
Artificial Intelligence-Driven Drug Toxicity Prediction: Advances, Challenges, and Future Directions

Abstract
1. Introduction
2. Drug Toxicity Prediction: Databases and Tools
2.1. Toxicity Database
2.1.1. Toxicology Resources for Intelligent Computation
2.1.2. Integrated Chemical Environment (ICE)
2.1.3. Distributed Structure-Searchable Toxicity (DSSTox) Database
2.1.4. DrugBank
2.1.5. ChEMBL
2.1.6. Online Chemical Modeling Environment (OCHEM)
2.1.7. PubChem
2.2. Biological Experimental Data
2.2.1. In Vitro Cytotoxicity Test Data
2.2.2. Animal Experiment Data
2.3. Clinical Data
2.3.1. Food and Drug Administration (FDA) Adverse Event Reporting System
2.3.2. Electronic Medical Record System

{kind=link}
{kind=link}
| Number | Database Name | Database Content | Characteristic | Developer | Website |
|---|---|---|---|---|---|
| 1 | Tox21 [21] | It contains data on more than 10,000 chemicals, as well as nuclear receptors and stress response pathways, generating more than 150 million data points. | High throughput screening data; Multiple toxicological toxicity endpoints; Data visualization and open access | FDA | https://tripod.nih.gov/tox21/challenge/ |
| 2 | Comp Tox Dashboard [22] | It contains data on more than 1.2 million chemicals and provides information about chemical structure, environmental behavior, and biological activity. | Real-time prediction; Batch Search; Advanced search; Data integration | Environmental Protection Agency (EPA) | https://comptox.epa.gov/dashboard |
| 3 | Toxicity Reference Database (ToxRefDB) [23] | It contains in vivo research data of more than 5900 guideline or class guideline studies from more than 1100 chemicals and provides quantitative dose-response data of each dose treatment group, including data of the control group, as well as dose, effect value, and variance information. | High-quality data; All-purpose; Data integration; Distinguishing between missing and negative endpoints | EPA | https://www.epa.gov/chemical-research/downloadable-computational-toxicology-data |
| 4 | Toxin and Toxin Target Database (T3DB) [24] | It contains 3678 toxins, described by 41,602 synonyms, including pollutants, pesticides, drugs, and food toxins. These toxins were associated with 2073 corresponding toxin target records, with a total of 42,374 toxin target associations. | Data diversity and accessibility; Potential applications; Data integration | The Chinese University of Hong Kong | http://www.t3db.ca/ |
| 5 | Therapeutic Target Database (TTD) [25] | TTD contains more than 3500 drug targets and nearly 40,000 drug molecules. The database provides information about target-related diseases and helps researchers understand the potential role of targets in disease treatment. | Reliable data source; Clear target classification; Powerful retrieval function; Rich auxiliary functions | National University of Singapore | http://db.idrblab.net/ttd/ |
| 6 | Side Effect Resource (SIDER) [26] | It contains information on 1430 drugs and 5868 side effects, covering a variety of treatment areas. The database provides frequency data of each side effect, allowing users to assess the possibility of specific adverse reactions. | Comprehensiveness; Reliable data source; Multilingual support; Structured data | Technical University of Berlin | http://sideeffects.embl.de/ |
| 7 | The Marker [27] | It contains 218 efficacy biomarkers, 624 safety biomarkers, 104 monitoring biomarkers, 15,893 predictive biomarkers, and 103 alternative endpoints. These data cover a large number of drugs and a wide range of disease categories, not limited to anti-cancer therapy. | Systematic organization; Rich data; User-friendliness; Free access | IDRBLAB | https://idrblab.org/themarker |
| 8 | Comparative Toxicogenomics Database (CTD) [28] | CTD contains the interaction information between chemical substances and genes/proteins, as well as the interaction information between chemical substances and phenotypes. | User-friendly interface; The database is searchable, accessible, interoperable, and reusable; Non-redundant data; Comparative analysis is available | North Carolina State University | https://ctdbase.org |
| 9 | Gene Expression Omnibus (GEO) [29] | GEO’s significance for ML models stems from its vast repository of gene expression data, encompassing both microarray and RNA-seq datasets. These datasets enable the training of models to predict how chemicals or drugs alter gene expression, thereby providing insights into their potential molecular-level toxic effects. | High data quality; Various forms of data storage; Data visualization; Easy data retrieval; Timely data update | National Center for Biotechnology Information (NCBI) | https://www.ncbi.nlm.nih.gov/geo/ |
| 10 | Drug Matrix [30] | It contains the comprehensive results of thousands of highly controlled and standardized toxicological experiments involving rat or primary rat hepatocytes, which are systematically treated with therapeutic, industrial, and environmental chemicals, including non-toxic doses and toxic doses | High-quality data; Multiple data types; User-friendliness; Data analysis tools | National Toxicology Program (NTP) | https://norecopa.no/3r-guide/drugmatrix https://ntp.niehs.nih.gov/drugmatrix/index.html |
