Honey as a Source of Environmental DNA for the Detection and Monitoring of Honey Bee Pathogens and Parasites



Abstract
1. Introduction
2. Materials and Methods
2.1. Honey Samples
2.2. DNA Extraction from Honey Samples
2.3. PCR Analyses
2.4. Sequencing and Data Analysis
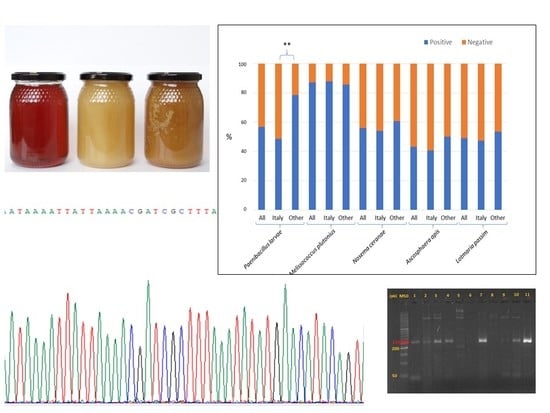
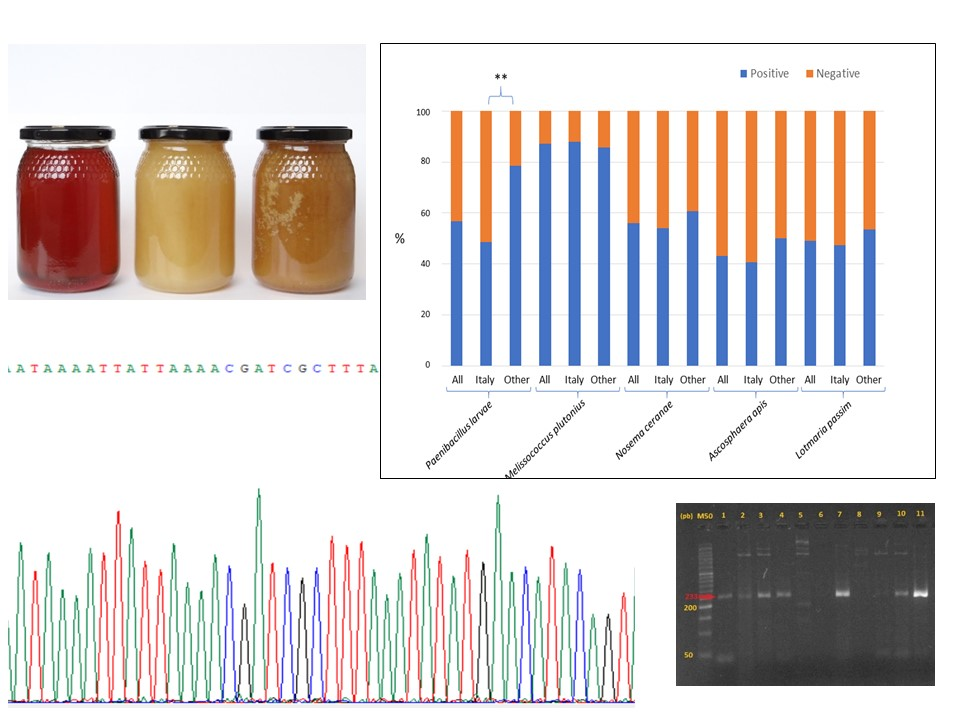
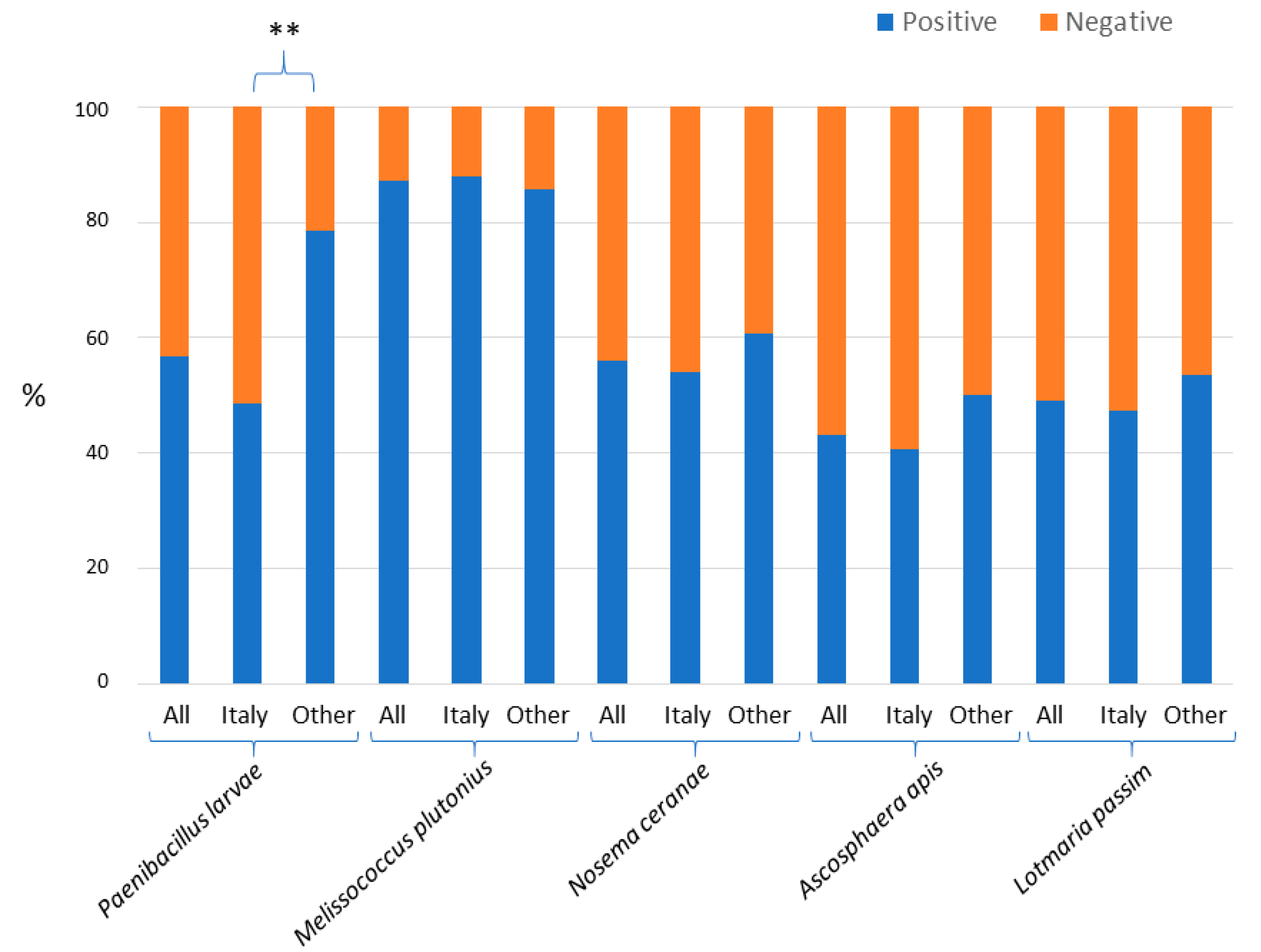
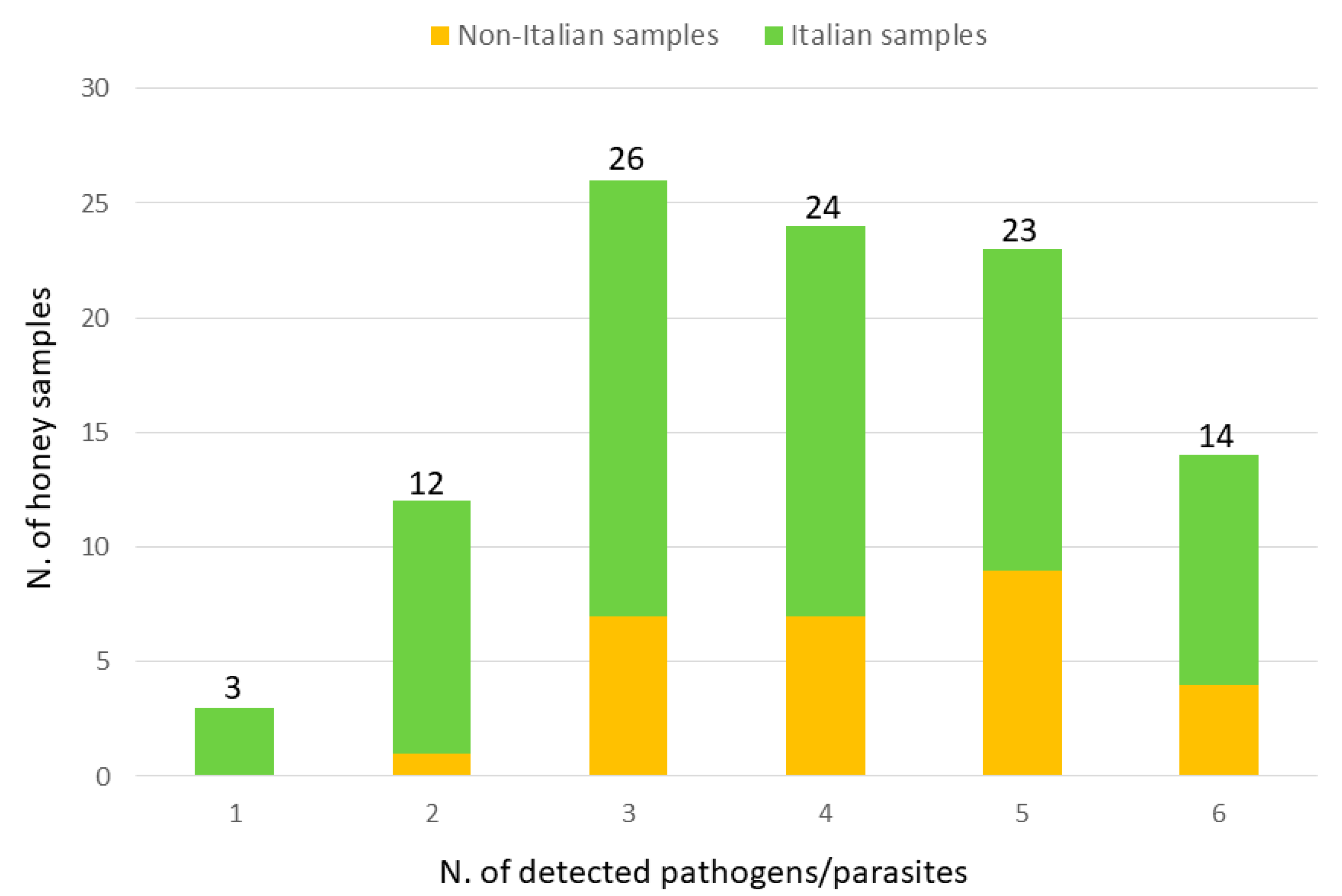
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Taberlet, P.; Coissac, E.; Hajibabaei, M.; Rieseberg, L.H. Environmental DNA. Mol. Ecol. 2012, 21, 1789–1793. [Google Scholar] [CrossRef] [PubMed]
- Bohmann, K.; Evans, A.; Gilbert, M.T.; Carvalho, G.R.; Creer, S.; Knapp, M.; Yu, D.W.; De Bruyn, M. Environmental DNA for wildlife biology and biodiversity monitoring. Trends Ecol. Evol. 2014, 29, 358–367. [Google Scholar] [CrossRef]
- Jerde, C.L.; Mahon, A.R.; Chadderton, W.L.; Lodge, D.M. “Sight-unseen” detection of rare aquatic species using environmental DNA. Conserv. Lett. 2011, 4, 150–157. [Google Scholar] [CrossRef]
- Thomsen, P.F.; Kielgast, J.; Iversen, L.L.; Wiuf, C.; Rasmussen, M.; Gilbert, M.T.; Orlando, L.; Willerslev, E. Monitoring endangered freshwater biodiversity using environmental DNA. Mol. Ecol. 2012, 21, 2565–2573. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, T.M.; McKelvey, K.S.; Young, M.K.; Jane, S.F.; Lowe, W.H.; Whiteley, A.R.; Schwartz, M.K. Robust detection of rare species using environmental DNA: The importance of primer specificity. PLoS ONE 2013, 8, e59520. [Google Scholar] [CrossRef] [PubMed]
- Bovo, S.; Ribani, A.; Utzeri, V.J.; Schiavo, G.; Bertolini, F.; Fontanesi, L. Shotgun metagenomics of honey DNA: Evaluation of a methodological approach to describe a multi-kingdom honey bee derived environmental DNA signature. PLoS ONE 2018, 13, e0205575. [Google Scholar] [CrossRef]
- Bovo, S.; Utzeri, V.J.; Ribani, A.; Cabbri, R.; Fontanesi, L. Shotgun sequencing of honey DNA can describe honey bee derived environmental signatures and the honey bee hologenome complexity. Sci. Rep. 2020, 10, 9279. [Google Scholar] [CrossRef]
- Dainat, B.; Evans, J.D.; Chen, Y.P.; Gauthier, L.; Neumann, P. Predictive markers of honey bee colony collapse. PLoS ONE 2012, 7, e32151. [Google Scholar] [CrossRef]
- Lee, K.; Steinhauer, N.; Travis, D.A.; Meixner, M.D.; Deen, J. Honey bee surveillance: A tool for understanding and improving honey bee health. Curr. Opin. Insect Sci. 2015, 10, 37–44. [Google Scholar] [CrossRef]
