
Beta-Blockers as Potential Adjuvants in Melanoma Treatment

 , ,
, ,  and
and 
Abstract
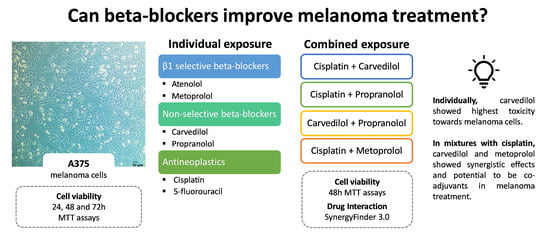
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Biological Model
2.3. Experimental Design
2.3.1. Individual Exposures
2.3.2. Combined Exposures
2.4. Cell Viability Assay
2.5. Data Analysis
3. Results
3.1. Individual Exposure
3.1.1. β-Blockers
3.1.2. Antineoplastic Drugs
3.2. Combined Exposures
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Ervik, M.; Lam, F.; Laversanne, M.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today; International Agency for Research on Cancer: Lyon, France, 2024; Available online: https://gco.iarc.who.int/today (accessed on 10 February 2025).
- SEER*Explorer: An Interactive Website for SEER Cancer Statistics. Surveillance Research Program, National Cancer Institute. Available online: https://seer.cancer.gov/explorer/ (accessed on 31 March 2023).
- Ferlay, J.; Laversanne, M.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Tomorrow (version 1.1); International Agency for Research on Cancer: Lyon, France, 2024; Available online: https://gco.iarc.who.int/tomorrow (accessed on 10 February 2025).
- Lazovich, D.; Vogel, R.I.; Berwick, M.; Weinstock, M.A.; Anderson, K.E.; Warshaw, E.M. Indoor tanning and risk of melanoma: A case-control study in a highly exposed population. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1557–1568. [Google Scholar] [CrossRef]
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Barsouk, A. Epidemiology of Melanoma. Med. Sci. 2021, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.J.; Johnson, D.B.; Sosman, J.A.; Chandra, S. Melanoma: What do all the mutations mean? Cancer 2018, 124, 3490–3499. [Google Scholar] [CrossRef]
- Natarelli, N.; Aleman, S.J.; Mark, I.M.; Tran, J.T.; Kwak, S.; Botto, E.; Aflatooni, S.; Diaz, M.J.; Lipner, S.R. A Review of Current and Pipeline Drugs for Treatment of Melanoma. Pharmaceuticals 2024, 17, 214. [Google Scholar] [CrossRef] [PubMed]
- Pham, J.P.; Joshua, A.M.; da Silva, I.P.; Dummer, R.; Goldinger, S.M. Chemotherapy in Cutaneous Melanoma: Is There Still a Role? Curr. Oncol. Rep. 2023, 25, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Eddy, K.; Chen, S. Overcoming Immune Evasion in Melanoma. Int. J. Mol. Sci. 2020, 21, 8984. [Google Scholar] [CrossRef]
- Lopes, J.; Rodrigues, C.M.P.; Gaspar, M.M.; Reis, C.P. Melanoma Management: From Epidemiology to Treatment and Latest Advances. Cancers 2022, 14, 4652. [Google Scholar] [CrossRef]
- Kulkarni, V.S.; Alagarsamy, V.; Solomon, V.R.; Jose, P.A.; Murugesan, S. Drug Repurposing: An Effective Tool in Modern Drug Discovery. Russ. J. Bioorg. Chem. 2023, 49, 157–166. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xia, Y.; Sun, M.; Huang, H.; Jin, W.L. Drug repurposing for cancer therapy. Signal Transduct. Target. Ther. 2024, 9, 92. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Farzam, K.; Jan, A. Beta Blockers. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK532906/ (accessed on 26 June 2024).