| 11 | Kyoto Encyclopedia of Genes and Genomes (KEGG) [31] | KEGG is an encyclopedia of genes and genomes, which offers structured information on metabolic pathways, molecular interactions, and gene functions. | Highly organized data structure; Multiple data types; Cross-species comparison; Multiple applications | Kanehisa Laboratory | https://www.genome.jp/kegg |
| 12 | Universal Protein Database (UniProt) [32] | It is a comprehensive database of protein sequence and function information. It provides detailed annotation of protein function, including protein function, subcellular localization, post-translational modification, protein–protein interaction, and pathway information. | Functional notes; Data consolidation; Sequence similarity search; Proteomic analysis | European Bioinformatics Institute (EBI) | https://www.uniprot.org/ |
| 13 | Therapeutics Data Commons (TDC) [33] | TDC covers a wide range of learning tasks, including target discovery, activity screening, efficacy, safety, and manufacturing, involving small molecules, antibodies, vaccines, and other biomedical products. | Three-tier structure; Rich datasets; Machine learning tasks; Data processing and evaluation | Harvard University | https://tdcommons.ai/ |
| 14 | Toxbank [34] | Dedicated database for toxicity data management and modeling repository of “gold” compounds and selected test compounds, as well as reference resources of cells, cell lines, and tissues related to in vitro toxicity studies. | Data management; Interdisciplinary cooperation; Data sharing | EU FP7 project | https://toxbank.net/ |
| 15 | LiverTox: Clinical and Research Information on Drug-Induced Liver Injury (LIVERTOX) [35] | LiverTox provides the clinical characteristics of drug-induced liver injury, disease severity classification, and causality evaluation scale. The database contains detailed records of more than 1400 drugs, herbs, and dietary supplements. The records of each drug include background information, hepatotoxicity description, case reports, references, etc. | Free use; Timely update; Comprehensiveness; Clinical and research support | National Library of Medicine (NIH) | https://livertox.nih.gov/ |
| 16 | Gene Expression Nebulas (GEN) [36] | Gen database integrates 323 high-quality transcriptome datasets, covering 50,500 samples of 30 species and 15,540,169 cells, and provides transcriptional maps under six biological scenarios: baseline reference, genetics, phenotype, environment, time, and space. | Data quality control and standardization; Analysis and visualization tools; User-friendliness | National Biological Information Center | https://ngdc.cncb.ac.cn/gen |
| 17 | The Cancer Genome Atlas (TCGA) [37] | The TCGA project conducted molecular characterization analysis on more than 20,000 primary cancers and matched normal samples, covering 33 cancer categories. It also contains detailed clinical data, such as patient survival time, treatment response, etc. These data are of great value for the study of clinical characteristics and treatment effects of cancer | Openness and sharing of data; Diversity and comprehensiveness of data; High quality and standardization of data | National Cancer Institute (NCI) | https://www.cancer.gov/ccg/research/genome-sequencing/tcga |
| 18 | GeneCards [38] | Genecards provide detailed information about all known and predicted human genes, including genome location, function, expression pattern, genetic variation, clinical relevance, and functional annotation of genes. It also integrates a variety of biological pathway information, providing the role and relationship of genes in different biological pathways | Data consolidation; User-friendliness; Analytical tools; Data access and mining | Weizmann Institute of Science | https://www.genecards.org/ |
3. Application of Artificial Intelligence and Core Algorithm Technology
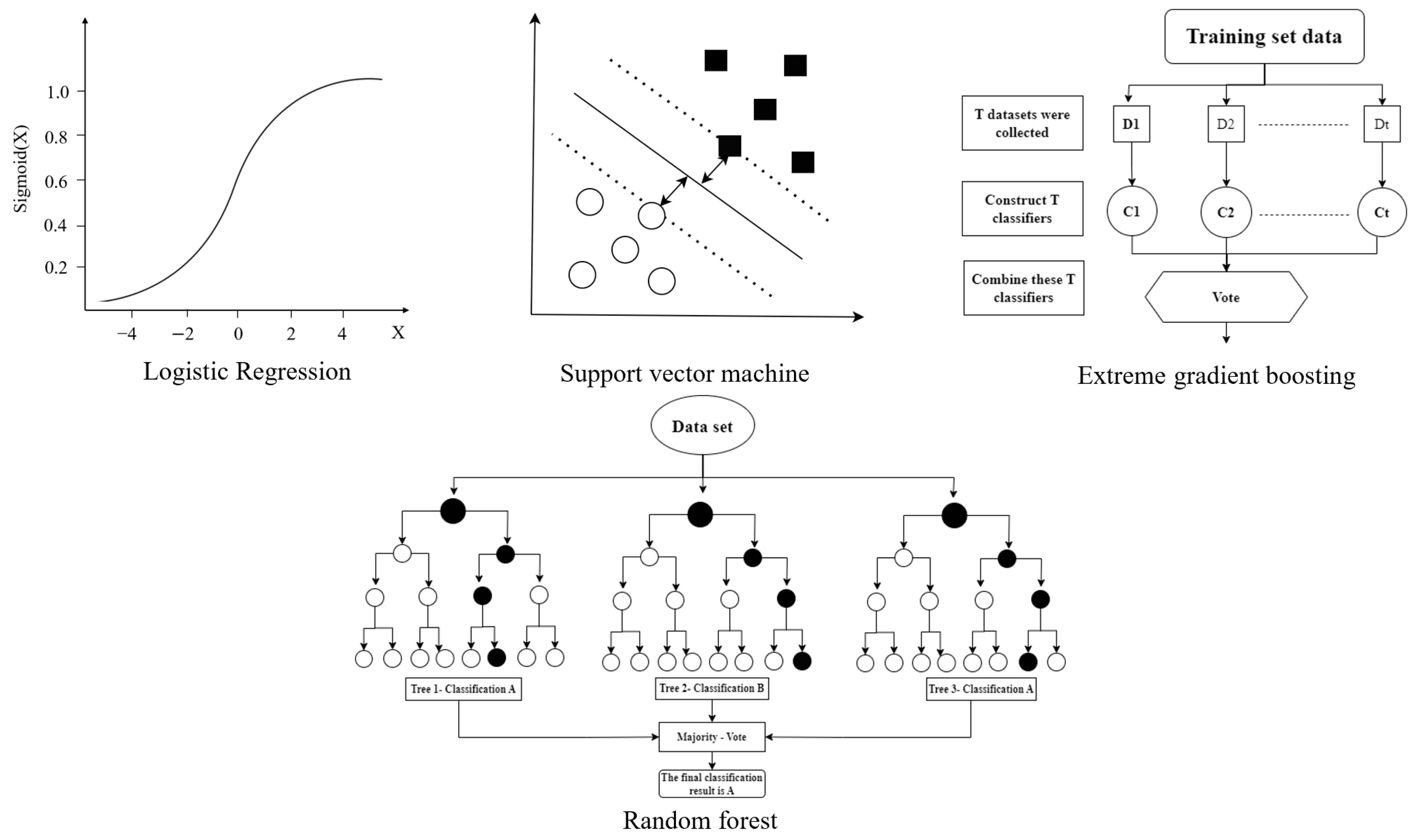
3.1. Machine Learning Algorithm
3.1.1. Traditional Machine Learning Algorithm
3.1.2. Deep Learning Algorithm
3.2. Molecular Representation
3.2.1. Molecular Fingerprint
3.2.2. Molecular Descriptor
3.2.3. Molecular Diagram
4. Research Progress of Different Toxicity Prediction
4.1. Acute Toxicity Prediction
4.1.1. Definition and Evaluation Index of Acute Toxicity
4.1.2. Relevant Research Results and Methods
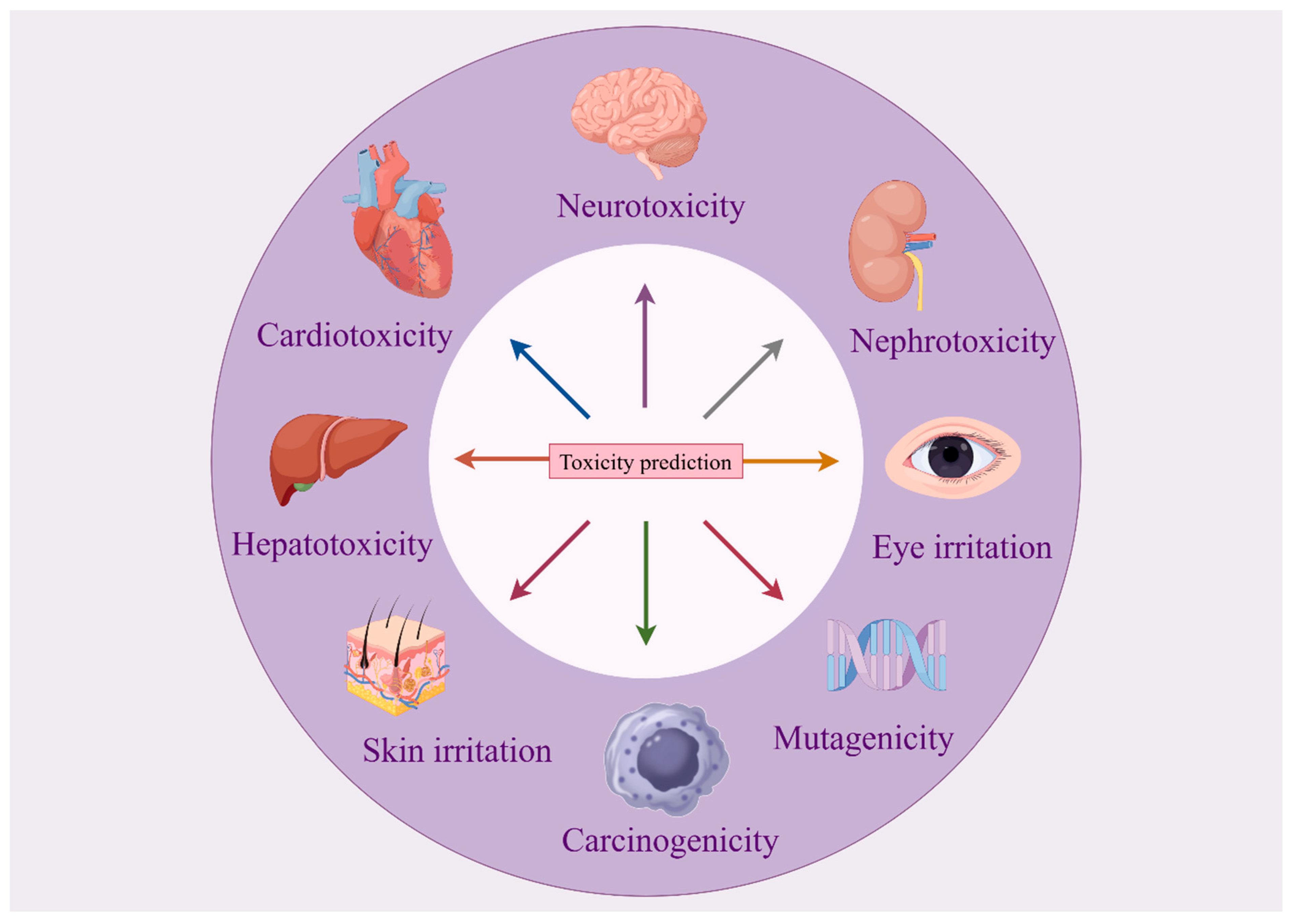
4.2. Organ-Specific Toxicity Prediction
4.2.1. Hepatotoxicity
4.2.2. Nephrotoxicity
4.2.3. Cardiotoxicity
4.2.4. Neurotoxicity
4.2.5. Other Toxicities
5. Challenges and Prospects
5.1. Expanding Sample Sizes
5.2. Improving Data Quality and Integrating Diverse Sources
5.3. Enhancing ML Models for Diverse Toxicity Detection
5.4. Enhance the Interpretability of ML in the Prediction of Drug Toxicity
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
List of Abbreviations
| Full Term | Abbreviation |
| Absorption, Distribution, Metabolism, Excretion and Toxicity | ADMET |
| Application Programming Interface | API |
