MicroRNA-451 Inhibits Migration of Glioblastoma while Making It More Susceptible to Conventional Therapy

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
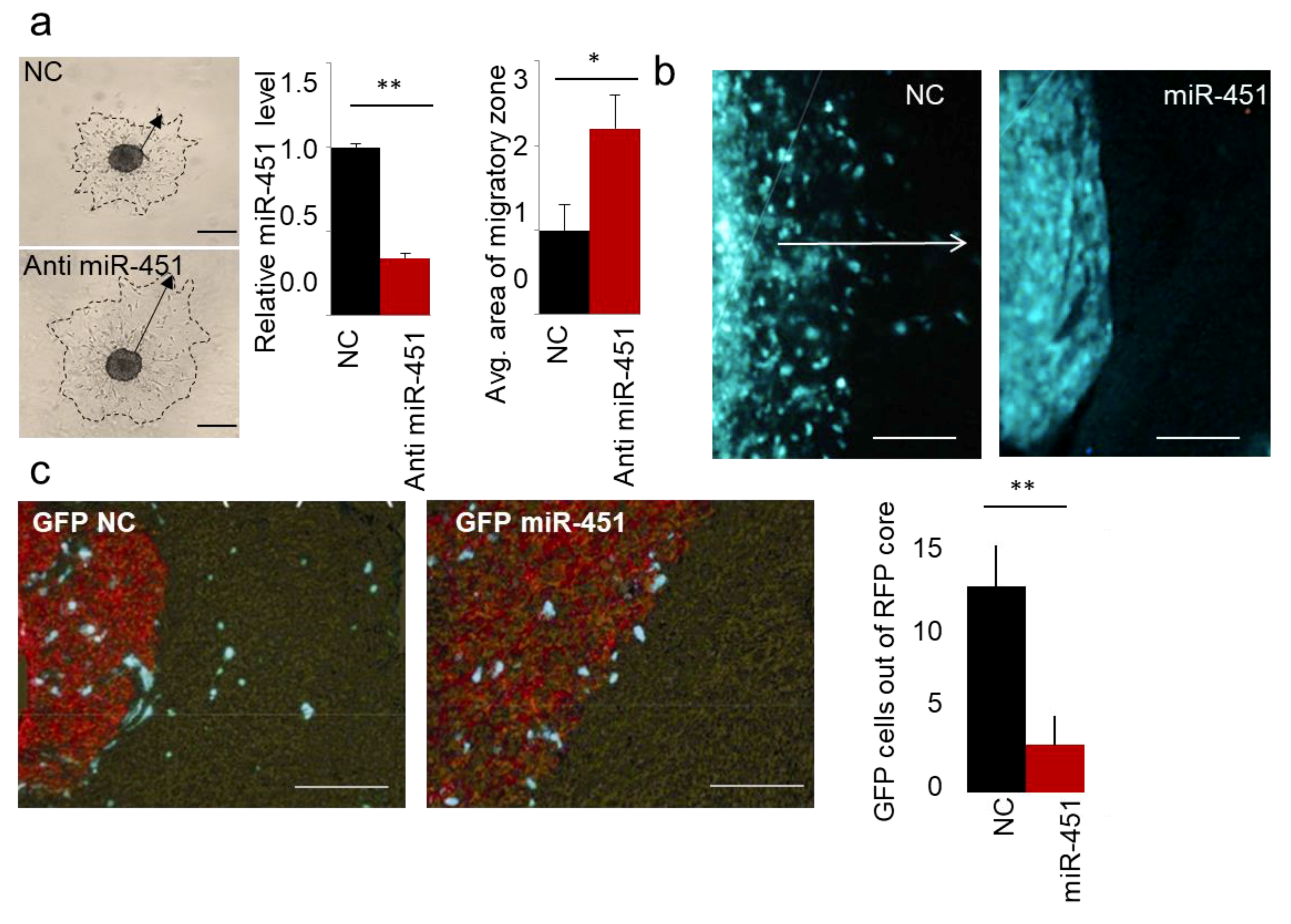
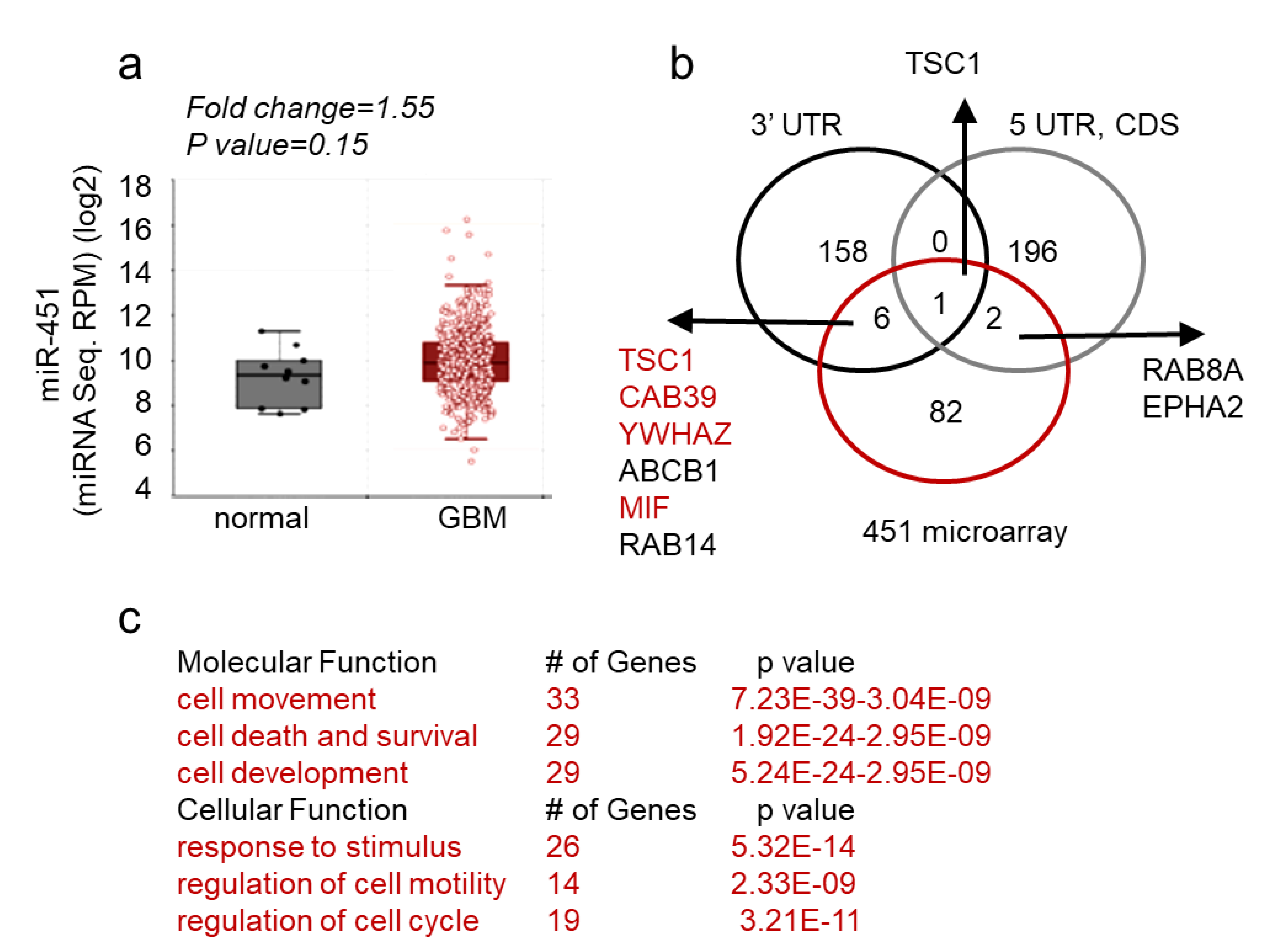
2.1. MicroRNA-451 Is a Potent Inhibitor of GBM Cell Motility/Migration In Vitro and Invasiveness In Vivo
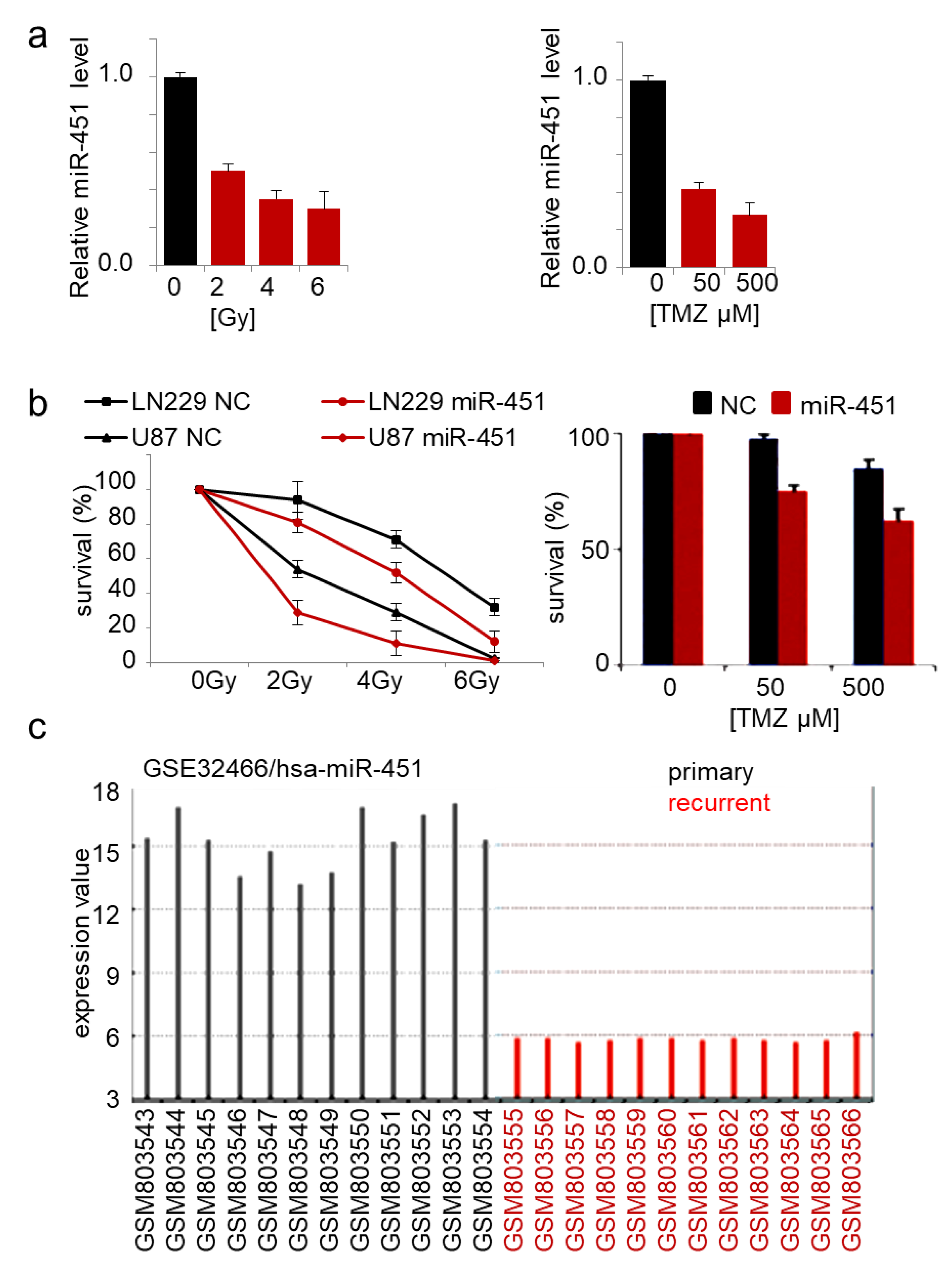
2.2. MicroRNA-451 Sensitizes GBM Cells to Conventional Therapy
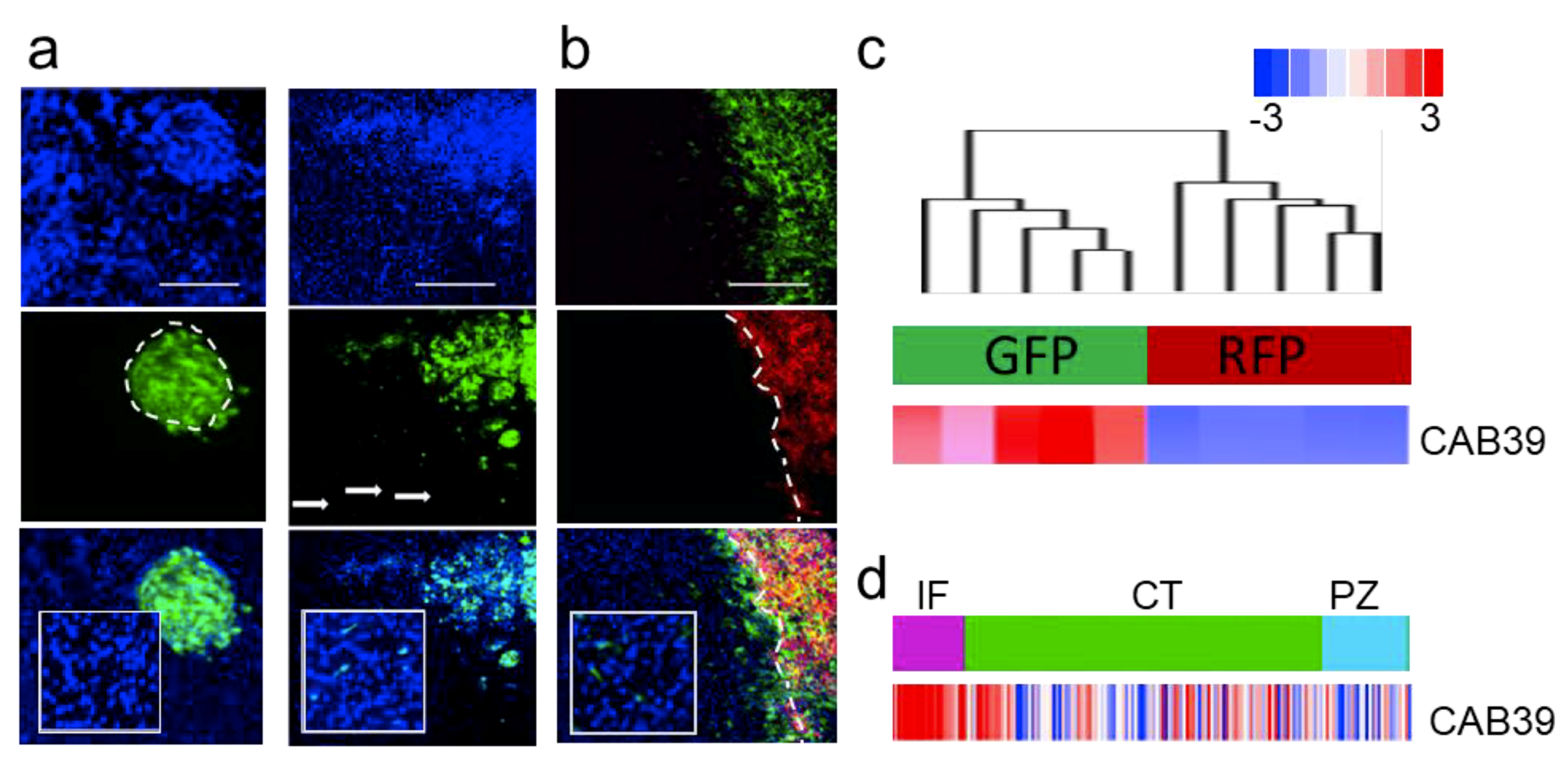
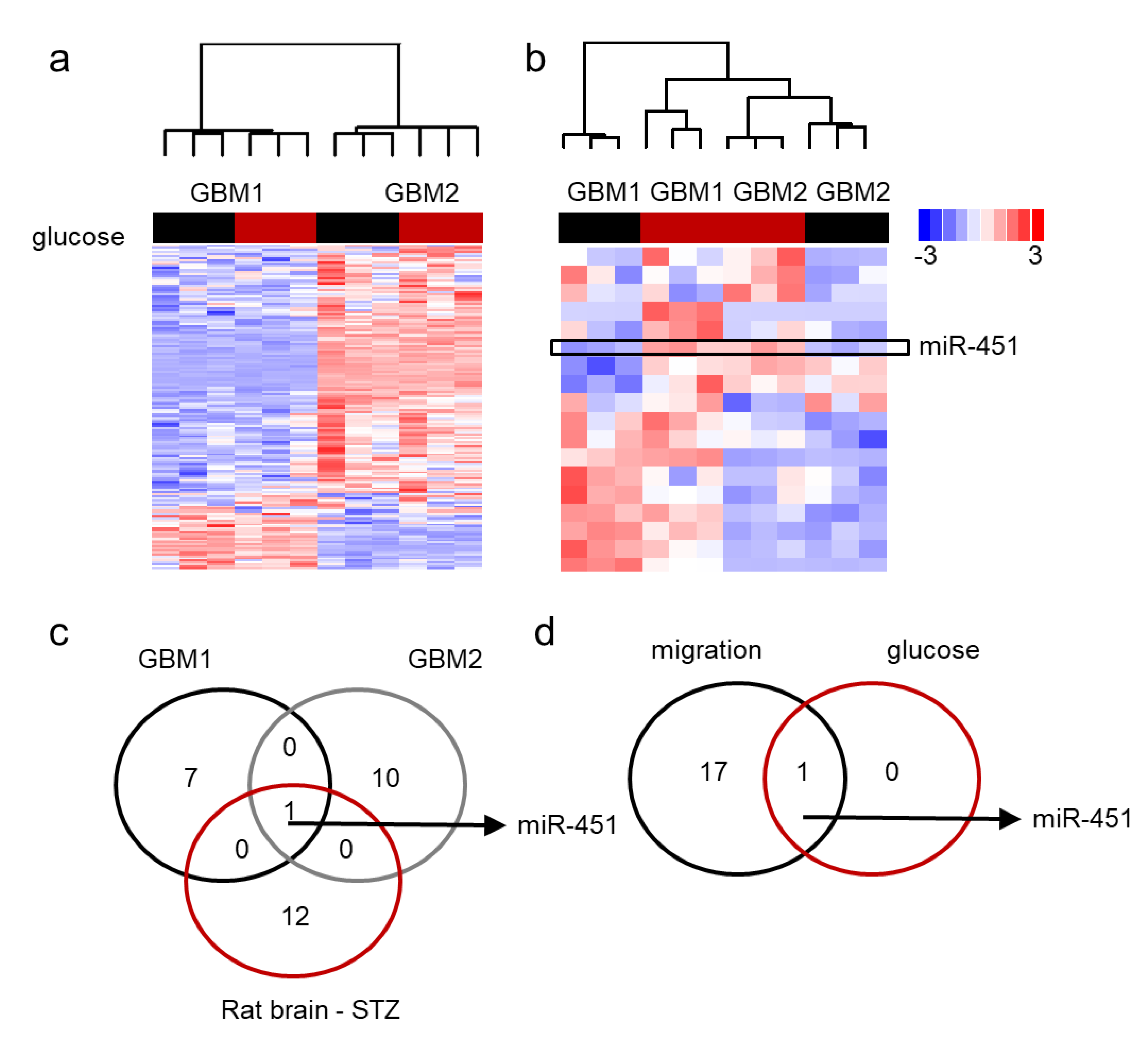
2.3. MiR-451 and Its Effector Network Are Linked to Cellular Response to Stress via AMPK Signaling to Drive the Microenvironmental Adaptation of GBM Cells/GSCs
3. Discussion
4. Materials and Methods
4.1. Statistical Analysis
4.2. Bioinformatic Analysis
4.3. Transcriptome Analysis
4.4. In Vivo Experiments
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Ricklefs, F.; Mineo, M.; Rooj, A.K.; Nakano, I.; Charest, A.; Weissleder, R.; Breakefield, X.O.; Chiocca, E.A.; Godlewski, J.; Bronisz, A. Extracellular Vesicles from High-Grade Glioma Exchange Diverse Pro-oncogenic Signals That Maintain Intratumoral Heterogeneity. Cancer Res. 2016, 76, 2876–2881. [Google Scholar] [CrossRef] [PubMed]
- Mineo, M.; Ricklefs, F.; Rooj, A.K.; Lyons, S.M.; Ivanov, P.; Ansari, K.I.; Nakano, I.; Chiocca, E.A.; Godlewski, J.; Bronisz, A. The Long Non-coding RNA HIF1A-AS2 Facilitates the Maintenance of Mesenchymal Glioblastoma Stem-like Cells in Hypoxic Niches. Cell Rep. 2016, 15, 2500–2509. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Wu, Q.; Hitomi, M.; Rahim, N.; Kim, Y.; Sloan, A.E.; Weil, R.J.; Nakano, I.; Sarkaria, J.N.; Stringer, B.W.; et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nature Neurosci. 2013, 16, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nature Rev. Cancer 2015, 15, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Wen, P.Y. Glioma in 2014: Unravelling tumour heterogeneity-implications for therapy. Nat. Rev. Clin. Oncol. 2015, 12, 69–70. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, T.C.; Bjerkvig, R. Molecular mechanisms of temozolomide resistance in glioblastoma multiforme. Expert Rev. Anticancer Ther. 2012, 12, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Haar, C.P.; Hebbar, P.; Wallace, G.C.T.; Das, A.; Vandergrift, W.A., 3rd; Smith, J.A.; Giglio, P.; Patel, S.J.; Ray, S.K.; Banik, N.L. Drug resistance in glioblastoma: A mini review. Neurochem. Res. 2012, 37, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- Beier, D.; Schulz, J.B.; Beier, C.P. Chemoresistance of glioblastoma cancer stem cells--much more complex than expected. Mol. Cancer 2011, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Squatrito, M.; Holland, E.C. DNA damage response and growth factor signaling pathways in gliomagenesis and therapeutic resistance. Cancer Res. 2011, 71, 5945–5949. [Google Scholar] [CrossRef] [PubMed]