- Oliver, E.; Mayor, F., Jr.; D’Ocon, P. Beta-blockers: Historical Perspective and Mechanisms of Action. Rev. Esp. Cardiol. 2019, 72, 853–862. [Google Scholar] [CrossRef]
- Peixoto, R.; Pereira, M.L.; Oliveira, M. Beta-Blockers and Cancer: Where Are We? Pharmaceuticals 2020, 13, 105. [Google Scholar] [CrossRef] [PubMed]
- Massalee, R.; Cao, X. Repurposing beta-blockers for combinatory cancer treatment: Effects on conventional and immune therapies. Front. Pharmacol. 2024, 14, 1325050. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhao, B.; Ban, F.; Li, Y.; Shi, Q.; Guo, S.; Yi, X.; Wang, H.; Gao, T.; Li, C.; Zhu, G. Exploiting mitochondrial dysfunction to overcome BRAF inhibitor resistance in advanced melanoma: The role of disulfiram as a copper ionophore. Cell Death Dis. 2025, 16, 482. [Google Scholar] [CrossRef] [PubMed]
- Calvani, M.; Bruno, G.; Dabraio, A.; Subbiani, A.; Bianchini, F.; Fontani, F.; Casazza, G.; Vignoli, M.; De Logu, F.; Frenos, S.; et al. β3-Adrenoreceptor Blockade Induces Stem Cells Differentiation in Melanoma Microenvironment. Int. J. Mol. Sci. 2020, 21, 1420. [Google Scholar] [CrossRef]
- Cole, S.W.; Sood, A.K. Molecular pathways: Beta-adrenergic signaling in cancer. Clin. Cancer Res. 2012, 18, 1201–1206. [Google Scholar] [CrossRef]
- Batalla-Covello, J.; Ali, S.; Xie, T.; Amit, M. β-Adrenergic signaling in skin cancer. FASEB BioAdv. 2022, 4, 225–234. [Google Scholar] [CrossRef]
- Sharma, A.E.; Chan, S.; Komorowski, A.S.; Cao, X.; Gao, Y.; Kshatri, K.; Desai, K.; Kuksis, M.; Rosen, M.; Sachdeva, A.; et al. The Impact of Beta Blockers on Survival in Cancer Patients: A Systematic Review and Meta-Analysis. Cancers 2025, 17, 1357. [Google Scholar] [CrossRef]
- O’Logbon, J.; Tarantola, L.; Williams, N.R.; Mehta, S.; Ahmed, A.; Davies, E.A. Does propranolol have a role in cancer treatment? A systematic review of the epidemiological and clinical trial literature on beta-blockers. J. Cancer Res. Clin. Oncol. 2025, 151, 212. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, Y.; Liu, F.; Li, Y.; Liu, X.; Ren, X.; Yuan, X. Impact of beta blockers on cancer neuroimmunology: A systematic review and meta-analysis of survival outcomes and immune modulation. Front. Immunol. 2025, 16, 1635331. [Google Scholar] [CrossRef]
- Fateeva, A.; Eddy, K.; Chen, S. Current State of Melanoma Therapy and Next Steps: Battling Therapeutic Resistance. Cancers 2024, 16, 1571. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Muller, N.M.; Tan, S.X.; Vipulaguna, N.; Zhou, C.; Hughes, M.C.B.; Soyer, H.P.; von Schuckmann, L.; Khosrotehrani, K. Beta-blockers and cutaneous melanoma outcomes: A systematic review and random-effects meta-analysis. Pigment Cell Melanoma Res. 2025, 38, e13225. [Google Scholar] [CrossRef]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell Viability Assays. In Assay Guidance Manual; Markossian, S., Grossman, A., Brimacombe, K., Arkin, M., Auld, D., Austin, C., Baell, J., Brimacombe, K., Chung, T.D.Y., Coussens, N.P., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. Available online: https://www.ncbi.nlm.nih.gov/books/NBK144065/ (accessed on 26 June 2024).