- Ward, L.; Brown, M.; Neumann, P.; Wilkins, S.; Pettis, J.; Boonham, N. A DNA method for screening hive debris for the presence of small hive beetle (Aethina tumida). Apidologie 2007, 38, 272–280. [Google Scholar] [CrossRef]
- Ryba, S.; Titera, D.; Haklova, M.; Stopka, P. A PCR method of detecting American foulbrood (Paenibacillus larvae) in winter beehive wax debris. Vet. Microbiol. 2009, 139, 193–196. [Google Scholar] [CrossRef]
- Ryba, S.; Kindlmann, P.; Titera, D.; Haklova, M.; Stopka, P. A new low-cost procedure for detecting nucleic acids in low-incidence samples: A case study of detecting spores of Paenibacillus larvae from bee debris. J. Econ. Entomol. 2012, 105, 1487–1491. [Google Scholar] [CrossRef] [PubMed]
- Forsgren, E.; Laugen, A.T. Prognostic value of using bee and hive debris samples for the detection of American foulbrood disease in honey bee colonies. Apidologie 2014, 45, 10–20. [Google Scholar] [CrossRef]
- Bakonyi, T.; Derakhshifar, I.; Grabensteiner, E.; Nowotny, N. Development and evaluation of PCR assays for the detection of Paenibacillus larvae in honey samples: Comparison with isolation and biochemical characterization. Appl. Environ. Microbiol. 2003, 69, 1504–1510. [Google Scholar] [CrossRef] [PubMed]
- Lauro, F.M.; Favaretto, M.; Covolo, L.; Rassu, M.; Bertoloni, G. Rapid detection of Paenibacillus larvae from honey and hive samples with a novel nested PCR protocol. Int. J. Food Microbiol. 2003, 81, 195–201. [Google Scholar] [CrossRef]
- McKee, B.; Djordjevic, S.; Goodman, R.; Hornitzky, M. The detection of Melissococcus pluton in honey bees (Apis mellifera) and their products using a hemi-nested PCR. Apidologie 2003, 34, 19–27. [Google Scholar] [CrossRef]
- D’Alessandro, B.; Antúnez, K.; Piccini, C.; Zunino, P. DNA extraction and PCR detection of Paenibacillus larvae spores from naturally contaminated honey and bees using spore-decoating and freeze-thawing techniques. World J. Microbiol. Biotech. 2007, 23, 593–597. [Google Scholar] [CrossRef]
- Forsgren, E. European foulbrood in honey bees. J. Invertebr. Pathol. 2010, 103, S5–S9. [Google Scholar] [CrossRef]
- Genersch, E. American Foulbrood in honeybees and its causative agent, Paenibacillus larvae. J. Invertebr. Pathol. 2010, 103, S10–S19. [Google Scholar] [CrossRef]