| Artificial Intelligence | AI |
| Classification Read-Across Structure-Activity Relationship | RASAR |
| Comparative Toxicogenomics Database | CTD |
| Convolutional Neural Network | CNN |
| Deep Learning | DL |
| Deep Neural Network | DNN |
| Deoxyribonucleic Acid | DNA |
| Distributed Structure-Searchable Toxicity Database | DSSTox |
| Drug-Induced Liver Injury | DILI |
| Electronic Medical Record | EMR |
| Environmental Protection Agency | EPA |
| European Bioinformatics Institute | EBI |
| Extreme Gradient Boosting | XGB |
| FDA Adverse Event Reporting System | FAERS |
| Federated Learning | FL |
| Food and Drug Administration | FDA |
| Gene Expression Nebulas | GEN |
| Gene Expression Omnibus | GEO |
| Globally Harmonized System | GHS |
| Gradient Boosting Machines | GBM |
| Gradient Boosting Trees | GBT |
| Graph Convolutional Networks | GCN |
| Graph Neural Network | GNN |
| Half Maximal Inhibitory Concentration | IC50 |
| Integrated Chemical Environment | ICE |
| K-Nearest Neighbor | KNN |
| Kyoto Encyclopedia of Genes and Genomes | KEGG |
| LiverTox: Clinical and Research Information on Drug-Induced Liver Injury | LIVERTOX |
| Logistic Regression | LR |
| Machine Learning | ML |
| Matthews Correlation Coefficient | MCC |
| Median Lethal Dose | LD50 |
| Molecular Access System | MACCS |
| National Cancer Institute | NCI |
| National Institute of Technology and Evaluation | NITE |
| National Library of Medicine | NIH |
| National Toxicology Program | NTP |
| Natural Language Processing | NLP |
| Neural Network | NN |
| Online Chemical Modeling Environment | OCHEM |
| Quantitative Structure-Activity Relationship | QSAR |
| Radio Therapy | RT |
| Random Forest | RF |
| Root Mean Square Errors | RMSEs |
| SHapley Additive exPlanations | SHAP |
| Side Effect Resource | SIDER |
| Structural Alarms | SA |
| Support Vector Machine | SVM |
| The Cancer Genome Atlas | TCGA |
| Therapeutic Target Database | TTD |
| Therapeutics Data Commons | TDC |
| Toxicity Reference Database | ToxRefDB |
| Toxicology Resources for Intelligent Computation | TOXRIC |
| Toxin and Toxin Target Database | T3DB |
| Trihalomethanes | THMs |
| Universal Protein Database | UniProt |
| World Health Organization | WHO |
References
- Basile, A.O.; Yahi, A.; Tatonetti, N.P. Artificial Intelligence for Drug Toxicity and Safety. Trends Pharmacol. Sci. 2019, 40, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Raies, A.B.; Bajic, V.B. In silico toxicology: Computational methods for the prediction of chemical toxicity. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2016, 6, 147–172. [Google Scholar] [CrossRef] [PubMed]
- Onakpoya, I.J.; Heneghan, C.J.; Aronson, J.K. Worldwide withdrawal of medicinal products because of adverse drug reactions: A systematic review and analysis. Crit. Rev. Toxicol. 2016, 46, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Zhou, X.; Geng, X.; Drewell, C.; Hubner, J.; Li, Z.; Zhang, Y.; Xue, M.; Marx, U.; Li, B. Repeated dose multi-drug testing using a microfluidic chip-based coculture of human liver and kidney proximal tubules equivalents. Sci. Rep. 2020, 10, 8879. [Google Scholar] [CrossRef]
- Handelman, G.S.; Kok, H.K.; Chandra, R.V.; Razavi, A.H.; Lee, M.J.; Asadi, H. eDoctor: Machine learning and the future of medicine. J. Intern. Med. 2018, 284, 603–619. [Google Scholar] [CrossRef]
- Ali, H.; Shroff, A.; Fulop, T.; Molnar, M.Z.; Sharif, A.; Burke, B.; Shroff, S.; Briggs, D.; Krishnan, N. Artificial intelligence assisted risk prediction in organ transplantation: A UK Live-Donor Kidney Transplant Outcome Prediction tool. Ren. Fail. 2025, 47, 2431147. [Google Scholar] [CrossRef]
- Miotto, R.; Wang, F.; Wang, S.; Jiang, X.; Dudley, J.T. Deep learning for healthcare: Review, opportunities and challenges. Brief. Bioinform. 2018, 19, 1236–1246. [Google Scholar] [CrossRef]
- Mullowney, M.W.; Duncan, K.R.; Elsayed, S.S.; Garg, N.; van der Hooft, J.J.J.; Martin, N.I.; Meijer, D.; Terlouw, B.R.; Biermann, F.; Blin, K.; et al. Artificial intelligence for natural product drug discovery. Nat. Rev. Drug Discov. 2023, 22, 895–916. [Google Scholar] [CrossRef]
- Wu, L.; Yan, B.; Han, J.; Li, R.; Xiao, J.; He, S.; Bo, X. TOXRIC: A comprehensive database of toxicological data and benchmarks. Nucleic Acids Res. 2023, 51, D1432–D1445. [Google Scholar] [CrossRef]
- Bell, S.M.; Phillips, J.; Sedykh, A.; Tandon, A.; Sprankle, C.; Morefield, S.Q.; Shapiro, A.; Allen, D.; Shah, R.; Maull, E.A.; et al. An Integrated Chemical Environment to Support 21st-Century Toxicology. Environ. Health Perspect. 2017, 125, 054501. [Google Scholar] [CrossRef]
- Daniel, A.B.; Choksi, N.; Abedini, J.; Bell, S.; Ceger, P.; Cook, B.; Karmaus, A.L.; Rooney, J.; To, K.T.; Allen, D.; et al. Data curation to support toxicity assessments using the Integrated Chemical Environment. Front. Toxicol. 2022, 4, 987848. [Google Scholar] [CrossRef]
- Grulke, C.M.; Williams, A.J.; Thillanadarajah, I.; Richard, A.M. EPA’s DSSTox database: History of development of a curated chemistry resource supporting computational toxicology research. Comput. Toxicol. 2019, 12, 100096. [Google Scholar] [CrossRef] [PubMed]
