synthesis of functional nanostructured materials and devices: Surface growth and aggregation. Proc. Energy Combust. Sci. 2017, 36, 29–50. [Google Scholar] [CrossRef]
- Strobel, R.; Pratsinis, S.E. Flame aerosol synthesis of smart nanostructured materials. J. Mater. Chem. 2007, 17, 4743–4756. [Google Scholar] [CrossRef]
- Teoh, W.Y.; Amal, R.; Mädler, L. Flame spray pyrolysis: An enabling technology for nanoparticles design and fabrication. Nanoscale 2010, 2, 1324–1347. [Google Scholar] [CrossRef] [PubMed]
- Dobbins, A.; Megaridis, C.M. Morphology of Flame-Generated Soot As Determined by Thermophoretic Sampling. Langmuir 1987, 3, 254–259. [Google Scholar] [CrossRef]
- Castro, C. Mécanismes de Croissance de Nanotubes de Carbone Alignés: Relation Catalyseur—Nanotube. Ph.D. Thesis, Paris XI Orsay University, Orsay, France, 2009. [Google Scholar]
- Jaouen, F.; Proietti, E.; Lefèvre, M.; Chentiz, R.; Dodelet, J.-P.; Wu, G.; Chung, H.T.; Johnston, P.; Zelenay, C.M. Recent advances in non-precious metal catalysis for oxygen-reduction reaction in polymer electrolyte fuel cells. Energy Environ. Sci. 2011, 4, 114–130. [Google Scholar] [CrossRef]
- Jia, Q.; Ramaswamy, N.; Tylus, U.; Strickland, K.; Li, J.; Serov, A.; Artyushkova, K.; Atanassov, P.; Anibal, J.; Gumeci, C.; et al. Spectroscopic insights into the nature of active sites in iron–nitrogen–carbon electrocatalysts for oxygen reduction in acid. Nano Energy 2016, 29, 65–82. [Google Scholar] [CrossRef]
- Jia, Q.; Ramaswamy, N.; Hafiz, H.; Tylus, U.; Strickland, K.; Wu, G.; Barbiellini, B.; Bansil, A.; Holby, E.F.; Zelenay, P.; et al. Experimental Observation of Redox-Induced Fe–N Switching Behavior as a Determinant Role for Oxygen Reduction Activity. ACS Nano 2015, 9, 12496–12505. [Google Scholar] [CrossRef] [PubMed]
- Daems, N.; Sheng, X.; Vankelecom, I.F.J.; Pescarmona, P. Metal-free doped carbon materials as electrocatalysts for the oxygen reduction reaction. J. Mater. Chem. A 2014, 2, 4085–4110. [Google Scholar] [CrossRef]
- Guo, D.; Shibuya, R.; Akiba, C.; Saji, S.; Kondo, T.; Nakamura, J. Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts. Science 2016, 351, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Jensen, J.O.; Zhang, W.; Cleemann, L.N.; Xing, W.; Bjerrum, N.J.; Li, Q. Hollow Spheres of Iron Carbide Nanoparticles Encased in Graphitic Layers as Oxygen Reduction Catalysts. Angew. Chem. Int. Ed. 2014, 53, 3675–3679. [Google Scholar] [CrossRef] [PubMed]
- Kramm, U.I.; Herrman-Geppert, I.; Bogdanoff, P.; Fiechter, S. Effect of an ammonia treatment on structure, composition and oxygen reduction reaction activity of Fe–N–C catalysts. J. Phys. Chem. C 2011, 115, 23417. [Google Scholar] [CrossRef]
- Ron, M.; Mathalone, Z. Hyperfine Interactions of 57Fe in Fe3C. Phys. Rev. B 1971, 4, 774–777. [Google Scholar] [CrossRef]
- Pasternak, M.P.; Taylor, R.D.; Jeanloz, R.; Li, X.; Nguyen, J.H.; McCammon, C.A. High Pressure Collapse of Magnetism in Fe0.94O: Mössbauer Spectroscopy Beyond 100 GPa. Phys. Rev. Lett. 1997, 79, 5046–5049. [Google Scholar] [CrossRef]
- Serov, A.; Artyushkova, K.; Niangar, E.; Wang, C.; Dale, N.; Jaouen, F.; Sougrati, M.-T.; Jia, Q.; Mukerjee, S.; Atanassov, P. Nano-Structured Non-Platinum Catalysts for Automotive Fuel Cell Application. Nano Energy 2015, 16, 293–300. [Google Scholar] [CrossRef]












| LPM-1 | LPM-1 | LPM-2 | LPM-2 | |||||
|---|---|---|---|---|---|---|---|---|
| R = 0.61 | R = 0.14 | R = 0.68 | R = 0.29 | |||||
| N at % | Fe at % | N at % | Fe at % | N at % | Fe at % | N at % | Fe at % | |
| As-prepared materials | 6.7 | 3.7 | 4.6 | 0.05 | 8.1 | 9.4 | 4.9 | 0.6 |
| H2SO4 treated | 2.0 | 0.3 | 4.2 | 0 | 6.0 | 3.0 | 4.2 | 0.3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez, H.; Jorda, V.; Bonville, P.; Vigneron, J.; Frégnaux, M.; Etcheberry, A.; Quinsac, A.; Habert, A.; Leconte, Y. Synthesis and Characterization of Carbon/Nitrogen/Iron Based Nanoparticles by Laser Pyrolysis as Non-Noble Metal Electrocatalysts for Oxygen Reduction. C 2018, 4, 43. https://doi.org/10.3390/c4030043
Perez H, Jorda V, Bonville P, Vigneron J, Frégnaux M, Etcheberry A, Quinsac A, Habert A, Leconte Y. Synthesis and Characterization of Carbon/Nitrogen/Iron Based Nanoparticles by Laser Pyrolysis as Non-Noble Metal Electrocatalysts for Oxygen Reduction. C. 2018; 4(3):43. https://doi.org/10.3390/c4030043
Chicago/Turabian StylePerez, Henri, Virginie Jorda, Pierre Bonville, Jackie Vigneron, Mathieu Frégnaux, Arnaud Etcheberry, Axelle Quinsac, Aurélie Habert, and Yann Leconte. 2018. "Synthesis and Characterization of Carbon/Nitrogen/Iron Based Nanoparticles by Laser Pyrolysis as Non-Noble Metal Electrocatalysts for Oxygen Reduction" C 4, no. 3: 43. https://doi.org/10.3390/c4030043
APA StylePerez, H., Jorda, V., Bonville, P., Vigneron, J., Frégnaux, M., Etcheberry, A., Quinsac, A., Habert, A., & Leconte, Y. (2018). Synthesis and Characterization of Carbon/Nitrogen/Iron Based Nanoparticles by Laser Pyrolysis as Non-Noble Metal Electrocatalysts for Oxygen Reduction. C, 4(3), 43. https://doi.org/10.3390/c4030043