Analysis of the Variability in the Non-Coding Regions of Influenza A Viruses


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. NCR Sequence Determination
2.2. Sequence Analyses
3. Results
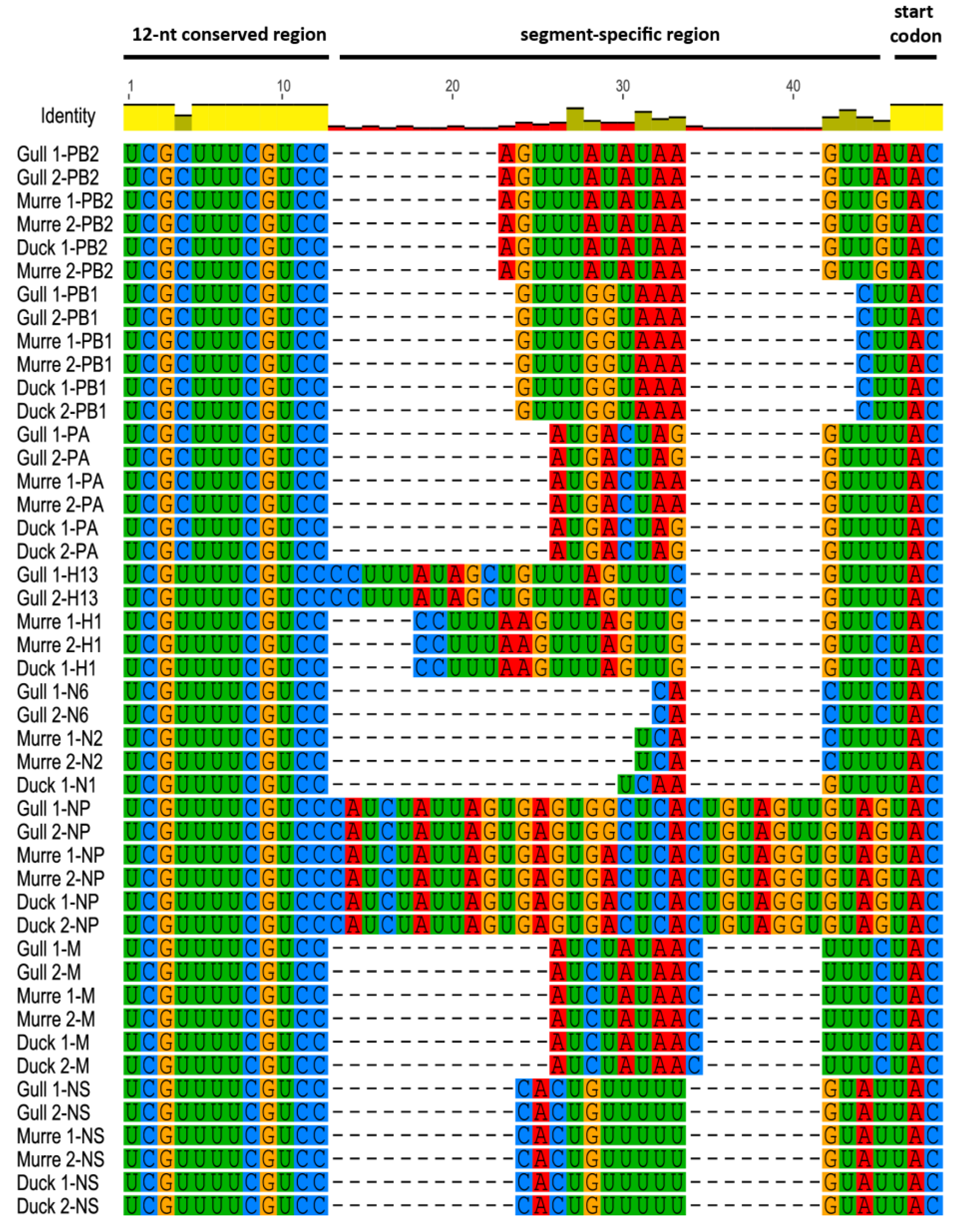
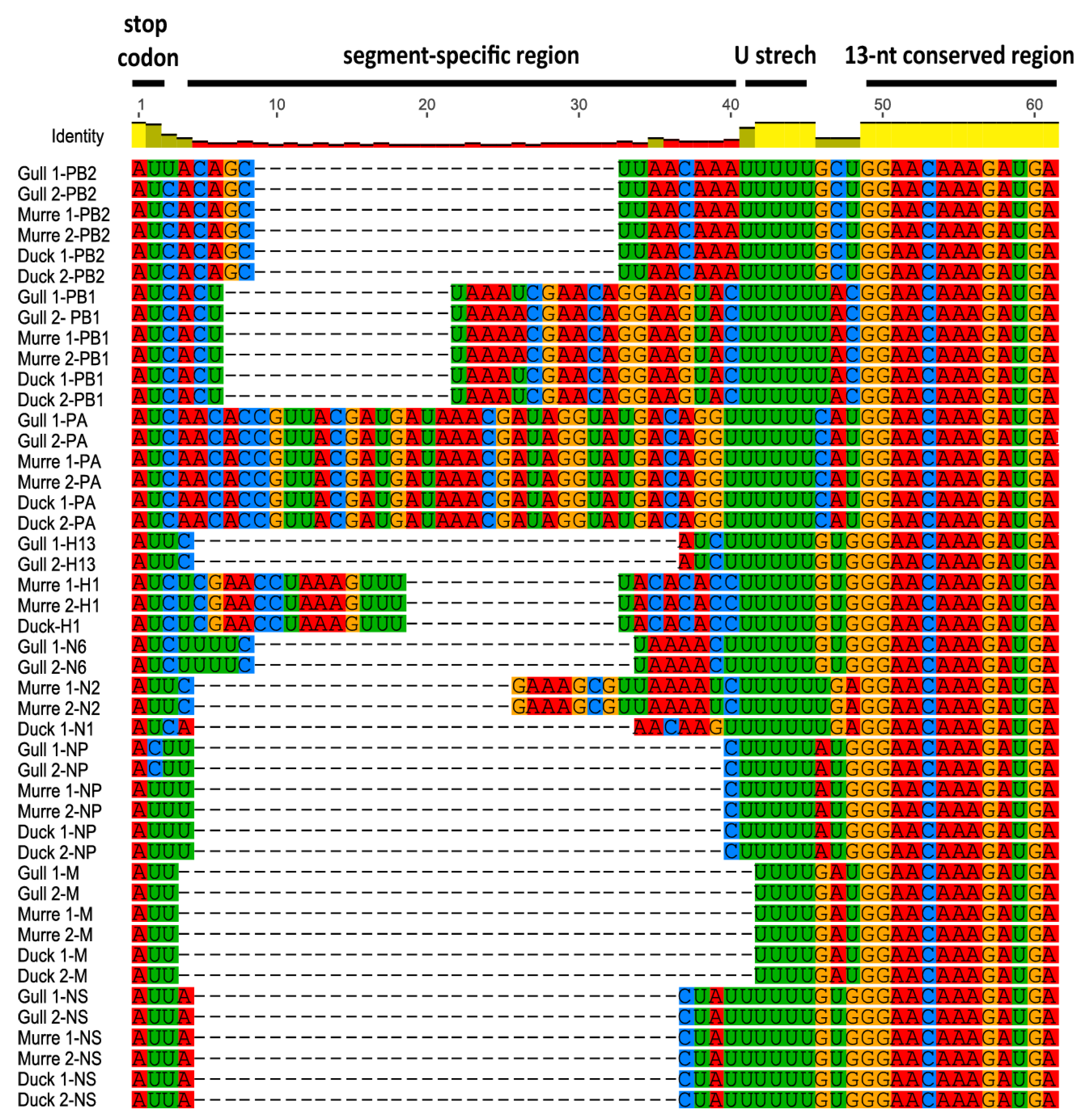
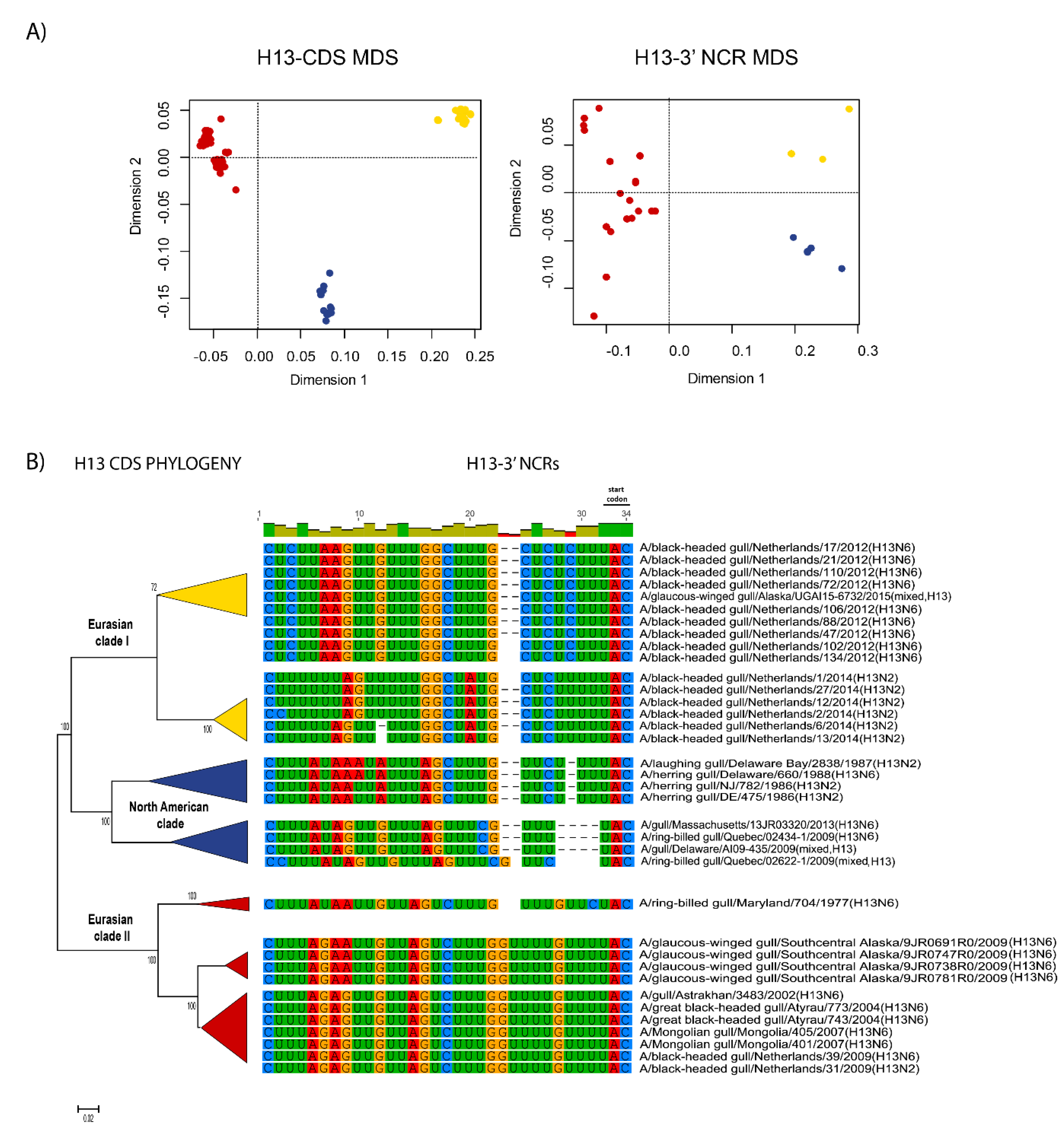
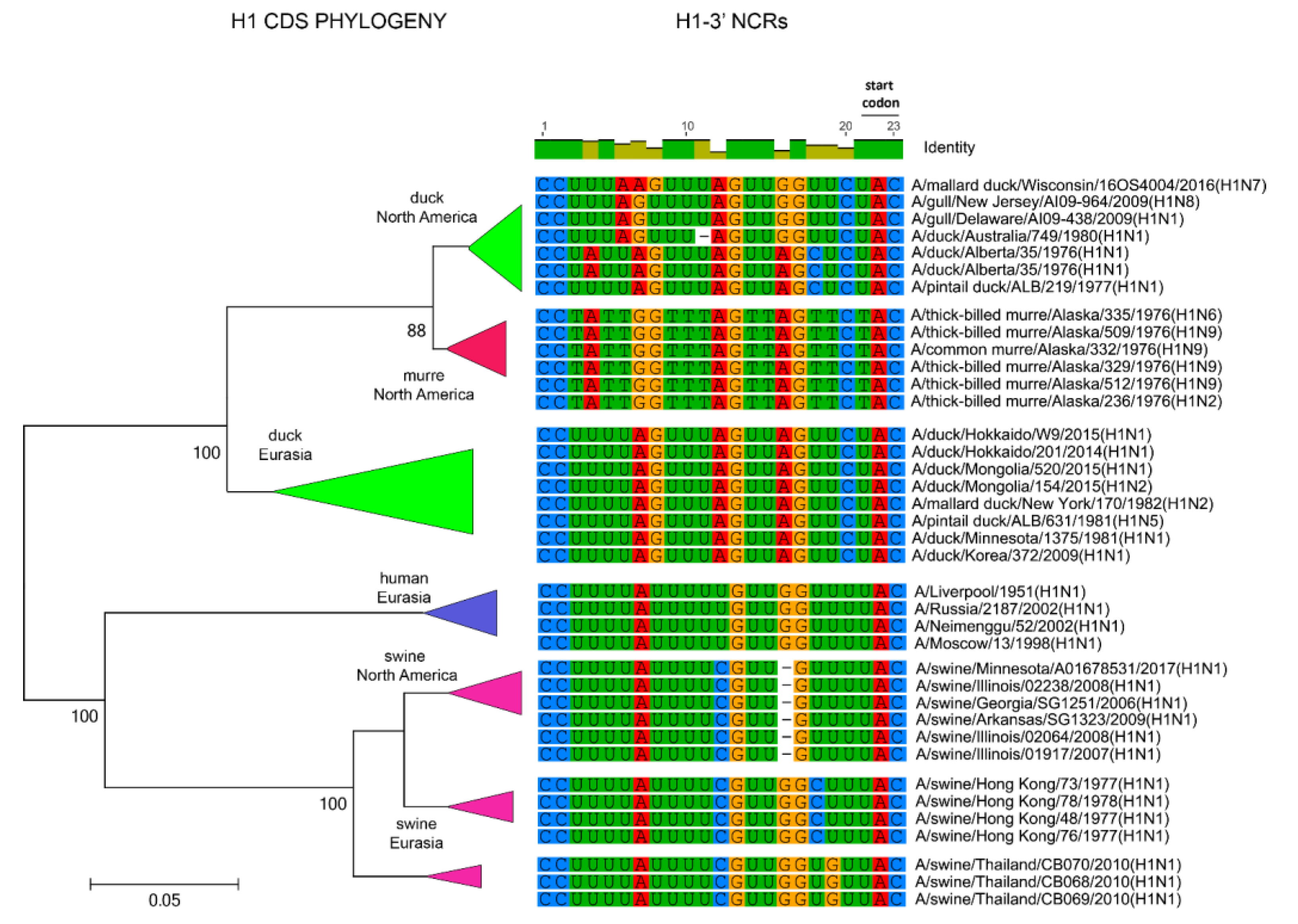
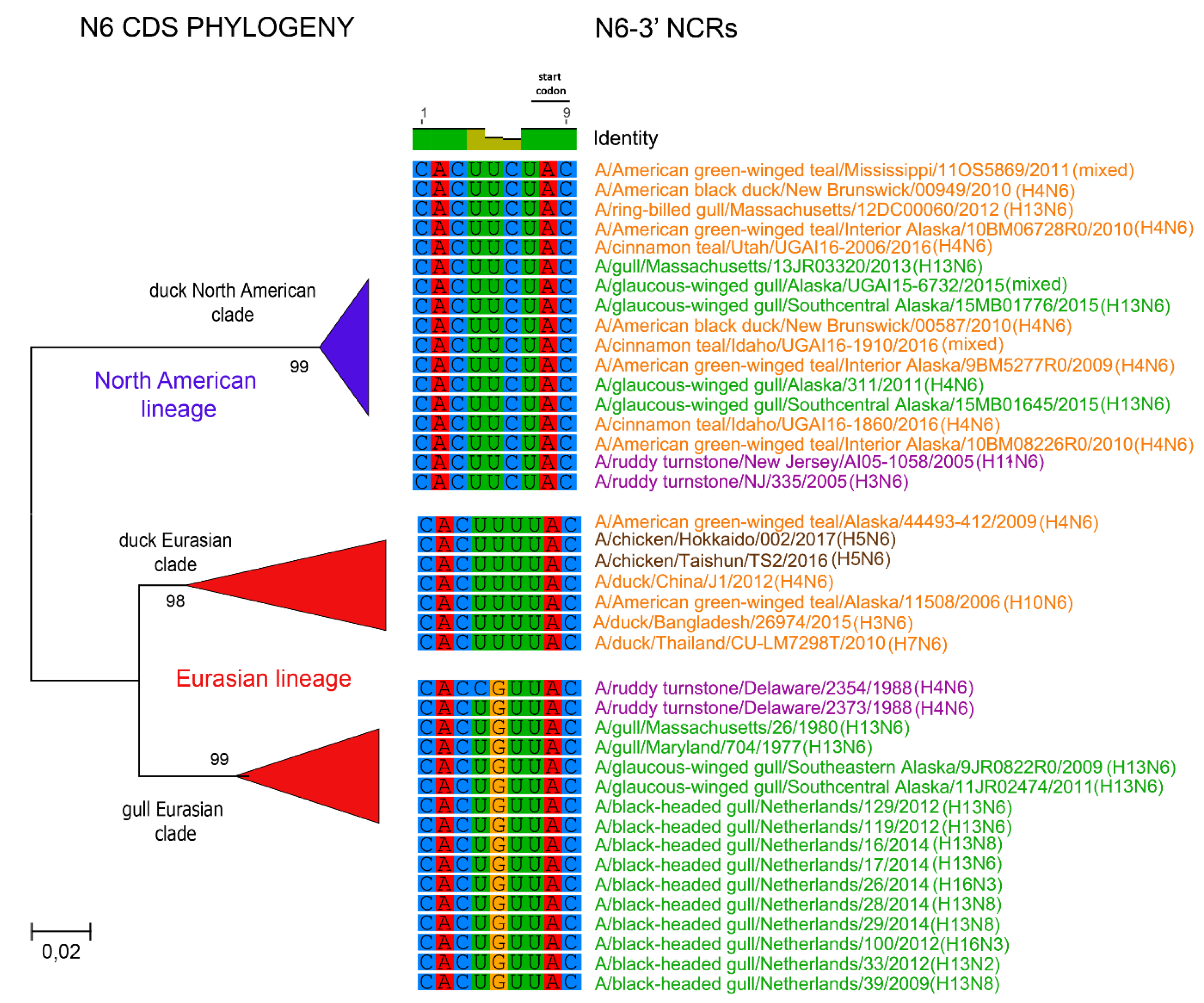
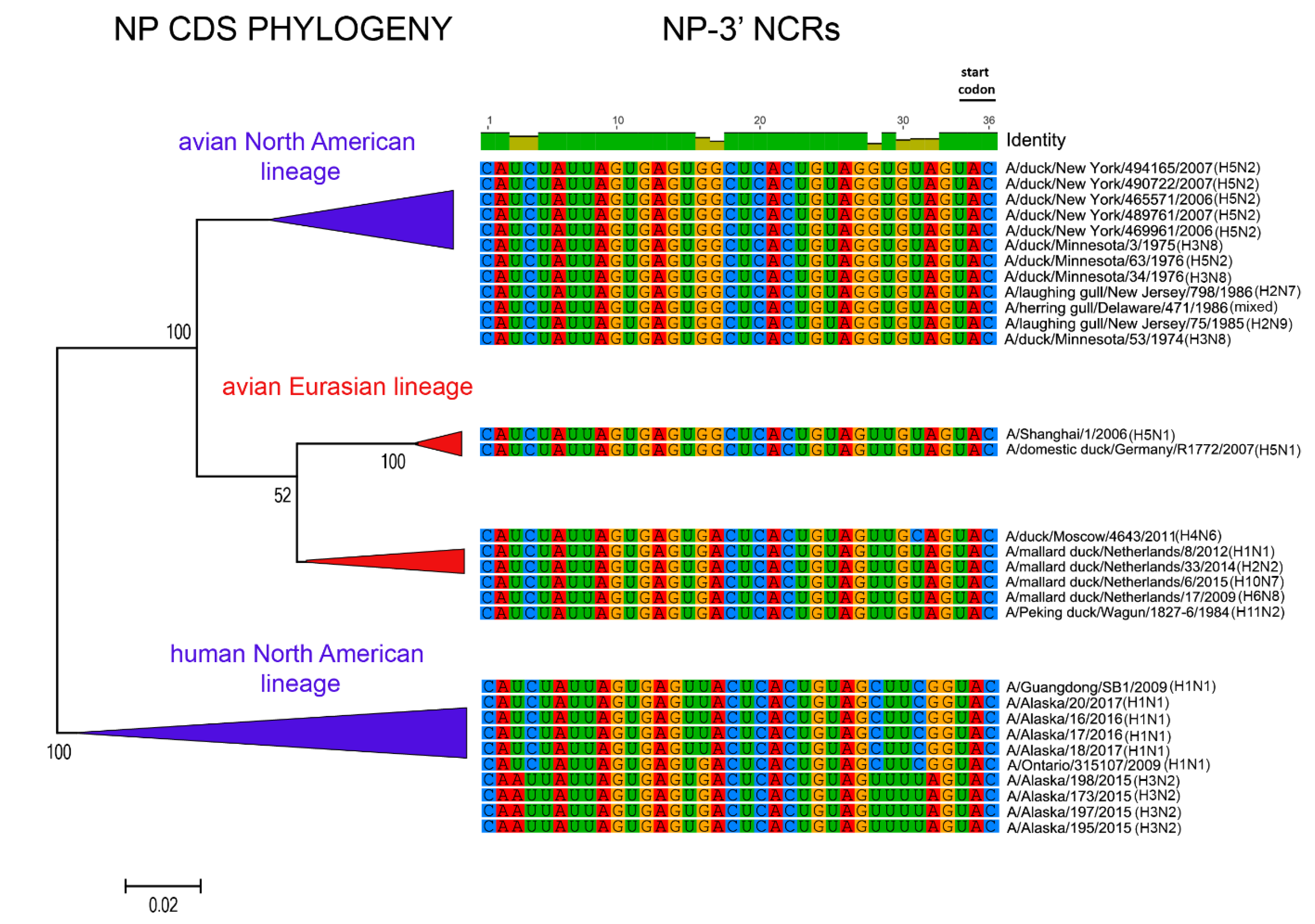
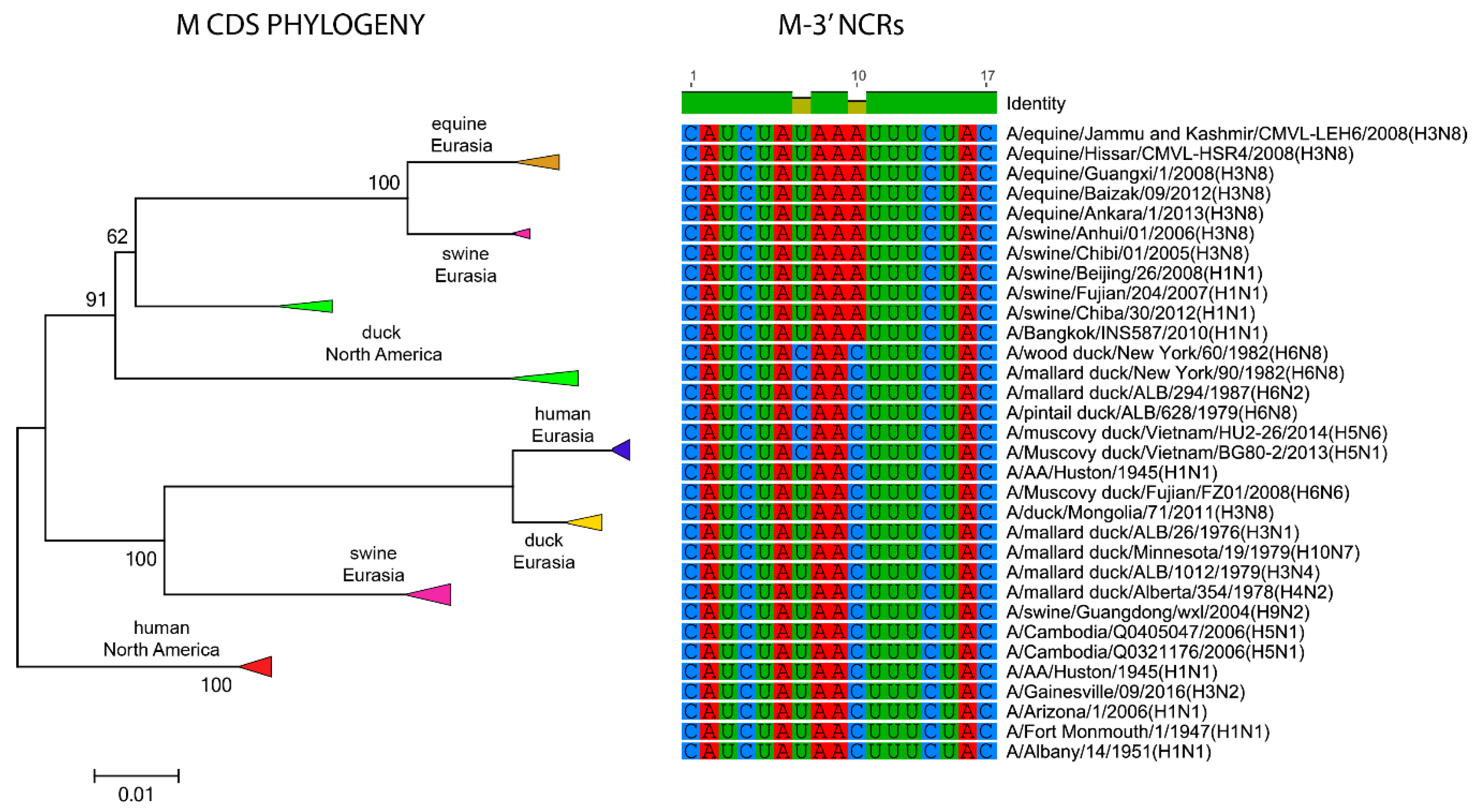
3.1. Characterization of NCRs from Different Wild Bird Viruses
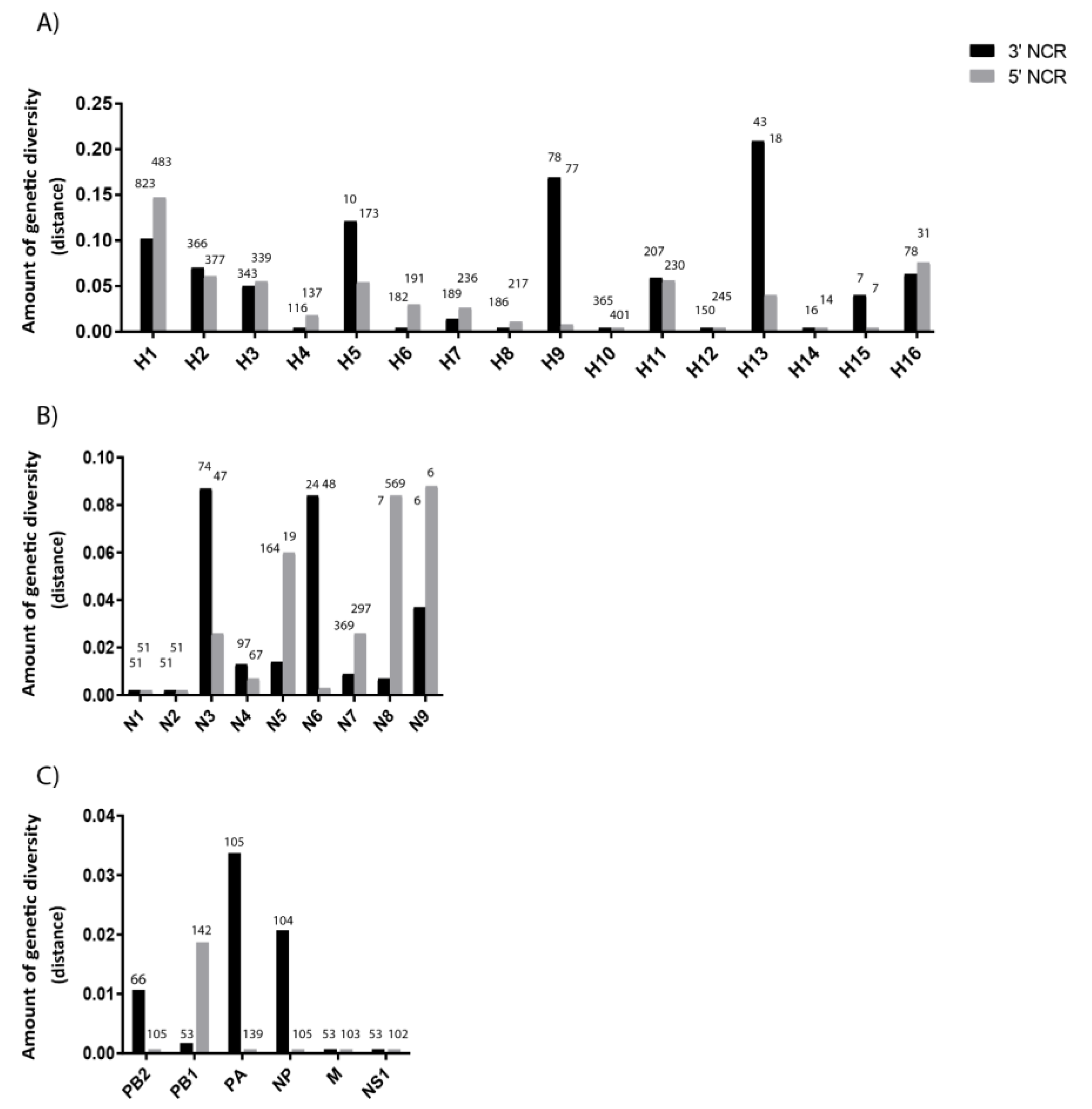
3.2. The Variability in Segment-Specific NCR Sequences Differs among Segments
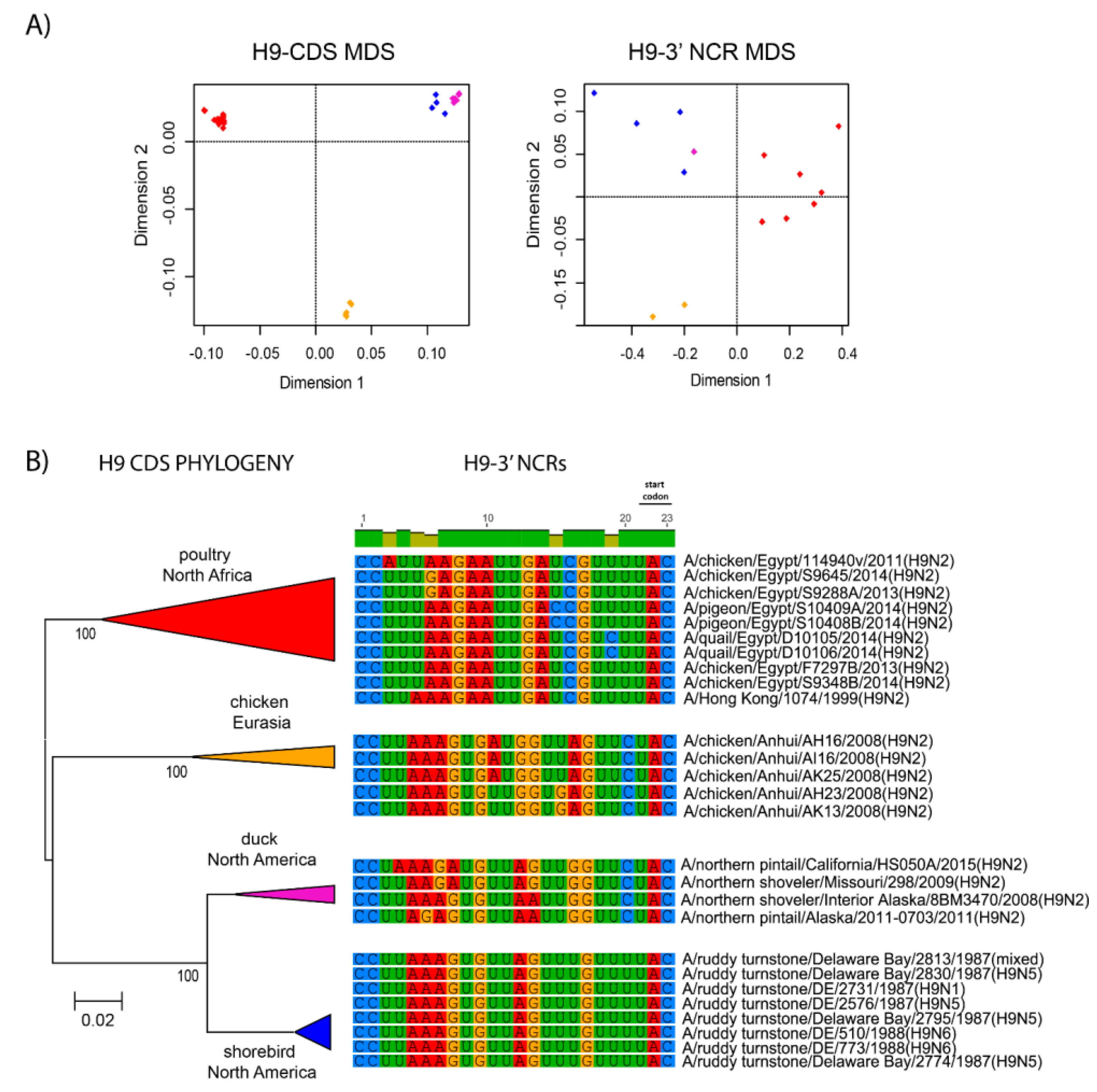
3.3. Patterns of Variability within NCRs Can Be Explained by Viral Host Species and Geographic Origins
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Robertson, J.S. 5’ and 3’ terminal nucleotide sequences of the RNA genome segments of influenza virus. Nucleic Acids Res. 1979, 6, 3745–3757. [Google Scholar] [CrossRef] [PubMed]