- Burkhardt, J.K.; Shin, B.J.; Boockvar, J.A. Neural stem cells and glioma stem-like cells respond differently to chemotherapeutic drugs: Selectivity at the cellular level. Neurosurgery 2011, 68, N21–N22. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mannino, M.; Chalmers, A.J. Radioresistance of glioma stem cells: Intrinsic characteristic or property of the microenvironment-stem cell unit? Mol. Oncol. 2011, 5, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.; Schweiger, U.; Pellerin, L.; Hubold, C.; Oltmanns, K.M.; Conrad, M.; Schultes, B.; Born, J.; Fehm, H.L. The selfish brain: Competition for energy resources. Neurosci. Biobehav. Rev. 2004, 28, 143–180. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMPK--sensing energy while talking to other signaling pathways. Cell Metab. 2014, 20, 939–952. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.M.; Hay, N. The double-edged sword of AMPK signaling in cancer and its therapeutic implications. Arch. Pharm. Res. 2015, 38, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Sanli, T.; Steinberg, G.R.; Singh, G.; Tsakiridis, T. AMP-activated protein kinase (AMPK) beyond metabolism: A novel genomic stress sensor participating in the DNA damage response pathway. Cancer Biol. Ther. 2014, 15, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.M.; Hay, N. The dark face of AMPK as an essential tumor promoter. Cell. Logist. 2012, 2, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Sanli, T.; Storozhuk, Y.; Linher-Melville, K.; Bristow, R.G.; Laderout, K.; Viollet, B.; Wright, J.; Singh, G.; Tsakiridis, T. Ionizing radiation regulates the expression of AMP-activated protein kinase (AMPK) in epithelial cancer cells: Modulation of cellular signals regulating cell cycle and survival. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2012, 102, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Sanli, T.; Rashid, A.; Liu, C.; Harding, S.; Bristow, R.G.; Cutz, J.C.; Singh, G.; Wright, J.; Tsakiridis, T. Ionizing radiation activates AMP-activated kinase (AMPK): A target for radiosensitization of human cancer cells. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.C.; Mao, M.; de Abreu, A.L.; Ansenberger-Fricano, K.; Ekoue, D.N.; Ganini, D.; Kajdacsy-Balla, A.; Diamond, A.M.; Minshall, R.D.; Consolaro, M.E.; et al. MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat. Commun. 2015, 6, 6053. [Google Scholar] [CrossRef] [PubMed]
- Rios, M.; Foretz, M.; Viollet, B.; Prieto, A.; Fraga, M.; Garcia-Caballero, T.; Costoya, J.A.; Senaris, R. Lipoprotein internalisation induced by oncogenic AMPK activation is essential to maintain glioblastoma cell growth. Eur. J. Cancer 2014, 50, 3187–3197. [Google Scholar] [CrossRef] [PubMed]
- Hindupur, S.K.; Balaji, S.A.; Saxena, M.; Pandey, S.; Sravan, G.S.; Heda, N.; Kumar, M.V.; Mukherjee, G.; Dey, D.; Rangarajan, A. Identification of a novel AMPK-PEA15 axis in the anoikis-resistant growth of mammary cells. Breast Cancer Res. BCR 2014, 16, 420. [Google Scholar] [CrossRef] [PubMed]
- Laderoute, K.R.; Calaoagan, J.M.; Chao, W.R.; Dinh, D.; Denko, N.; Duellman, S.; Kalra, J.; Liu, X.; Papandreou, I.; Sambucetti, L.; et al. 5′-AMP-activated protein kinase (AMPK) supports the growth of aggressive experimental human breast cancer tumors. J. Biol. Chem. 2014, 289, 22850–22864. [Google Scholar] [CrossRef] [PubMed]
- Rios, M.; Foretz, M.; Viollet, B.; Prieto, A.; Fraga, M.; Costoya, J.A.; Senaris, R. AMPK activation by oncogenesis is required to maintain cancer cell proliferation in astrocytic tumors. Cancer Res. 2013, 73, 2628–2638. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, V.W.; Chiu, P.M.; Chan, D.W.; Ngan, H.Y. Over-expressions of AMPK subunits in ovarian carcinomas with significant clinical implications. BMC Cancer 2012, 12, 357. [Google Scholar] [CrossRef] [PubMed]