- Giersch, T.; Berg, T.; Galea, F.; Hornitzky, M. Nosema ceranae infects honey bees (Apis mellifera) and contaminates honey in Australia. Apidologie 2009, 40, 117–123. [Google Scholar] [CrossRef]
- Cox-Foster, D.L.; Conlan, S.; Holmes, E.C.; Palacios, G.; Evans, J.D.; Moran, N.A.; Quan, P.L.; Briese, T.; Hornig, M.; Geiser, D.M.; et al. A metagenomic survey of microbes in honey bee colony collapse disorder. Science 2007, 318, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Vanengelsdorp, D.; Evans, J.D.; Saegerman, C.; Mullin, C.; Haubruge, E.; Nguyen, B.K.; Frazier, M.; Frazier, J.; Cox-Foster, D.; Chen, Y.; et al. Colony collapse disorder: A descriptive study. PLoS ONE 2009, 4, e6481. [Google Scholar] [CrossRef] [PubMed]
- Cornman, R.S.; Tarpy, D.R.; Chen, Y.; Jeffreys, L.; Lopez, D.; Pettis, J.S.; Van Engelsdorp, D.; Evans, J.D. Pathogen webs in collapsing honey bee colonies. PLoS ONE 2012, 7, e43562. [Google Scholar] [CrossRef] [PubMed]
- Higes, M.; Martín-Hernández, R.; Botías, C.; Bailón, E.G.; González-Porto, A.V.; Barrios, L.; Del Nozal, M.J.; Bernal, J.L.; Jiménez, J.J.; Palencia, P.G.; et al. How natural infection by Nosema ceranae causes honeybee colony collapse. Environ. Microbiol. 2008, 10, 2659–2669. [Google Scholar] [CrossRef] [PubMed]
- Higes, M.; Martín-Hernández, R.; Garrido-Bailón, E.; González-Porto, A.V.; García-Palencia, P.; Meana, A.; Del Nozal, M.J.; Mayo, R.; Bernal, J.L. Honeybee colony collapse due to Nosema ceranae in professional apiaries. Environ. Microbiol. Rep. 2009, 1, 110–113. [Google Scholar] [CrossRef]
- Klee, J.; Besana, A.M.; Genersch, E.; Gisder, S.; Nanetti, A.; Tam, D.Q.; Chinh, T.X.; Puerta, F.; Ruz, J.M.; Kryger, P.; et al. Widespread dispersal of the microsporidian Nosema ceranae, an emergent pathogen of the western honey bee, Apis mellifera. J. Invertebr. Pathol. 2007, 96, 1–10. [Google Scholar] [CrossRef]
- Higes, M.; Meana, A.; Bartolomé, C.; Botías, C.; Martín-Hernández, R. Nosema ceranae (Microsporidia), a controversial 21st century honey bee pathogen. Environ. Microbiol. Rep. 2013, 5, 17–29. [Google Scholar] [CrossRef]
- Branchiccela, B.; Arredondo, D.; Higes, M.; Invernizzi, C.; Martín-Hernández, R.; Tomasco, I.; Zunino, P.; Antúnez, K. Characterization of Nosema ceranae genetic variants from different geographic origins. Microb. Ecol. 2017, 73, 978–987. [Google Scholar] [CrossRef]
- Aronstein, D.A.; Murray, K.D. Chalkbrood disease in honeybees. J. Invertebr. Pathol. 2010, 103, 20–29. [Google Scholar] [CrossRef]