- Knox, C.; Wilson, M.; Klinger, C.M.; Franklin, M.; Oler, E.; Wilson, A.; Pon, A.; Cox, J.; Chin, N.E.L.; Strawbridge, S.A.; et al. DrugBank 6.0: The DrugBank Knowledgebase for 2024. Nucleic Acids Res. 2024, 52, D1265–D1275. [Google Scholar] [CrossRef] [PubMed]
- Zdrazil, B.; Felix, E.; Hunter, F.; Manners, E.J.; Blackshaw, J.; Corbett, S.; de Veij, M.; Ioannidis, H.; Lopez, D.M.; Mosquera, J.F.; et al. The ChEMBL Database in 2023: A drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic Acids Res. 2024, 52, D1180–D1192. [Google Scholar] [CrossRef]
- Kovalishyn, V.; Zyabrev, V.; Kachaeva, M.; Ziabrev, K.; Keith, K.; Harden, E.; Hartline, C.; James, S.H.; Brovarets, V. Design of new imidazole derivatives with anti-HCMV activity: QSAR modeling, synthesis and biological testing. J. Comput. Aided Mol. Des. 2021, 35, 1177–1187. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef]
- Ito, M.; Codony-Servat, C.; Codony-Servat, J.; Llige, D.; Chaib, I.; Sun, X.; Miao, J.; Sun, R.; Cai, X.; Verlicchi, A.; et al. Targeting PKCiota-PAK1 signaling pathways in EGFR and KRAS mutant adenocarcinoma and lung squamous cell carcinoma. Cell Commun. Signal 2019, 17, 137. [Google Scholar] [CrossRef]
- Pu, X.; Ye, Q.; Cai, J.; Yang, X.; Fu, Y.; Fan, X.; Wu, H.; Chen, J.; Qiu, Y.; Yue, S. Typing FGFR2 translocation determines the response to targeted therapy of intrahepatic cholangiocarcinomas. Cell Death Dis. 2021, 12, 256. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yang, Q.; Li, Z.; Yan, S.; Ming, S. Systematic analysis of sugammadex-related adverse drug reaction signals using FAERS database. Int. J. Surg. 2025, 111, 1988–1994. [Google Scholar] [CrossRef]
- Elizabeth, R.M.C.; Sattari, F.; Lefsrud, L.; Gue, B. Visualizing what’s missing: Using deep learning and Bow-Tie diagrams to identify and visualize missing leading indicators in industrial construction. J. Saf. Res. 2025, 93, 1–11. [Google Scholar] [CrossRef]
- Feshuk, M.; Kolaczkowski, L.; Dunham, K.; Davidson-Fritz, S.E.; Carstens, K.E.; Brown, J.; Judson, R.S.; Paul Friedman, K. The ToxCast pipeline: Updates to curve-fitting approaches and database structure. Front. Toxicol. 2023, 5, 1275980. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.J.; Grulke, C.M.; Edwards, J.; McEachran, A.D.; Mansouri, K.; Baker, N.C.; Patlewicz, G.; Shah, I.; Wambaugh, J.F.; Judson, R.S.; et al. The CompTox Chemistry Dashboard: A community data resource for environmental chemistry. J. Cheminform. 2017, 9, 61. [Google Scholar] [CrossRef]
- Feshuk, M.; Kolaczkowski, L.; Watford, S.; Paul Friedman, K. ToxRefDB v2.1: Update to curated in vivo study data in the Toxicity Reference Database. Front. Toxicol. 2023, 5, 1260305. [Google Scholar] [CrossRef]
- Wishart, D.; Arndt, D.; Pon, A.; Sajed, T.; Guo, A.C.; Djoumbou, Y.; Knox, C.; Wilson, M.; Liang, Y.; Grant, J.; et al. T3DB: The toxic exposome database. Nucleic Acids Res. 2015, 43, D928–D934. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Y.; Zhao, D.; Yu, X.; Shen, X.; Zhou, Y.; Wang, S.; Qiu, Y.; Chen, Y.; Zhu, F. TTD: Therapeutic Target Database describing target druggability information. Nucleic Acids Res. 2024, 52, D1465–D1477. [Google Scholar] [CrossRef]
- Kuhn, M.; Letunic, I.; Jensen, L.J.; Bork, P. The SIDER database of drugs and side effects. Nucleic Acids Res. 2016, 44, D1075–D1079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, Y.; Zhou, Y.; Yu, X.; Shen, X.; Hong, Y.; Zhang, Y.; Wang, S.; Mou, M.; Zhang, J.; et al. TheMarker: A comprehensive database of therapeutic biomarkers. Nucleic Acids Res. 2024, 52, D1450–D1464. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.P.; Wiegers, T.C.; Johnson, R.J.; Sciaky, D.; Wiegers, J.; Mattingly, C.J. Comparative Toxicogenomics Database (CTD): Update 2023. Nucleic Acids Res. 2023, 51, D1257–D1262. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Lin, L.; Wan, L.; He, H.; Liu, W. Drug vector representation: A tool for drug similarity analysis. Mol. Genet. Genom. 2020, 295, 1055–1062. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Huang, K.; Fu, T.; Gao, W.; Zhao, Y.; Roohani, Y.; Leskovec, J.; Coley, C.W.; Xiao, C.; Sun, J.; Zitnik, M. Artificial intelligence foundation for therapeutic science. Nat. Chem. Biol. 2022, 18, 1033–1036. [Google Scholar] [CrossRef]
- Kohonen, P.; Benfenati, E.; Bower, D.; Ceder, R.; Crump, M.; Cross, K.; Grafstrom, R.C.; Healy, L.; Helma, C.; Jeliazkova, N.; et al. The ToxBank Data Warehouse: Supporting the Replacement of In Vivo Repeated Dose Systemic Toxicity Testing. Mol. Inform. 2013, 32, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Kelleci Celi, K.F.; Karaduman, G. Machine Learning-Based Prediction of Drug-Induced Hepatotoxicity: An OvA-QSTR Approach. J. Chem. Inf. Model. 2023, 63, 4602–4614. [Google Scholar] [CrossRef]