- Skehel, J.J.; Hay, A.J. Nucleotide sequences at the 5’ termini of influenza virus RNAs and their transcripts. Nucleic Acids Res. 1978, 5, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Desselberger, U.; Racaniello, V.R.; Zazra, J.J.; Palese, P. The 3’ and 5’-terminal sequences of influenza A, B and C virus RNA segments are highly conserved and show partial inverted complementarity. Gene 1980, 8, 315–328. [Google Scholar] [CrossRef]
- Hsu, M.T.; Parvin, J.D.; Gupta, S.; Krystal, M.; Palese, P. Genomic RNAs of influenza viruses are held in a circular conformation in virions and in infected cells by a terminal panhandle. Proc. Natl. Acad. Sci. USA 1987, 84, 8140–8144. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, M.; Muster, T. Mutations in the nonconserved noncoding sequences of the influenza A virus segments affect viral vRNA formation. Virus Res. 1996, 44, 23–31. [Google Scholar] [CrossRef]
- Kim, H.J.; Fodor, E.; Brownlee, G.G.; Seong, B.L. Mutational analysis of the RNA-fork model of the influenza A virus vRNA promoter in vivo. J. Gen. Virol. 1997, 78, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Seong, B.L.; Brownlee, G.G. Nucleotides 9 to 11 of the influenza A virion RNA promoter are crucial for activity in vitro. J. Gen. Virol. 1992, 73, 3115–3124. [Google Scholar] [CrossRef] [PubMed]
- Fodor, E.; Pritlove, D.C.; Brownlee, G.G. The influenza virus panhandle is involved in the initiation of transcription. J. Virol. 1994, 68, 4092–4096. [Google Scholar] [PubMed]
- Flick, R.; Neumann, G.; Hoffmann, E.; Neumeier, E.; Hobom, G. Promoter elements in the influenza vRNA terminal structure. RNA 1996, 2, 1046–1057. [Google Scholar] [PubMed]
- Fodor, E.; Seong, B.L.; Brownlee, G.G. Photochemical cross-linking of influenza A polymerase to its virion RNA promoter defines a polymerase binding site at residues 9 to 12 of the promoter. J. Gen. Virol. 1993, 74, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Palese, P. Mutational analysis of the promoter required for influenza virus virion RNA synthesis. J. Virol. 1992, 66, 4331–4338. [Google Scholar] [PubMed]