- Hedtke, K.; Jensen, P.M.; Jensen, A.B.; Genersch, E. Evidence for emerging parasites and pathogens influencing outbreaks of stress-related diseases like chalkbrood. J. Invertebr. Pathol. 2011, 108, 167–173. [Google Scholar] [CrossRef]
- Ravoet, J.; Maharramov, J.; Meeus, I.; De Smet, L.; Wenseleers, T.; Smagghe, G.; De Graaf, D.C. Comprehensive bee pathogen screening in Belgium reveals Crithidia mellificae as a new contributory factor to winter mortality. PLoS ONE 2013, 8, e72443. [Google Scholar] [CrossRef] [PubMed]
- Ravoet, J.; Schwarz, R.S.; Descamps, T.; Yañez, O.; Tozkar, C.O.; Martin-Hernandez, R.; Bartolomé, C.; De Smet, L.; Higes, M.; Wenseleers, T.; et al. Differential diagnosis of the honey bee trypanosomatids Crithidia mellificae and Lotmaria passim. J. Invertebr. Pathol. 2015, 130, 21–27. [Google Scholar] [CrossRef]
- Schwarz, R.S.; Bauchan, G.; Murphy, C.; Ravoet, J.; De Graaf, D.C.; Evans, J.D. Characterization of two species of Trypanosomatidae from the honey bee Apis mellifera: Crithidia mellificae Langridge and McGhee, 1967 and Lotmaria passim n. gen., n. sp. J. Eukaryot. Microbiol. 2015, 62, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Stevanovic, J.; Schwarz, R.S.; Vejnovic, B.; Evans, J.D.; Irwin, R.E.; Glavinic, U.; Stanimirovic, Z. Species-specific diagnostics of Apis mellifera trypanosomatids: A nine-year survey (2007–2015) for trypanosomatids and microsporidians in Serbian honey bees. J. Invertebr. Pathol. 2016, 139, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Sammataro, D.; Gerson, U.; Needham, G. Parasitic mites of honey bees: Life history, implications, and impact. Ann. Rev. Entomol. 2000, 45, 519–548. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Bailón, E.; Higes, M.; Martínez-Salvador, A.; Antúnez, K.; Botías, C.; Meana, A.; Prieto, L.; Martín-Hernández, R. The prevalence of the honeybee brood pathogens Ascosphaera apis, Paenibacillus larvae and Melissococcus plutonius in Spanish apiaries determined with a new multiplex PCR assay. Microb. Biotechnol. 2013, 6, 731–739. [Google Scholar]
- Guzmán-Novoa, E.; Eccles, L.; Calvete, Y.; Mcgowan, J.; Kelly, P.G.; Correa-Benítez, A. Varroa destructor is the main culprit for the death and reduced populations of overwintered honey bee (Apis mellifera) colonies in Ontario, Canada. Apidologie 2010, 41, 443–450. [Google Scholar] [CrossRef]