- Zhang, Y.; Zou, D.; Zhu, T.; Xu, T.; Chen, M.; Niu, G.; Zong, W.; Pan, R.; Jing, W.; Sang, J.; et al. Gene Expression Nebulas (GEN): A comprehensive data portal integrating transcriptomic profiles across multiple species at both bulk and single-cell levels. Nucleic Acids Res. 2022, 50, D1016–D1024. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Wu, X.; Wei, D.; Zhou, Y.; Cao, Q.; Han, G.; Han, E.; Chen, Z.; Guo, Y.; Huo, W.; Wang, C.; et al. Pesticide exposures and 10-year atherosclerotic cardiovascular disease risk: Integrated epidemiological and bioinformatics analysis. J. Hazard. Mater. 2025, 485, 136835. [Google Scholar] [CrossRef]
- Li, X.; Cui, Y.; Gao, S.; Zhang, Q.; Dai, Y.; Wang, S.; Wu, J.; Li, G.; Song, J. Development and validation of a score model for predicting the risk of first esophagogastric variceal hemorrhage and mortality in patients with hepatocellular carcinoma. Ann. Med. 2025, 57, 2490210. [Google Scholar] [CrossRef]
- Albrich, W.C.; Just, N.; Kahlert, C.; Casanova, C.; Baty, F.; Hilty, M. Serotype epidemiology and case-fatality risk of invasive pneumococcal disease: A nationwide population study from Switzerland, 2012-2022. Emerg. Microbes Infect. 2025, 14, 2488189. [Google Scholar] [CrossRef]
- Ai, S.; Liu, X.; Zhao, N.; Miao, Q.; Qin, Y.; Li, X. Risk factors of infective endocarditis-associated acute kidney injury: Benefits of low-dose amikacin and surgery. Ann. Med. 2025, 57, 2482023. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Ding, W.; Yang, X.; Bai, H.; Jiao, F.; Guo, Y.; Zhang, T.; Zou, X.; Wang, Y. Construction and validation of a predictive model for meningoencephalitis in pediatric scrub typhus based on machine learning algorithms. Emerg. Microbes Infect. 2025, 14, 2469651. [Google Scholar] [CrossRef]
- Hwang, J.; Youm, C.; Park, H.; Kim, B.; Choi, H.; Cheon, S.M. Machine learning for early detection and severity classification in people with Parkinson’s disease. Sci. Rep. 2025, 15, 234. [Google Scholar] [CrossRef] [PubMed]
- Cakir, A.; Tuncer, M.; Taymaz-Nikerel, H.; Ulucan, O. Side effect prediction based on drug-induced gene expression profiles and random forest with iterative feature selection. Pharmacogenom. J. 2021, 21, 673–681. [Google Scholar] [CrossRef]
- Schoning, V.; Krahenbuhl, S.; Drewe, J. The hepatotoxic potential of protein kinase inhibitors predicted with Random Forest and Artificial Neural Networks. Toxicol. Lett. 2018, 299, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Lesinski, W.; Mnich, K.; Golinska, A.K.; Rudnicki, W.R. Integration of human cell lines gene expression and chemical properties of drugs for Drug Induced Liver Injury prediction. Biol. Direct 2021, 16, 2. [Google Scholar] [CrossRef]
- Fourches, D.; Barnes, J.C.; Day, N.C.; Bradley, P.; Reed, J.Z.; Tropsha, A. Cheminformatics analysis of assertions mined from literature that describe drug-induced liver injury in different species. Chem. Res. Toxicol. 2010, 23, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Mulliner, D.; Schmidt, F.; Stolte, M.; Spirkl, H.P.; Czich, A.; Amberg, A. Computational Models for Human and Animal Hepatotoxicity with a Global Application Scope. Chem. Res. Toxicol. 2016, 29, 757–767. [Google Scholar] [CrossRef]
- Lee, S.; Kang, Y.M.; Park, H.; Dong, M.S.; Shin, J.M.; No, K.T. Human nephrotoxicity prediction models for three types of kidney injury based on data sets of pharmacological compounds and their metabolites. Chem. Res. Toxicol. 2013, 26, 1652–1659. [Google Scholar] [CrossRef]
- Cai, C.; Guo, P.; Zhou, Y.; Zhou, J.; Wang, Q.; Zhang, F.; Fang, J.; Cheng, F. Deep Learning-Based Prediction of Drug-Induced Cardiotoxicity. J. Chem. Inf. Model. 2019, 59, 1073–1084. [Google Scholar] [CrossRef]
- Weber, S.; Erhardt, F.; Allgeier, J.; Saka, D.; Donga, N.; Neumann, J.; Lange, C.M.; Gerbes, A.L. Drug-Induced Liver Injury Caused by Metamizole: Identification of a Characteristic Injury Pattern. Liver Int. 2025, 45, e70012. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yu, P.; Zhang, T.G.; Kang, Y.L.; Zhao, X.; Li, Y.Y.; He, J.H.; Zhang, J. In silico prediction of drug-induced myelotoxicity by using Naive Bayes method. Mol. Divers. 2015, 19, 945–953. [Google Scholar] [CrossRef]
- Kokori, E.; Patel, R.; Olatunji, G.; Ukoaka, B.M.; Abraham, I.C.; Ajekiigbe, V.O.; Kwape, J.M.; Babalola, A.E.; Udam, N.G.; Aderinto, N. Machine learning in predicting heart failure survival: A review of current models and future prospects. Heart Fail. Rev. 2025, 30, 431–442. [Google Scholar] [CrossRef]
- Vo, A.H.; Van Vleet, T.R.; Gupta, R.R.; Liguori, M.J.; Rao, M.S. An Overview of Machine Learning and Big Data for Drug Toxicity Evaluation. Chem. Res. Toxicol. 2020, 33, 20–37. [Google Scholar] [CrossRef]
- Raies, A.B.; Bajic, V.B. In silico toxicology: Comprehensive benchmarking of multi-label classification methods applied to chemical toxicity data. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1352. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, F.; Luo, L.; Zhang, J. Predicting drug side effects by multi-label learning and ensemble learning. BMC Bioinform. 2015, 16, 365. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Shin, M. An integrative model of multi-organ drug-induced toxicity prediction using gene-expression data. BMC Bioinform. 2014, 15 (Suppl 16), S2. [Google Scholar] [CrossRef]