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 3.0: An interactive analysis and consensus interpretation of multi-drug synergies across multiple samples. Nucleic Acids Res. 2022, 50, W739–W743. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wawszczyk, J.; Wolan, R.; Smolik, S.; Kapral, M. In vitro and in silico study on the effect of carvedilol and sorafenib alone and in combination on the growth and inflammatory response of melanoma cells. Saudi Pharm. J. 2023, 31, 1306–1316. [Google Scholar] [CrossRef]
- Stanojkovic, T.P.; Zizak, Z.; Mihailovic-Stanojevic, N.; Petrovic, T.; Juranic, Z. Inhibition of proliferation on some neoplastic cell lines-act of carvedilol and captopril. J. Exp. Clin. Cancer Res. CR 2005, 24, 387–395. [Google Scholar]
- Dezong, G.; Zhongbing, M.; Qinye, F.; Zhigang, Y. Carvedilol suppresses migration and invasion of malignant breast cells by inactivating Src involving cAMP/PKA and PKCδ signaling pathway. J. Cancer Res. Ther. 2014, 10, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Gillis, R.D.; Botteri, E.; Chang, A.; Ziegler, A.I.; Chung, N.C.; Pon, C.K.; Shackleford, D.M.; Andreassen, B.K.; Halls, M.L.; Baker, J.G.; et al. Carvedilol blocks neural regulation of breast cancer progression in vivo and is associated with reduced breast cancer mortality in patients. Eur. J. Cancer 2021, 147, 106–116. [Google Scholar] [CrossRef]
- Farhoumand, L.S.; Fiorentzis, M.; Kraemer, M.M.; Sak, A.; Stuschke, M.; Rassaf, T.; Hendgen-Cotta, U.; Bechrakis, N.E.; Berchner-Pfannschmidt, U. The Adrenergic Receptor Antagonist Carvedilol Elicits Anti-Tumor Responses in Uveal Melanoma 3D Tumor Spheroids and May Serve as Co-Adjuvant Therapy with Radiation. Cancers 2022, 14, 3097. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Lin, W.S.; Lin, C.L.; Kao, C.H. Carvedilol use is associated with reduced cancer risk: A nationwide population-based cohort study. Int. J. Cardiol. 2015, 184, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Montoya, A.; Amaya, C.N.; Belmont, A.; Diab, N.; Trevino, R.; Villanueva, G.; Rains, S.; Sanchez, L.A.; Badri, N.; Otoukesh, S.; et al. Use of non-selective β-blockers is associated with decreased tumor proliferative indices in early stage breast cancer. Oncotarget 2017, 8, 6446–6460. [Google Scholar] [CrossRef]
- Wang, F.; Liu, H.; Wang, F.; Xu, R.; Wang, P.; Tang, F.; Zhang, X.; Zhu, Z.; Lv, H.; Han, T. Propranolol suppresses the proliferation and induces the apoptosis of liver cancer cells. Mol. Med. Rep. 2018, 17, 5213–5221. [Google Scholar] [CrossRef]
- Brohée, L.; Peulen, O.; Nusgens, B.; Castronovo, V.; Thiry, M.; Colige, A.C.; Deroanne, C.F. Propranolol sensitizes prostate cancer cells to glucose metabolism inhibition and prevents cancer progression. Sci. Rep. 2018, 8, 7050. [Google Scholar] [CrossRef] [PubMed]
- Terzi, M.Y.; Urhan-Kucuk, M. Anti-proliferative effects of beta-blocker propranolol on human lung cancer and noncancer cells. Bratisl. Med. J. 2023, 124, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.; Takahashi, T.; Kurokawa, Y.; Kobayashi, T.; Saito, T.; Ishida, T.; Serada, S.; Fujimoto, M.; Naka, T.; Wada, N.; et al. Propranolol suppresses gastric cancer cell growth by regulating proliferation and apoptosis. Gastric Cancer 2021, 24, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Chen, X.; Zeng, W.; Peng, C.; Huang, G.; Li, X.; Ouyang, Z.; Luo, Y.; Xu, X.; Xu, B.; et al. Propranolol induced G0/G1/S phase arrest and apoptosis in melanoma cells via AKT/MAPK pathway. Oncotarget 2016, 7, 68314–68327. [Google Scholar] [CrossRef]