- Pritlove, D.C.; Fodor, E.; Seong, B.L.; Brownlee, G.G. In vitro transcription and polymerase binding studies of the termini of influenza A virus cRNA: Evidence for a cRNA panhandle. J. Gen. Virol. 1995, 76, 2205–2213. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.X.; Luytjes, W.; Enami, M.; Palese, P. The polyadenylation signal of influenza virus RNA involves a stretch of uridines followed by the RNA duplex of the panhandle structure. J. Virol. 1991, 65, 2861–2867. [Google Scholar] [PubMed]
- Li, X.; Palese, P. Characterization of the polyadenylation signal of influenza virus RNA. J. Virol. 1994, 68, 1245–1249. [Google Scholar] [PubMed]
- Poon, L.L.; Pritlove, D.C.; Fodor, E.; Brownlee, G.G. Direct evidence that the poly(A) tail of influenza A virus mRNA is synthesized by reiterative copying of a U track in the virion RNA template. J. Virol. 1999, 73, 3473–3476. [Google Scholar] [PubMed]
- Pritlove, D.C.; Poon, L.L.; Fodor, E.; Sharps, J.; Brownlee, G.G. Polyadenylation of influenza virus mRNA transcribed in vitro from model virion RNA templates: Requirement for 5’ conserved sequences. J. Virol. 1998, 72, 1280–1286. [Google Scholar] [PubMed]
- Lee, M.K.; Bae, S.H.; Park, C.J.; Cheong, H.K.; Cheong, C.; Choi, B.S. A single-nucleotide natural variation (U4 to C4) in an influenza A virus promoter exhibits a large structural change: Implications for differential viral RNA synthesis by RNA-dependent RNA polymerase. Nucleic Acids Res. 2003, 31, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Palese, P.; Garcia-Sastre, A. Nonconserved nucleotides at the 3’ and 5’ ends of an influenza A virus RNA play an important role in viral RNA replication. Virology 1996, 217, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo-Chaigne, B.; Barbezange, C.V.; Léandri, S.; Roquin, C.; Berthault, C.; Van Der Werf, S. Incorporation of the influenza A virus NA segment into virions does not require cognate non-coding sequences. Sci. Rep. 2017, 7, 43462. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Palese, P. Rewiring the RNAs of influenza virus to prevent reassortment. Proc. Natl. Acad. Sci. USA 2009, 106, 15891. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Fujii, Y.; Noda, T.; Muramoto, Y.; Watanabe, T.; Takada, A.; Goto, H.; Horimoto, T.; Kawaoka, Y. Importance of both the coding and the segment-specific noncoding regions of the influenza A virus NS segment for its efficient incorporation into virions. J. Virol. 2005, 79, 3766–3774. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Peng, Y.; Zhou, K.; Cao, M.; Wang, J.; Wang, X.; Jiang, T.; Deng, T. New Insights into the Nonconserved Noncoding Region of the Subtype-Determinant Hemagglutinin and Neuraminidase Segments of Influenza A Viruses. J. Virol. 2014, 88, 11493–11503. [Google Scholar] [CrossRef] [PubMed]
- White, M.C.; Steel, J.; Lowen, A.C. Heterologous Packaging Signals on Segment 4, but Not Segment 6 or Segment 8, Limit Influenza A Virus Reassortment. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.; Stech, J.; Guan, Y.; Webster, R.G.; Perez, D.R. Universal primer set for the full-length amplification of all influenza A viruses. Arch. Virol. 2001, 146, 2275–2289. [Google Scholar] [CrossRef] [PubMed]