- Le Conte, Y.; Ellis, M.; Ritter, W. Varroa mites and honey bee health: Can Varroa explain part of the colony losses. Apidologie 2010, 41, 353–363. [Google Scholar] [CrossRef]
- Anderson, D.L.; Morgan, M.J. Genetic and morphological variation of bee-parasitic Tropilaelaps mites (Acari: Laelapidae): New and re-defined species. Exp. Appl. Acarol. 2007, 43, 1–24. [Google Scholar] [CrossRef]
- De Guzman, L.I.; Williams, G.R.; Khongphinitbunjong, K.; Chantawannakul, P. Ecology, life history, and management of Tropilaelaps mites. J. Econ. Entomol. 2017, 110, 319–332. [Google Scholar] [CrossRef]
- Traynor, K.S.; Rennich, K.; Forsgren, E.; Rose, R.; Pettis, J.; Kunkel, G.; Madella, S.; Evans, J.; Lopez, D.; Van Engelsdorp, D. Multiyear survey targeting disease incidence in US honey bees. Apidologie 2016, 47, 325–347. [Google Scholar] [CrossRef]
- Pettis, J.S.; Rose, R.; Lichtenberg, E.M.; Chantawannakul, P.; Buawangpong, N.; Somana, W.; Sukumalanand, P.; Vanengelsdorp, D. A rapid survey technique for Tropilaelaps mite (Mesostigmata: Laelapidae) detection. J. Econ. Entomol. 2013, 106, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Utzeri, V.J.; Ribani, A.; Schiavo, G.; Bertolini, F.; Bovo, S.; Fontanesi, L. Application of next generation semiconductor based sequencing to detect the botanical composition of monofloral, polyfloral and honeydew honey. Food Control 2018, 86, 342–349. [Google Scholar] [CrossRef]
- Utzeri, V.J.; Ribani, A.; Fontanesi, L. Authentication of honey based on a DNA method to differentiate Apis mellifera subspecies: Application to Sicilian honey bee (A. m. siciliana) and Iberian honey bee (A. m. iberiensis) honeys. Food Control 2018, 91, 294–301. [Google Scholar] [CrossRef]
- Utzeri, V.J.; Schiavo, G.; Ribani, A.; Bertolini, F.; Bovo, S.; Fontanesi, L. A next generation sequencing approach for targeted Varroa destructor (Acari: Varroidae) mitochondrial DNA analysis based on honey derived environmental DNA. J. Invertebr. Pathol. 2019, 161, 47–53. [Google Scholar] [CrossRef]
- Han, S.H.; Lee, D.B.; Lee, D.W.; Kim, E.H.; Yoon, B.S. Ultra-rapid real-time PCR for the detection of Paenibacillus larvae, the causative agent of American foulbrood (AFB). J. Invertebr. Pathol. 2008, 99, 8–13. [Google Scholar] [CrossRef]
- Arai, R.; Miyoshi-Akiyama, T.; Okumura, Y.; Morinaga, K.; Wu, M.; Sugimura, Y.; Yoshiyama, M.; Okura, M.; Kirikae, T.; Takamatsu, D. Development of duplex PCR assay for detection and differentiation of typical and atypical Melissococcus plutonius strains. J. Vet. Med. Sci. 2014, 76, 491–498. [Google Scholar] [CrossRef]