- Wrobel, L.J.; Le Gal, F.A. Inhibition of human melanoma growth by a non-cardioselective β-blocker. J. Investig. Dermatol. 2015, 135, 525–531. [Google Scholar] [CrossRef]
- Coelho, M.; Moz, M.; Correia, G.; Teixeira, A.; Medeiros, R.; Ribeiro, L. Antiproliferative effects of β-blockers on human colorectal cancer cells. Oncol. Rep. 2015, 33, 2513–2520. [Google Scholar] [CrossRef]
- Talarico, G.; Orecchioni, S.; Dallaglio, K.; Reggiani, F.; Mancuso, P.; Calleri, A.; Gregato, G.; Labanca, V.; Rossi, T.; Noonan, D.M.; et al. Aspirin and atenolol enhance metformin activity against breast cancer by targeting both neoplastic and microenvironment cells. Sci. Rep. 2016, 6, 18673. [Google Scholar] [CrossRef]
- Hajatbeigi, B.; Hajighasemi, F. Cytotoxicity of Metoprolol on Leukemic Cells in Vitro. IJBC 2018, 10, 124–129. [Google Scholar]
- He, D.; Hu, J.; Li, Y.; Zeng, X. Preventive use of beta-blockers for anthracycline-induced cardiotoxicity: A network meta-analysis. Front. Cardiovasc. Med. 2022, 9, 968534. [Google Scholar] [CrossRef]
- Ma, Y.; Bai, F.; Qin, F.; Li, J.; Liu, N.; Li, D.; Li, T.; Xie, H.; Liu, D.; Zhou, S.; et al. Beta-blockers for the primary prevention of anthracycline-induced cardiotoxicity: A meta-analysis of randomized controlled trials. BMC Pharmacol. Toxicol. 2019, 20, 18. [Google Scholar] [CrossRef]
- Moretti, S.; Massi, D.; Farini, V.; Baroni, G.; Parri, M.; Innocenti, S.; Cecchi, R.; Chiarugi, P. β-adrenoceptors are upregulated in human melanoma and their activation releases pro-tumorigenic cytokines and metalloproteases in melanoma cell lines. Lab. Investig. 2013, 93, 279–290. [Google Scholar] [CrossRef]
- Weng, C.H.; Wu, C.S.; Wu, J.C.; Kung, M.L.; Wu, M.H.; Tai, M.H. Cisplatin-Induced Giant Cells Formation Is Involved in Chemoresistance of Melanoma Cells. Int. J. Mol. Sci. 2020, 21, 7892. [Google Scholar] [CrossRef]
- Shrikhande, S.S.; Jain, D.S.; Athawale, R.B.; Bajaj, A.N.; Goel, P.; Kamran, Z.; Nikam, Y.; Gude, R. Evaluation of anti-metastatic potential of Cisplatin polymeric nanocarriers on B16F10 melanoma cells. Saudi Pharm. J. 2015, 23, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Kim, S.Y.; Lee, E.; Lee, S.; Oh, S.; Jung, J.; Kim, K.S.; Moon, A. Sex-Dependent Adverse Drug Reactions to 5-Fluorouracil in Colorectal Cancer. Biol. Pharm. Bull. 2019, 42, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Huang, S.; Wei, Y.; Cao, S.; Pi, C.; Feng, T.; Liang, J.; Zhao, L.; Ren, G. Curcumin Enhances the Anticancer Effect Of 5-fluorouracil against Gastric Cancer through Down-Regulation of COX-2 and NF- κB Signaling Pathways. J. Cancer 2017, 8, 3697–3706. [Google Scholar] [CrossRef] [PubMed]
- Majounie, E.; Wee, K.; Williamson, L.M.; Jones, M.R.; Pleasance, E.; Lim, H.J.; Ho, C.; Renouf, D.J.; Yip, S.; Jones, S.J.M.; et al. Fluorouracil sensitivity in a head and neck squamous cell carcinoma with a somatic DPYD structural variant. Mol. Case Stud. 2020, 6, a004713. [Google Scholar] [CrossRef]