- Park, H.U.; Suy, S.; Danner, M.; Dailey, V.; Zhang, Y.; Li, H.; Hyduke, D.R.; Collins, B.T.; Gagnon, G.; Kallakury, B.; et al. AMP-activated protein kinase promotes human prostate cancer cell growth and survival. Mol. Cancer Ther. 2009, 8, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ulbrich, J.; Muller, J.; Wustefeld, T.; Aeberhard, L.; Kress, T.R.; Muthalagu, N.; Rycak, L.; Rudalska, R.; Moll, R.; et al. Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nature 2012, 483, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Zhong, D.; Liu, X.; Khuri, F.R.; Sun, S.Y.; Vertino, P.M.; Zhou, W. LKB1 is necessary for Akt-mediated phosphorylation of proapoptotic proteins. Cancer Res. 2008, 68, 7270–7277. [Google Scholar] [CrossRef] [PubMed]
- Laderoute, K.R.; Amin, K.; Calaoagan, J.M.; Knapp, M.; Le, T.; Orduna, J.; Foretz, M.; Viollet, B. 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol. Cell. Biol. 2006, 26, 5336–5347. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Lu, J.; Kusakai, G.; Kishimoto, A.; Ogura, T.; Esumi, H. ARK5 is a tumor invasion-associated factor downstream of Akt signaling. Mol. Cell. Biol. 2004, 24, 3526–3535. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Jimnez, F.J.; Alastrue-Agudo, A.; Erceg, S.; Stojkovic, M.; Moreno-Manzano, V. FM19G11 favors spinal cord injury regeneration and stem cell self-renewal by mitochondrial uncoupling and glucose metabolism induction. Stem Cells 2012, 30, 2221–2233. [Google Scholar] [CrossRef] [PubMed]
- Zeve, D.; Seo, J.; Suh, J.M.; Stenesen, D.; Tang, W.; Berglund, E.D.; Wan, Y.; Williams, L.J.; Lim, A.; Martinez, M.J.; et al. Wnt signaling activation in adipose progenitors promotes insulin-independent muscle glucose uptake. Cell Metab. 2012, 15, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Ge, W.; Ren, J. mTOR-STAT3-notch signalling contributes to ALDH2-induced protection against cardiac contractile dysfunction and autophagy under alcoholism. J. Cell. Mol. Med. 2012, 16, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Connolly, N.M.; Dussmann, H.; Anilkumar, U.; Huber, H.J.; Prehn, J.H. Single-cell imaging of bioenergetic responses to neuronal excitotoxicity and oxygen and glucose deprivation. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 10192–10205. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Vellon, L.; Oliveras-Ferraros, C.; Cufi, S.; Vazquez-Martin, A. mTOR-regulated senescence and autophagy during reprogramming of somatic cells to pluripotency: A roadmap from energy metabolism to stem cell renewal and aging. Cell Cycle 2011, 10, 3658–3677. [Google Scholar] [CrossRef] [PubMed]
- Zaugg, K.; Yao, Y.; Reilly, P.T.; Kannan, K.; Kiarash, R.; Mason, J.; Huang, P.; Sawyer, S.K.; Fuerth, B.; Faubert, B.; et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011, 25, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Weisova, P.; Concannon, C.G.; Devocelle, M.; Prehn, J.H.; Ward, M.W. Regulation of glucose transporter 3 surface expression by the AMP-activated protein kinase mediates tolerance to glutamate excitation in neurons. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 2997–3008. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Pan, D.A. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem. Soc. Trans. 2002, 30, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chhipa, R.R.; Nakano, I.; Dasgupta, B. The AMPK inhibitor compound C is a potent AMPK-independent antiglioma agent. Mol. Cancer Ther. 2014, 13, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D. Adaptation to starvation: Translating a matter of life or death. Cancer Cell 2013, 23, 713–715. [Google Scholar] [CrossRef] [PubMed]