- Chen, Y.; Evans, J.D.; Zhou, L.; Boncristiani, H.; Kimura, K.; Xiao, T.; Litkowski, A.M.; Pettis, J.S. Asymmetrical coexistence of Nosema ceranae and Nosema apis in honey bees. J. Invertebr. Pathol. 2009, 101, 204–209. [Google Scholar] [CrossRef]
- Kojima, Y.; Yoshiyama, M.; Kimura, K.; Kadowaki, T. PCR-based detection of a tracheal mite of the honey bee Acarapis woodi. J. Invertebr. Pathol. 2011, 108, 135–137. [Google Scholar] [CrossRef]
- Garrido-Bailón, E.; Bartolomé, C.; Prieto, L.; Botías, C.; Martínez-Salvador, A.; Meana, A.; Martín-Hernández, R.; Higes, M. The prevalence of Acarapis woodi in Spanish honey bee (Apis mellifera) colonies. Exp. Parasitol. 2012, 132, 530–536. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Ellis, J.D.; Munn, P.A. The worldwide health status of honey bees. Bee World 2005, 86, 88–101. [Google Scholar] [CrossRef]
- Kojima, Y.; Toki, T.; Morimoto, T.; Yoshiyama, M.; Kimura, K.; Kadowaki, T. Infestation of Japanese native honey bees by tracheal mite and virus from non-native European honey bees in Japan. Microb. Ecol. 2011, 62, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Çakmak, I.; Aydın, L.; Gulegen, E.; Wells, H. Varroa (Varroa destructor) and tracheal mite (Acarapis woodi) incidence in the Republic of Turkey. J. Apic. Res. 2003, 42, 57–60. [Google Scholar] [CrossRef]
- Adjlane, N.; Assia, H.C.; Haddad, N.; Ahcen, B. Prevalence of acariosis in honeybee colonies of Apis mellifera intermissa in Algeria. J. Entomol. Res. 2018, 42, 451–454. [Google Scholar] [CrossRef]
- Ahn, A.J.; Ahn, K.S.; Noh, J.H.; Kim, Y.H.; Yoo, M.S.; Kang, S.W.; Yu, D.H.; Shin, S.S. Molecular prevalence of Acarapis mite infestations in honey bees in Korea. Korean J. Parasitol. 2015, 53, 315–320. [Google Scholar] [CrossRef]
- Martín-Hernández, R.; Botías, C.; Bailón, E.G.; Martínez-Salvador, A.; Prieto, L.; Meana, A.; Higes, M. Microsporidia infecting Apis mellifera: Coexistence or competition. Is Nosema ceranae replacing Nosema apis. Environ. Microbiol. 2012, 14, 2127–2138. [Google Scholar] [CrossRef]
- Natsopoulou, M.E.; McMahon, D.P.; Doublet, V.; Bryden, J.; Paxton, R.J. Interspecific competition in honeybee intracellular gut parasites is asymmetric and favours the spread of an emerging infectious disease. Proc. R. Soc. B Biol. Sci. 2015, 282, 20141896. [Google Scholar] [CrossRef]