- Zhong, S.; Guan, X. Count-Based Morgan Fingerprint: A More Efficient and Interpretable Molecular Representation in Developing Machine Learning-Based Predictive Regression Models for Water Contaminants’ Activities and Properties. Environ. Sci. Technol. 2023, 57, 18193–18202. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.M.; Atta, S.; Naz, I.; Fatima, H.; Haq, I.U. Toxicological assessment of standardized Artemisia roxburghiana wall. ex Besser aqueous leaf extract to acute and subacute exposure in Albino mice. J. Ethnopharmacol. 2025, 344, 119531. [Google Scholar] [CrossRef]
- Lou, S.; Yu, Z.; Huang, Z.; Wang, H.; Pan, F.; Li, W.; Liu, G.; Tang, Y. In Silico Prediction of Chemical Acute Dermal Toxicity Using Explainable Machine Learning Methods. Chem. Res. Toxicol. 2024, 37, 513–524. [Google Scholar] [CrossRef]
- Li, Y.; Wang, B.; Ma, F.; Fan, W.; Wang, Y.; Chen, L.; Dong, Z. Using the super-learner to predict the chemical acute toxicity on rats. J. Hazard. Mater. 2024, 480, 136311. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhang, Y.; Mai, X.; Ai, P.; Xie, L.; Xue, B.; Zheng, Q. Excellent absorption-dominant electromagnetic interference shielding performances of asymmetric gradient layered composite films exploited with assistance of machine learning. J. Colloid. Interface Sci. 2025, 697, 137927. [Google Scholar] [CrossRef]
- Feitosa, F.L.; Victoria, F.C.; Sanches, I.H.; Silva-Mendonca, S.; Borba, J.; Braga, R.C.; Andrade, C.H. Cyto-Safe: A Machine Learning Tool for Early Identification of Cytotoxic Compounds in Drug Discovery. J. Chem. Inf. Model. 2024, 64, 9056–9062. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Jiang, Y.; Zhang, X.; Zheng, R.; Qiu, R.; Sun, Y.; Zhao, C.; Shang, H. ResNet18DNN: Prediction approach of drug-induced liver injury by deep neural network with ResNet18. Brief. Bioinform. 2022, 23, bbab503. [Google Scholar] [CrossRef]
- Fuzi, B.; Mathai, N.; Kirchmair, J.; Ecker, G.F. Toxicity prediction using target, interactome, and pathway profiles as descriptors. Toxicol. Lett. 2023, 381, 20–26. [Google Scholar] [CrossRef]
- Xu, Y.; Dai, Z.; Chen, F.; Gao, S.; Pei, J.; Lai, L. Deep Learning for Drug-Induced Liver Injury. J. Chem. Inf. Model. 2015, 55, 2085–2093. [Google Scholar] [CrossRef]
- Lee, S.; Yoo, S. InterDILI: Interpretable prediction of drug-induced liver injury through permutation feature importance and attention mechanism. J. Cheminform. 2024, 16, 1. [Google Scholar] [CrossRef]
- Gadaleta, D.; Garcia de Lomana, M.; Serrano-Candelas, E.; Ortega-Vallbona, R.; Gozalbes, R.; Roncaglioni, A.; Benfenati, E. Quantitative structure-activity relationships of chemical bioactivity toward proteins associated with molecular initiating events of organ-specific toxicity. J. Cheminform. 2024, 16, 122. [Google Scholar] [CrossRef] [PubMed]
- Jochum, K.; Miccoli, A.; Sommersdorf, C.; Poetz, O.; Braeuning, A.; Tralau, T.; Marx-Stoelting, P. Comparative case study on NAMs: Towards enhancing specific target organ toxicity analysis. Arch. Toxicol. 2024, 98, 3641–3658. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, Y.; Cao, J.; Lao, J.; Wang, B.; Li, S.; Huang, X.; Tang, F.; Li, X. The incidence and risk factors analysis of acute kidney injury in hospitalized patients received diuretics: A single-center retrospective study. Front. Pharmacol. 2022, 13, 924173. [Google Scholar] [CrossRef]
- Song, M.H.; Xiang, B.X.; Yang, C.Y.; Lee, C.H.; Yan, Y.X.; Yang, Q.J.; Yin, W.J.; Zhou, Y.; Zuo, X.C.; Xie, Y.L. A pilot clinical risk model to predict polymyxin-induced nephrotoxicity: A real-world, retrospective cohort study. J. Antimicrob. Chemother. 2024, 79, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Bao, P.; Sun, Y.; Qiu, P.; Li, X. Development and validation of a nomogram to predict the risk of vancomycin-related acute kidney injury in critical care patients. Front. Pharmacol. 2024, 15, 1389140. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Q.; Gong, X. Unveiling the Mysteries of Contrast-Induced Acute Kidney Injury: New Horizons in Pathogenesis and Prevention. Toxics 2024, 12, 620. [Google Scholar] [CrossRef]
- Rao, M.; Nassiri, V.; Srivastava, S.; Yang, A.; Brar, S.; McDuffie, E.; Sachs, C. Artificial Intelligence and Machine Learning Models for Predicting Drug-Induced Kidney Injury in Small Molecules. Pharmaceuticals 2024, 17, 1550. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Zheng, X.; Luo, J.; Zhang, Y.; Gao, X.; Jin, L.; Liu, W.; Zhang, C.; Gao, D.; Xu, B.; et al. Machine learning accelerates the discovery of epitope-based dual-bioactive peptides against skin infections. Int. J. Antimicrob. Agents 2024, 64, 107371. [Google Scholar] [CrossRef]
- Banerjee, A.; Roy, K. Machine learning assisted classification RASAR modeling for the nephrotoxicity potential of a curated set of orally active drugs. Sci. Rep. 2025, 15, 808. [Google Scholar] [CrossRef]
- Heo, S.; Kang, E.A.; Yu, J.Y.; Kim, H.R.; Lee, S.; Kim, K.; Hwangbo, Y.; Park, R.W.; Shin, H.; Ryu, K.; et al. Time Series AI Model for Acute Kidney Injury Detection Based on a Multicenter Distributed Research Network: Development and Verification Study. JMIR Med. Inform. 2024, 12, e47693. [Google Scholar] [CrossRef]