- Leprivier, G.; Remke, M.; Rotblat, B.; Dubuc, A.; Mateo, A.R.; Kool, M.; Agnihotri, S.; El-Naggar, A.; Yu, B.; Somasekharan, S.P.; et al. The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell 2013, 153, 1064–1079. [Google Scholar] [CrossRef] [PubMed]
- Jang, T.; Calaoagan, J.M.; Kwon, E.; Samuelsson, S.; Recht, L.; Laderoute, K.R. 5′-AMP-activated protein kinase activity is elevated early during primary brain tumor development in the rat. Int. J. Cancer 2011, 128, 2230–2239. [Google Scholar] [CrossRef] [PubMed]
- Qiu, B.; Simon, M.C. Oncogenes strike a balance between cellular growth and homeostasis. Semin. Cell Dev. Biol. 2015, 43, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Di Leva, G.; Croce, C.M. The Role of microRNAs in the Tumorigenesis of Ovarian Cancer. Front. Oncol. 2013, 3, 153. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, M.; Croce, C.M. MicroRNAs as therapeutic targets in chemoresistance. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2013, 16, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Croce, C.M. microRNA involvement in human cancer. Carcinogenesis 2012, 33, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Bronisz, A.; Wang, Y.; Nowicki, M.O.; Peruzzi, P.; Ansari, K.; Ogawa, D.; Balaj, L.; De Rienzo, G.; Mineo, M.; Nakano, I.; et al. Extracellular vesicles modulate the glioblastoma microenvironment via a tumor suppression signaling network directed by miR-1. Cancer Res. 2014, 74, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Peruzzi, P.; Bronisz, A.; Nowicki, M.O.; Wang, Y.; Ogawa, D.; Price, R.; Nakano, I.; Kwon, C.H.; Hayes, J.; Lawler, S.E.; et al. MicroRNA-128 coordinately targets Polycomb Repressor Complexes in glioma stem cells. Neuro-Oncol. 2013, 15, 1212–1224. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Williams, S.; Otsuki, A.; Nuovo, G.; Raychaudhury, A.; Newton, H.B.; Chiocca, E.A.; Lawler, S. Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res. 2008, 68, 9125–9130. [Google Scholar] [CrossRef] [PubMed]
- Kefas, B.; Comeau, L.; Floyd, D.H.; Seleverstov, O.; Godlewski, J.; Schmittgen, T.; Jiang, J.; diPierro, C.G.; Li, Y.; Chiocca, E.A.; et al. The neuronal microRNA miR-326 acts in a feedback loop with notch and has therapeutic potential against brain tumors. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 15161–15168. [Google Scholar] [CrossRef] [PubMed]
- Kefas, B.; Godlewski, J.; Comeau, L.; Li, Y.; Abounader, R.; Hawkinson, M.; Lee, J.; Fine, H.; Chiocca, E.A.; Lawler, S.; et al. microRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma. Cancer Res. 2008, 68, 3566–3572. [Google Scholar] [CrossRef] [PubMed]
- Teplyuk, N.M.; Uhlmann, E.J.; Wong, A.H.; Karmali, P.; Basu, M.; Gabriely, G.; Jain, A.; Wang, Y.; Chiocca, E.A.; Stephens, R.; et al. MicroRNA-10b inhibition reduces E2F1-mediated transcription and miR-15/16 activity in glioblastoma. Oncotarget 2015, 6, 3770–3783. [Google Scholar] [CrossRef] [PubMed]