- Emsen, B.; Guzman-Novoa, E.; Hamiduzzaman, M.M.; Eccles, L.; Lacey, B.; Ruiz-Pérez, R.A.; Nasr, M. Higher prevalence and levels of Nosema ceranae than Nosema apis infections in Canadian honey bee colonies. Parasitol. Res. 2016, 115, 175–181. [Google Scholar] [CrossRef]
- Tritschler, M.; Retschnig, G.; Yañez, O.; Williams, G.R.; Neumann, P. Host sharing by the honey bee parasites Lotmaria passim and Nosema ceranae. Ecol. Evol. 2017, 7, 1850–1857. [Google Scholar] [CrossRef] [PubMed]
- Forsgren, E.; Lundhagen, A.C.; Imdorf, A.; Fries, I. Distribution of Melissococcus plutonius in honeybee colonies with and without symptoms of European foulbrood. Microb. Ecol. 2005, 50, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Belloy, L.; Imdorf, A.; Fries, I.; Forsgren, E.; Berthoud, H.; Kuhn, R.; Charrière, J.D. Spatial distribution of Melissococcus plutonius in adult honey bees collected from apiaries and colonies with and without symptoms of European foulbrood. Apidologie 2007, 38, 136–140. [Google Scholar] [CrossRef]
- Haynes, E.; Helgason, T.; Young, J.P.W.; Thwaites, R.; Budge, G.E. A typing scheme for the honeybee pathogen Melissococcus plutonius allows detection of disease transmission events and a study of the distribution of variants. Environ. Microbiol. Rep. 2013, 5, 525–529. [Google Scholar] [CrossRef]
- Antúnez, K.; D’Alessandro, B.; Piccini, C.; Corbella, E.; Zunino, P. Paenibacillus larvae larvae spores in honey samples from Uruguay: A nationwide survey. J. Invertebr. Pathol. 2004, 86, 56–58. [Google Scholar] [CrossRef]
- Gillard, M.; Charriere, J.D.; Belloy, L. Distribution of Paenibacillus larvae spores inside honey bee colonies and its relevance for diagnosis. J. Invertebr. Pathol. 2008, 99, 92–95. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Species | Primer Name 1 | Primer Sequences (5’-3’): Forward and Reverse 2 | Size in Bp | Amplified Region | Ta °C 3 | Reference |
|---|---|---|---|---|---|---|
| Apis mellifera | AM_Forward AM_Reverse | GGCAGAATAAGTGCATTG TTAATATGAATTAAGTGGGG | C 85, M 139, A 153 4 | mtDNA COI-COII | 51 | [44] |
| Paenibacillus larvae | AF_6f AF_7r | GCAAGTCGAGCGGACCTTGT TCGTCGCCTTGGTAAGC | 237 | 16S rRNA | 63 | [14] |
| Paenibacillus larvae | Han233PaeLarv16S_F Han233PaeLarv16S_R | GTGTTTCCTTCGGGAGACG CTCTAGGTCGGCTACGCATC | 233 | 16S rRNA | 59 | [46] |
| Melissococcus plutonius | MeliFORa MeliREVa | GTTAAAAGGCGCTTTCGGGT GAGGAAAACAGTTACTCTTTCCCCTA | 281 | 16S rRNA | 63 | [36] |