- Feng, X.; Jin, X.; Zhou, R.; Jiang, Q.; Wang, Y.; Zhang, X.; Shang, K.; Zhang, J.; Yu, C.; Shou, J. Deep learning approach identified a gene signature predictive of the severity of renal damage caused by chronic cadmium accumulation. J. Hazard. Mater. 2022, 433, 128795. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Hua, Y.; Wang, B.; Zhang, R.; Li, X. In Silico Prediction and Insights Into the Structural Basis of Drug Induced Nephrotoxicity. Front. Pharmacol. 2021, 12, 793332. [Google Scholar] [CrossRef]
- Devalckeneer, A.; Bouviez, M.; Gautier, A.; Colet, J.M. Metabolomic Prediction of Cadmium Nephrotoxicity in the Snail Helix aspersa maxima. Metabolites 2024, 14, 455. [Google Scholar] [CrossRef]
- Mizuno, S.; Noda, T.; Mogushi, K.; Hase, T.; Iida, Y.; Takeuchi, K.; Ishiwata, Y.; Uchida, S.; Nagata, M. Prediction of Vancomycin-Associated Nephrotoxicity Based on the Area under the Concentration-Time Curve of Vancomycin: A Machine Learning Analysis. Biol. Pharm. Bull. 2024, 47, 1946–1952. [Google Scholar] [CrossRef] [PubMed]
- Dzidic-Krivic, A.; Sher, E.K.; Kusturica, J.; Farhat, E.K.; Nawaz, A.; Sher, F. Unveiling drug induced nephrotoxicity using novel biomarkers and cutting-edge preventive strategies. Chem. Biol. Interact. 2024, 388, 110838. [Google Scholar] [CrossRef] [PubMed]
- Chiu, L.W.; Ku, Y.E.; Chan, F.Y.; Lie, W.N.; Chao, H.J.; Wang, S.Y.; Shen, W.C.; Chen, H.Y. Machine learning algorithms to predict colistin-induced nephrotoxicity from electronic health records in patients with multidrug-resistant Gram-negative infection. Int. J. Antimicrob. Agents 2024, 64, 107175. [Google Scholar] [CrossRef]
- Su, R.; Yang, H.; Wei, L.; Chen, S.; Zou, Q. A multi-label learning model for predicting drug-induced pathology in multi-organ based on toxicogenomics data. PLoS Comput. Biol. 2022, 18, e1010402. [Google Scholar] [CrossRef]
- Mukherjee, S.; Swanson, K.; Walther, P.; Shivnaraine, R.V.; Leitz, J.; Pang, P.D.; Zou, J.; Wu, J.C. ADMET-AI Enables Interpretable Predictions of Drug-Induced Cardiotoxicity. Circulation 2025, 151, 285–287. [Google Scholar] [CrossRef]
- Yang, H.; Xiu, J.; Yan, W.; Liu, K.; Cui, H.; Wang, Z.; He, Q.; Gao, Y.; Han, W. Large Language Models as Tools for Molecular Toxicity Prediction: AI Insights into Cardiotoxicity. J. Chem. Inf. Model. 2025, 65, 2268–2282. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.S.; Lee, J.; Lee, Y.; Cho, D.; Oh, K.S.; Jang, J.; Nong, N.T.; Lee, H.M.; Na, D. hERGBoost: A gradient boosting model for quantitative IC(50) prediction of hERG channel blockers. Comput. Biol. Med. 2025, 184, 109416. [Google Scholar] [CrossRef]
- Chen, Z.; Li, N.; Zhang, P.; Li, Y.; Li, X. CardioDPi: An explainable deep-learning model for identifying cardiotoxic chemicals targeting hERG, Cav1.2, and Nav1.5 channels. J. Hazard. Mater. 2024, 474, 134724. [Google Scholar] [CrossRef]
- Au Yeung, V.P.W.; Obrezanova, O.; Zhou, J.; Yang, H.; Bowen, T.J.; Ivanov, D.; Saffadi, I.; Carter, A.S.; Subramanian, V.; Dillmann, I.; et al. Computational approaches identify a transcriptomic fingerprint of drug-induced structural cardiotoxicity. Cell Biol. Toxicol. 2024, 40, 50. [Google Scholar] [CrossRef]
- Zhu, Z.; Wu, R.; Luo, M.; Zeng, L.; Zhang, D.; Hu, N.; Hu, Y.; Li, Y. Two-Dimensional Deep Learning Frameworks for Drug-Induced Cardiotoxicity Detection. ACS Sens. 2024, 9, 3316–3326. [Google Scholar] [CrossRef]
- Huang, C.; Pei, J.; Li, D.; Liu, T.; Li, Z.; Zhang, G.; Chen, R.; Xu, X.; Li, B.; Lian, Z.; et al. Analysis and Validation of Critical Signatures and Immune Cell Infiltration Characteristics in Doxorubicin-Induced Cardiotoxicity by Integrating Bioinformatics and Machine Learning. J. Inflamm. Res. 2024, 17, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Long, M.; Alnoury, M.; Udupa, J.K.; Tong, Y.; Wu, C.; Poole, N.; Mannikeri, S.; Ky, B.; Feigenberg, S.J.; Zou, J.W.; et al. Prediction of Radiation Therapy Induced Cardiovascular Toxicity from Pretreatment CT Images in Patients with Thoracic Malignancy via an Optimal Biomarker Approach. Acad. Radiol. 2025, 32, 1895–1905. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, Y.; Zhang, R.; Chen, Z.; Hua, Y.; Zhang, P.; Guo, H.; Cui, X.; Huang, X.; Li, X. Machine Learning Modeling and Insights into the Structural Characteristics of Drug-Induced Neurotoxicity. J. Chem. Inf. Model. 2022, 62, 6035–6045. [Google Scholar] [CrossRef]
- Pang, X.; He, X.; Yang, Y.; Wang, L.; Sun, Y.; Cao, H.; Liang, Y. NeuTox 2.0: A hybrid deep learning architecture for screening potential neurotoxicity of chemicals based on multimodal feature fusion. Environ. Int. 2025, 195, 109244. [Google Scholar] [CrossRef]
- He, X.; Yang, Z.; Wang, L.; Sun, Y.; Cao, H.; Liang, Y. NeuTox: A weighted ensemble model for screening potential neuronal cytotoxicity of chemicals based on various types of molecular representations. J. Hazard. Mater. 2024, 465, 133443. [Google Scholar] [CrossRef]
- Bercu, J.; Trejo-Martin, A.; Chen, C.; Schuler, M.; Cheung, J.; Cheairs, T.; Lynch, A.M.; Thomas, D.; Czich, A.; Atrakchi, A.; et al. HESI GTTC ring trial: Concordance between Ames and rodent carcinogenicity outcomes for N-nitrosamines (NAs) with rat and hamster metabolic conditions. Regul. Toxicol. Pharmacol. 2025, 161, 105835. [Google Scholar] [CrossRef]