- Guessous, F.; Alvarado-Velez, M.; Marcinkiewicz, L.; Zhang, Y.; Kim, J.; Heister, S.; Kefas, B.; Godlewski, J.; Schiff, D.; Purow, B.; et al. Oncogenic effects of miR-10b in glioblastoma stem cells. J. Neuro-Oncol. 2013, 112, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, J.; Krichevsky, A.M.; Johnson, M.D.; Chiocca, E.A.; Bronisz, A. Belonging to a network--microRNAs, extracellular vesicles, and the glioblastoma microenvironment. Neuro-Oncol. 2015, 17, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, J.; Newton, H.B.; Chiocca, E.A.; Lawler, S.E. MicroRNAs and glioblastoma; the stem cell connection. Cell Death Differ. 2010, 17, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Bronisz, A.; Godlewski, J.; Wallace, J.A.; Merchant, A.S.; Nowicki, M.O.; Mathsyaraja, H.; Srinivasan, R.; Trimboli, A.J.; Martin, C.K.; Li, F.; et al. Reprogramming of the tumour microenvironment by stromal PTEN-regulated miR-320. Nat. Cell Biol. 2011, 14, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Bronisz, A.; Chiocca, E.A.; Godlewski, J. Response to energy depletion: miR-451/AMPK loop. Oncotarget 2015, 6, 17851–17852. [Google Scholar] [CrossRef] [PubMed]
- Ansari, K.I.; Ogawa, D.; Rooj, A.K.; Lawler, S.E.; Krichevsky, A.M.; Johnson, M.D.; Chiocca, E.A.; Bronisz, A.; Godlewski, J. Glucose-based regulation of miR-451/AMPK signaling depends on the OCT1 transcription factor. Cell Rep. 2015, 11, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, J.; Bronisz, A.; Nowicki, M.O.; Chiocca, E.A.; Lawler, S. microRNA-451: A conditional switch controlling glioma cell proliferation and migration. Cell Cycle 2010, 9, 2742–2748. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Nuovo, G.; Palatini, J.; De Lay, M.; Van Brocklyn, J.; Ostrowski, M.C.; Chiocca, E.A.; Lawler, S.E. MicroRNA-451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in glioma cells. Mol. Cell 2010, 37, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Nowicki, M.O.; Dmitrieva, N.; Stein, A.M.; Cutter, J.L.; Godlewski, J.; Saeki, Y.; Nita, M.; Berens, M.E.; Sander, L.M.; Newton, H.B.; et al. Lithium inhibits invasion of glioma cells; possible involvement of glycogen synthase kinase-3. Neuro-Oncol. 2008, 10, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Babapoor, S.; Fleming, E.; Wu, R.; Dadras, S.S. A novel miR-451a isomiR, associated with amelanotypic phenotype, acts as a tumor suppressor in melanoma by retarding cell migration and invasion. PLoS ONE 2014, 9, e107502. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Jiang, N.; Guo, R.; Jiang, W.; He, Q.M.; Xu, Y.F.; Li, Y.Q.; Tang, L.L.; Mao, Y.P.; Sun, Y.; et al. MiR-451 inhibits cell growth and invasion by targeting MIF and is associated with survival in nasopharyngeal carcinoma. Mol. Cancer 2013, 12, 123. [Google Scholar] [CrossRef] [PubMed]
- Rajasinghe, L.D.; Pindiprolu, R.H.; Gupta, S.V. Delta-tocotrienol inhibits non-small-cell lung cancer cell invasion via the inhibition of NF-kappaB, uPA activator, and MMP-9. OncoTargets Ther. 2018, 11, 4301–4314. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.Y.; Cui, J.Y.; Yuan, J.; Wang, X. MiR-451a suppressed cell migration and invasion in non-small cell lung cancer through targeting ATF2. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 5554–5561. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, X.; Shi, J.; Pan, Y.; Chen, Q.; Leng, P.; Wang, Y. miR-451 suppresses bladder cancer cell migration and invasion via directly targeting c-Myc. Oncol. Rep. 2016, 36, 2049–2058. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Peng, L.; Chao, C.; Fu, B.; Wang, G.; Wang, Y.; Zhu, X. miR-451 inhibits invasion and proliferation of bladder cancer by regulating EMT. Int. J. Clin. Exp. Pathol. 2014, 7, 7653–7662. [Google Scholar] [PubMed]