- Widjaja, I.; de Vries, E.; Rottier, P.J.; de Haan, C.A. Competition between Influenza A Virus Genome Segments. PLoS ONE 2012, 7, e47529. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Donnelly, M.E.; Scholes, D.T.; George, K.S.; Hatta, M.; Kawaoka, Y.; Wentworth, D.E. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and Swine origin human influenza a viruses. J. Virol. 2009, 83, 10309–10313. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Qu, Y.; Zhang, R.; Cui, P.; Song, G.; Duan, Z.; Lei, F. Evolutionary genomics of the pandemic 2009 H1N1 influenza viruses (pH1N 1v). Virol. J. 2011, 8, 250. [Google Scholar] [CrossRef] [PubMed]
- Bahl, J.; Vijaykrishna, D.; Holmes, E.C.; Smith, G.J.; Guan, Y. Gene flow and competitive exclusion of avian influenza A virus in natural reservoir hosts. Virology 2009, 390, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Furuse, Y.; Oshitani, H. Evolution of the influenza A virus untranslated regions. Infect. Genet. Evol. 2011, 11, 1150–1154. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benkaroun, J.; Robertson, G.J.; Whitney, H.; Lang, A.S. Analysis of the Variability in the Non-Coding Regions of Influenza A Viruses. Vet. Sci. 2018, 5, 76. https://doi.org/10.3390/vetsci5030076
Benkaroun J, Robertson GJ, Whitney H, Lang AS. Analysis of the Variability in the Non-Coding Regions of Influenza A Viruses. Veterinary Sciences. 2018; 5(3):76. https://doi.org/10.3390/vetsci5030076
Chicago/Turabian StyleBenkaroun, Jessica, Gregory J. Robertson, Hugh Whitney, and Andrew S. Lang. 2018. "Analysis of the Variability in the Non-Coding Regions of Influenza A Viruses" Veterinary Sciences 5, no. 3: 76. https://doi.org/10.3390/vetsci5030076
APA StyleBenkaroun, J., Robertson, G. J., Whitney, H., & Lang, A. S. (2018). Analysis of the Variability in the Non-Coding Regions of Influenza A Viruses. Veterinary Sciences, 5(3), 76. https://doi.org/10.3390/vetsci5030076