- Rane, R.; Satpute, B.; Kumar, D.; Suryawanshi, M.; Prabhune, A.G.; Gawade, B.; Mahajan, A.; Pawar, A.; Sakat, S. Mutagenic and genotoxic in silico QSAR prediction of dimer impurity of gliflozins; canagliflozin, dapaglifozin, and emphagliflozin and in vitro evaluation by Ames and micronucleus test. Drug Chem. Toxicol. 2025, 48, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ma, W.; Ouyang, Y.; Cheng, P.; Zhong, D. Prediction of trihalomethane occurrence and cancer risk using interpretable machine learning and virtual data augmentation. J. Hazard. Mater. 2025, 494, 138697. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Lou, S.; Wang, H.; Li, W.; Liu, G.; Tang, Y. AttentiveSkin: To Predict Skin Corrosion/Irritation Potentials of Chemicals via Explainable Machine Learning Methods. Chem. Res. Toxicol. 2024, 37, 361–373. [Google Scholar] [CrossRef]
- Di, P.; Zheng, M.; Yang, T.; Chen, G.; Ren, J.; Li, X.; Jiang, H. Prediction of serious eye damage or eye irritation potential of compounds via consensus labelling models and active learning models based on uncertainty strategies. Food Chem. Toxicol. 2022, 169, 113420. [Google Scholar] [CrossRef]
- Lynch, C.; Sakamuru, S.; Ooka, M.; Huang, R.; Klumpp-Thomas, C.; Shinn, P.; Gerhold, D.; Rossoshek, A.; Michael, S.; Casey, W.; et al. High-Throughput Screening to Advance In Vitro Toxicology: Accomplishments, Challenges, and Future Directions. Annu. Rev. Pharmacol. Toxicol. 2024, 64, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Dou, B.; Zhu, Z.; Merkurjev, E.; Ke, L.; Chen, L.; Jiang, J.; Zhu, Y.; Liu, J.; Zhang, B.; Wei, G.W. Machine Learning Methods for Small Data Challenges in Molecular Science. Chem. Rev. 2023, 123, 8736–8780. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.S.; Ying, R.Y.; Chen, X.T.; Yeh, Y.J.; Wei, C.C.; Chan, C.C. Integrating high-throughput phenotypic profiling and transcriptomic analyses to predict the hepatosteatosis effects induced by per- and polyfluoroalkyl substances. J. Hazard. Mater. 2024, 469, 133891. [Google Scholar] [CrossRef]
- Laban, B.B.; Ralevic, U.; Leskovac, A.; Petrovic, S.; Stoiljkovic, M.; Rosic, M.; Nikezic, A.V. Spectral and cytotoxicity studies of hybrid silver nanoparticles in human lymphocytes. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2025, 340, 126360. [Google Scholar] [CrossRef]
- Mukhopadhyay, D.; Cocco, P.; Orru, S.; Cherchi, R.; De Matteis, S. The role of MicroRNAs as early biomarkers of asbestos-related lung cancer: A systematic review and meta-analysis. Pulmonology 2025, 31, 2416792. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liang, Z.; Zhang, G. Using explainable machine learning to investigate the relationship between childhood maltreatment, positive psychological traits, and CPTSD symptoms. Eur. J. Psychotraumatol. 2025, 16, 2455800. [Google Scholar] [CrossRef]
- Son, A.; Park, J.; Kim, W.; Yoon, Y.; Lee, S.; Ji, J.; Kim, H. Recent Advances in Omics, Computational Models, and Advanced Screening Methods for Drug Safety and Efficacy. Toxics 2024, 12, 822. [Google Scholar] [CrossRef]
- Nelson, C.P.; Brown, P.; Fitzpatrick, S.; Ford, K.A.; Howard, P.C.; MacGill, T.; Margerrison, E.E.C.; O’Shaughnessy, J.; Patterson, T.A.; Raghuwanshi, R.; et al. Advancing alternative methods to reduce animal testing. Science 2024, 386, 724–726. [Google Scholar] [CrossRef]
- Bai, C.; Wu, L.; Li, R.; Cao, Y.; He, S.; Bo, X. Machine Learning-Enabled Drug-Induced Toxicity Prediction. Adv. Sci. 2025, 12, e2413405. [Google Scholar] [CrossRef]
| Category | Method | Core Principle | Key Features |
|---|---|---|---|
| Molecular Fingerprint | Morgan Fingerprint | Circular substructure expansion around each atom |
|
| MACCS Fingerprint | 166 predefined substructure keys |
| |
| RDKit Fingerprint | Hybrid of path-based and topological patterns |
| |
| Molecular Descriptor | Physicochemical | Mathematical quantification of properties |
|
| Topological | Graph-theory indices |
| |
| Electronic | Quantum chemical properties |
| |
| Molecular Graph | GNN | Atoms = Nodes, Bonds = Edges |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, R.; Wen, H.; Lin, Z.; Li, B.; Zhou, X. Artificial Intelligence-Driven Drug Toxicity Prediction: Advances, Challenges, and Future Directions. Toxics 2025, 13, 525. https://doi.org/10.3390/toxics13070525
Zhang R, Wen H, Lin Z, Li B, Zhou X. Artificial Intelligence-Driven Drug Toxicity Prediction: Advances, Challenges, and Future Directions. Toxics. 2025; 13(7):525. https://doi.org/10.3390/toxics13070525
Chicago/Turabian StyleZhang, Ruiqiu, Hairuo Wen, Zhi Lin, Bo Li, and Xiaobing Zhou. 2025. "Artificial Intelligence-Driven Drug Toxicity Prediction: Advances, Challenges, and Future Directions" Toxics 13, no. 7: 525. https://doi.org/10.3390/toxics13070525
APA StyleZhang, R., Wen, H., Lin, Z., Li, B., & Zhou, X. (2025). Artificial Intelligence-Driven Drug Toxicity Prediction: Advances, Challenges, and Future Directions. Toxics, 13(7), 525. https://doi.org/10.3390/toxics13070525