- Godlewski, J.; Ferrer-Luna, R.; Rooj, A.K.; Mineo, M.; Ricklefs, F.; Takeda, Y.S.; Nowicki, M.O.; Salinska, E.; Nakano, I.; Lee, H.; et al. MicroRNA Signatures and Molecular Subtypes of Glioblastoma: The Role of Extracellular Transfer. Stem Cell Rep. 2017, 8, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.Y.; Zhang, K.; Chen, D.Q.; Chen, J.; Feng, B.; Song, H.; Chen, Y.; Zhu, Z.; Lu, L.; De, W.; et al. MicroRNA-451: Epithelial-mesenchymal transition inhibitor and prognostic biomarker of hepatocelluar carcinoma. Oncotarget 2015, 6, 18613–18630. [Google Scholar] [CrossRef] [PubMed]
- Alural, B.; Ayyildiz, Z.O.; Tufekci, K.U.; Genc, S.; Genc, K. Erythropoietin Promotes Glioblastoma via miR-451 Suppression. Vitam. Horm. 2017, 105, 249–271. [Google Scholar] [CrossRef] [PubMed]
- Kovalchuk, O.; Filkowski, J.; Meservy, J.; Ilnytskyy, Y.; Tryndyak, V.P.; Chekhun, V.F.; Pogribny, I.P. Involvement of microRNA-451 in resistance of the MCF-7 breast cancer cells to chemotherapeutic drug doxorubicin. Mol. Cancer Ther. 2008, 7, 2152–2159. [Google Scholar] [CrossRef] [PubMed]
- Gal, H.; Pandi, G.; Kanner, A.A.; Ram, Z.; Lithwick-Yanai, G.; Amariglio, N.; Rechavi, G.; Givol, D. MIR-451 and Imatinib mesylate inhibit tumor growth of Glioblastoma stem cells. Biochem. Biophys. Res. Commun. 2008, 376, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Bian, H.B.; Pan, X.; Yang, J.S.; Wang, Z.X.; De, W. Upregulation of microRNA-451 increases cisplatin sensitivity of non-small cell lung cancer cell line (A549). J. Exp. Clin. Cancer Res. CR 2011, 30, 20. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Tian, H.; Zhang, Y.; Zhao, H.; Ma, K. miR-451 selectively increases sensitivity to cisplatin in ERCC1-high non-small cell lung cancer cells. J. Cell. Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Xu, Y.; Sun, C.; He, Z. MicroRNA-451 sensitizes lung cancer cells to cisplatin through regulation of Mcl-1. Mol. Cell. Biochem. 2016, 423, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chen, D.Q.; Huang, J.Y.; Zhang, K.; Feng, B.; Pan, B.Z.; Chen, J.; De, W.; Chen, L.B. Acquisition of radioresistance in docetaxel-resistant human lung adenocarcinoma cells is linked with dysregulation of miR-451/c-Myc-survivin/rad-51 signaling. Oncotarget 2014, 5, 6113–6129. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, A.; Katzenellenbogen, B.S. Tamoxifen downregulation of miR-451 increases 14-3-3zeta and promotes breast cancer cell survival and endocrine resistance. Oncogene 2012, 31, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, L.; Wang, Y.; Ding, Y.; Chen, T.; Wang, Y.; Wang, H.; Li, Y.; Duan, K.; Chen, S.; et al. Involvement of miR-451 in resistance to paclitaxel by regulating YWHAZ in breast cancer. Cell Death Dis. 2017, 8, e3071. [Google Scholar] [CrossRef] [PubMed]