| Melissococcus plutonius | Mp_Arai187_F Mp_Arai187_R | TGGTAGCTTAGGCGGAAAAC TGGAGCGATTAGAGTCGTTAGA | 187 | NapA | 59 | [47] |
| Nosema apis | Nose_apis_chen_F Nose_apis_chen_R | CCATTGCCGGATAAGAGAGT CCACCAAAAACTCCCAAGAG | 269 | SSUrRNA | 54–60 5 | [48] |
| Nosema ceranae | Nose_cera_chen_F Nose_cera_chen_R | CGGATAAAAGAGTCCGTTACC TGAGCAGGGTTCTAGGGAT | 250 | SSUrRNA | 58 | [48] |
| Lotmaria passim | LpCytb_F1 LpCytb_R1 | CGAAGTGCACATATATGCTTTAC GCCAAACACCAATAACTGGTACT | 247 | mtDNA cyt b | 59 | [34] |
| Ascosphaera apis | AscosFORa AscosREVa | TGTGTCTGTGCGGCTAGGTG GCTAGCCAGGGGGGAACTAA | 136 | 18S rRNA | 61 | [36] |
| Acarapis woodi | Acarap_cox1_F1 Acarap_cox1_R1 | CAGTAGGGCTAGATATCGATACCCGAGCTT TGAGCTACAACATAATATCTGTCATGAAGA | 247 | mtDNA Cox 1 | 55–62 5 | [49] |
| Acarapis woodi | Acarapis_cox1_F2 Acarapis_cox1_R2 | CGGGCCCGAGCTTATTTTACTGCTG GCGCCTGTCAATCCACCTACAGAAA | 162 | mtDNA Cox 1 | 55–62 5 | [50] |
| Varroa destructor | Varr_cox1_181_F Varr_cox1_181_R | GCCTTTATTTGTATGGTCTG TGGGTGTCCAAAAAATCA | 181 | mtDNA Cox 1 | 52 | [45] |
| Varroa destructor | Varr_cox1_148_F Varr_cox1_148_R | TGGTATTATTTCTCATGTAATTTG ATCAATATCTATTCCTACTGTAAA | 148 | mtDNA Cox 1 | 55 | [45] |
| Tropilaelaps clareae6 | Tropi_cox1_F Tropi_cox1_R | TATTTGTATGATCTGTCCTA ATAATACCAAATCCTGGTA | 214 | mtDNA Cox 1 | 54–60 5 | This study |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribani, A.; Utzeri, V.J.; Taurisano, V.; Fontanesi, L. Honey as a Source of Environmental DNA for the Detection and Monitoring of Honey Bee Pathogens and Parasites. Vet. Sci. 2020, 7, 113. https://doi.org/10.3390/vetsci7030113
Ribani A, Utzeri VJ, Taurisano V, Fontanesi L. Honey as a Source of Environmental DNA for the Detection and Monitoring of Honey Bee Pathogens and Parasites. Veterinary Sciences. 2020; 7(3):113. https://doi.org/10.3390/vetsci7030113
Chicago/Turabian StyleRibani, Anisa, Valerio Joe Utzeri, Valeria Taurisano, and Luca Fontanesi. 2020. "Honey as a Source of Environmental DNA for the Detection and Monitoring of Honey Bee Pathogens and Parasites" Veterinary Sciences 7, no. 3: 113. https://doi.org/10.3390/vetsci7030113
APA StyleRibani, A., Utzeri, V. J., Taurisano, V., & Fontanesi, L. (2020). Honey as a Source of Environmental DNA for the Detection and Monitoring of Honey Bee Pathogens and Parasites. Veterinary Sciences, 7(3), 113. https://doi.org/10.3390/vetsci7030113