- Bitarte, N.; Bandres, E.; Boni, V.; Zarate, R.; Rodriguez, J.; Gonzalez-Huarriz, M.; Lopez, I.; Javier Sola, J.; Alonso, M.M.; Fortes, P.; et al. MicroRNA-451 is involved in the self-renewal, tumorigenicity, and chemoresistance of colorectal cancer stem cells. Stem Cells 2011, 29, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Trog, D.; Yeghiazaryan, K.; Fountoulakis, M.; Friedlein, A.; Moenkemann, H.; Haertel, N.; Schueller, H.; Breipohl, W.; Schild, H.; Leppert, D.; et al. Pro-invasive gene regulating effect of irradiation and combined temozolomide-radiation treatment on surviving human malignant glioma cells. Eur. J. Pharmacol. 2006, 542, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Wild-Bode, C.; Weller, M.; Rimner, A.; Dichgans, J.; Wick, W. Sublethal irradiation promotes migration and invasiveness of glioma cells: Implications for radiotherapy of human glioblastoma. Cancer Res. 2001, 61, 2744–2750. [Google Scholar] [PubMed]
- Lenzen, S. The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia 2008, 51, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Su, M.; Wang, S.; Zou, Y.; Wang, X.; Wang, Y.; Cui, H.; Zhao, P.; Hui, R.; Wang, J. MiR-451 is decreased in hypertrophic cardiomyopathy and regulates autophagy by targeting TSC1. J. Cell. Mol. Med. 2014, 18, 2266–2274. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; dos Santos, C.O.; Zhao, G.; Jiang, J.; Amigo, J.D.; Khandros, E.; Dore, L.C.; Yao, Y.; D’Souza, J.; Zhang, Z.; et al. miR-451 protects against erythroid oxidant stress by repressing 14-3-3zeta. Genes Dev. 2010, 24, 1620–1633. [Google Scholar] [CrossRef] [PubMed]
- Patrick, D.M.; Zhang, C.C.; Tao, Y.; Yao, H.; Qi, X.; Schwartz, R.J.; Jun-Shen Huang, L.; Olson, E.N. Defective erythroid differentiation in miR-451 mutant mice mediated by 14-3-3zeta. Genes Dev. 2010, 24, 1614–1619. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMPK: Positive and negative regulation, and its role in whole-body energy homeostasis. Curr. Opin. Cell Biol. 2015, 33, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Shakya, A.; Tantin, D. Stem cells, stress, metabolism and cancer: A drama in two Octs. Trends Biochem. Sci. 2009, 34, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Boudeau, J.; Reid, J.L.; Mustard, K.J.; Udd, L.; Makela, T.P.; Alessi, D.R.; Hardie, D.G. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2003, 2, 28. [Google Scholar] [CrossRef] [PubMed]
- Lizcano, J.M.; Goransson, O.; Toth, R.; Deak, M.; Morrice, N.A.; Boudeau, J.; Hawley, S.A.; Udd, L.; Makela, T.P.; Hardie, D.G.; et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. The EMBO journal 2004, 23, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Andreelli, F. AMP-activated protein kinase and metabolic control. Handb. Exp. Pharmacol. 2011, 303–330. [Google Scholar] [CrossRef]
- de Los Reyes, V.A.; Jung, E.; Kim, Y. Optimal control strategies of eradicating invisible glioblastoma cells after conventional surgery. J. R. Soc. Interface 2015, 12. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Kemp, B.E. AMPK in Health and Disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Powathil, G.; Kang, H.; Trucu, D.; Kim, H.; Lawler, S.; Chaplain, M. Strategies of eradicating glioma cells: A multi-scale mathematical model with MiR-451-AMPK-mTOR control. PLoS ONE 2015, 10, e0114370. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y. Regulation of cell proliferation and migration in glioblastoma: New therapeutic approach. Front. Oncol. 2013, 3, 53. [Google Scholar] [CrossRef] [PubMed]
- Schuetz, T.A.; Becker, S.; Mang, A.; Toma, A.; Buzug, T.M. A computational multiscale model of glioblastoma growth: Regulation of cell migration and proliferation via microRNA-451, LKB1 and AMPK. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2012, 2012, 6620–6623. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Roh, S.; Lawler, S.; Friedman, A. miR451 and AMPK mutual antagonism in glioma cell migration and proliferation: A mathematical model. PLoS ONE 2011, 6, e28293. [Google Scholar] [CrossRef] [PubMed]
- Shakya, A.; Cooksey, R.; Cox, J.E.; Wang, V.; McClain, D.A.; Tantin, D. Oct1 loss of function induces a coordinate metabolic shift that opposes tumorigenicity. Nat. Cell Biol. 2009, 11, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Almeida, R.; Almeida, J.; Shoshkes, M.; Mendes, N.; Mesquita, P.; Silva, E.; Van Seuningen, I.; Reis, C.A.; Santos-Silva, F.; David, L. OCT-1 is over-expressed in intestinal metaplasia and intestinal gastric carcinomas and binds to, but does not transactivate, CDX2 in gastric cells. J. Pathol. 2005, 207, 396–401. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jin, T.; Branch, D.R.; Zhang, X.; Qi, S.; Youngson, B.; Goss, P.E. Examination of POU homeobox gene expression in human breast cancer cells. Int. J. Cancer 1999, 81, 104–112. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Goodman, B.; Zheng, Y.; Tantin, D. Dynamic regulation of Oct1 during mitosis by phosphorylation and ubiquitination. PLoS ONE 2011, 6, e23872. [Google Scholar] [CrossRef] [PubMed]
- Rooj, A.K.; Ricklefs, F.; Mineo, M.; Nakano, I.; Chiocca, E.A.; Bronisz, A.; Godlewski, J. MicroRNA-Mediated Dynamic Bidirectional Shift between the Subclasses of Glioblastoma Stem-like Cells. Cell Rep. 2017, 19, 2026–2032. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nowicki, M.O.; Wang, X.; Arnold, W.D.; Fernandez, S.A.; Mo, X.; Wechuk, J.; Krisky, D.; Goss, J.; Wolfe, D.; et al. Comparative effectiveness of antinociceptive gene therapies in animal models of diabetic neuropathic pain. Gene Ther. 2013, 20, 742–750. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Celiku, O.; Johnson, S.; Zhao, S.; Camphausen, K.; Shankavaram, U. Visualizing molecular profiles of glioblastoma with GBM-BioDP. PLoS ONE 2014, 9, e101239. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Han, C.; Lu, D.; Wu, T. miR-17-92 cluster promotes cholangiocarcinoma growth: Evidence for PTEN as downstream target and IL-6/Stat3 as upstream activator. Am. J. Pathol. 2014, 184, 2828–2839. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogawa, D.; Ansari, K.; Nowicki, M.O.; Salińska, E.; Bronisz, A.; Godlewski, J. MicroRNA-451 Inhibits Migration of Glioblastoma while Making It More Susceptible to Conventional Therapy. Non-Coding RNA 2019, 5, 25. https://doi.org/10.3390/ncrna5010025
Ogawa D, Ansari K, Nowicki MO, Salińska E, Bronisz A, Godlewski J. MicroRNA-451 Inhibits Migration of Glioblastoma while Making It More Susceptible to Conventional Therapy. Non-Coding RNA. 2019; 5(1):25. https://doi.org/10.3390/ncrna5010025
Chicago/Turabian StyleOgawa, Daisuke, Khairul Ansari, Michal O. Nowicki, Elżbieta Salińska, Agnieszka Bronisz, and Jakub Godlewski. 2019. "MicroRNA-451 Inhibits Migration of Glioblastoma while Making It More Susceptible to Conventional Therapy" Non-Coding RNA 5, no. 1: 25. https://doi.org/10.3390/ncrna5010025
APA StyleOgawa, D., Ansari, K., Nowicki, M. O., Salińska, E., Bronisz, A., & Godlewski, J. (2019). MicroRNA-451 Inhibits Migration of Glioblastoma while Making It More Susceptible to Conventional Therapy. Non-Coding RNA, 5(1), 25. https://doi.org/10.3390/ncrna5010025