- Ryan, R.F.; Krementz, E.T.; Litwin, M.S. A role for topical 5-fluorouracil therapy in melanoma. J. Surg. Oncol. 1988, 38, 250–256. [Google Scholar] [CrossRef]
- Focaccetti, C.; Bruno, A.; Magnani, E.; Bartolini, D.; Principi, E.; Dallaglio, K.; Bucci, E.O.; Finzi, G.; Sessa, F.; Noonan, D.M.; et al. Effects of 5-fluorouracil on morphology, cell cycle, proliferation, apoptosis, autophagy and ROS production in endothelial cells and cardiomyocytes. PLoS ONE 2015, 10, e0115686. [Google Scholar] [CrossRef]
- Chaudhary, K.R.; Yan, S.X.; Heilbroner, S.P.; Sonett, J.R.; Stoopler, M.B.; Shu, C.; Halmos, B.; Wang, T.J.C.; Hei, T.K.; Cheng, S.K. Effects of β-Adrenergic Antagonists on Chemoradiation Therapy for Locally Advanced Non-Small Cell Lung Cancer. J. Clin. Med. 2019, 8, 575. [Google Scholar] [CrossRef]
- Pasquier, E.; Ciccolini, J.; Carre, M.; Giacometti, S.; Fanciullino, R.; Pouchy, C.; Montero, M.P.; Serdjebi, C.; Kavallaris, M.; André, N. Propranolol potentiates the anti-angiogenic effects and anti-tumor efficacy of chemotherapy agents: Implication in breast cancer treatment. Oncotarget 2011, 2, 797–809. [Google Scholar] [CrossRef]
- Carvalho Rodrigues, M.A.; Gobe, G.; Santos, N.A.; Santos, A.C. Carvedilol protects against apoptotic cell death induced by cisplatin in renal tubular epithelial cells. J. Toxicol. Environ. Health Part A 2012, 75, 981–990. [Google Scholar] [CrossRef]
- Hildebrandt, J.; Bauerschlag, D.O.; Fricker, G.; Girreser, U.; Konukiewitz, B.; Kellers, F.; Maass, N.; Clement, B.; Flörkemeier, I. In Vivo and In Vitro Pharmacokinetic Studies of a Dual Topoisomerase I/II Inhibitor. ACS Pharmacol. Transl. Sci. 2025, 8, 1050–1071. [Google Scholar] [CrossRef]
- Archer, M.; Dogra, N.; Dovey, Z.; Ganta, T.; Jang, H.-S.; Khusid, J.A.; Lantz, A.; Mihalopoulos, M.; Stockert, J.A.; Zahalka, A.; et al. Role of α- and β-adrenergic signaling in phenotypic targeting: Significance in benign and malignant urologic disease. Cell Commun. Signal 2021, 19, 78. [Google Scholar] [CrossRef]
- Lin, Y.; Liu, Y.; Gao, Z.; Jing, D.; Bi, R.; Cui, X.; Cao, Q.; Zhao, Q.; Gao, R.; Su, Y.; et al. Beta-adrenergic receptor blocker propranolol triggers anti-tumor immunity and enhances irinotecan therapy in mice colorectal cancer. Eur. J. Pharmacol. 2023, 949, 175718. [Google Scholar] [CrossRef]
- Carnet Le Provost, K.; Kepp, O.; Kroemer, G.; Bezu, L. Trial watch: Beta-blockers in cancer therapy. Oncoimmunology 2023, 12, 2284486. [Google Scholar] [CrossRef]
- Dal Monte, M.; Calvani, M.; Cammalleri, M.; Favre, C.; Filippi, L.; Bagnoli, P. β-Adrenoceptors as drug targets in melanoma: Novel preclinical evidence for a role of β3-adrenoceptors. Br. J. Pharmacol. 2019, 176, 2496–2508. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (μM) | |||
|---|---|---|---|
| 24 h | 48 h | 72 h | |
| Carvedilol | 24.95 (13.06–47.26) | 22.97 (13.24–39.39) | 16.91 (15.47–18.99) |
| Propranolol | 96.04 (87.36–100.81) | 73.00 (68.67–77.05) | 58.03 (57.08–59.11) |
| Cisplatin | 14.01 (11.04–23.08) | 6.87 (5.58–8.48) | 2.46 (1.87–3.38) |
| 5-fluorouracil | NC | 15.10 (13.76–16.69) | 4.77 (4.48–5.07) |
| Drug Combination | Overall δ Score | Description | Peak Synergy Concentrations |
|---|---|---|---|
| Cisplatin + Propranolol | −3.028 ± 5.43 | Additive interaction with some antagonistic tendencies observed at higher concentrations of both drugs | No synergy peak |
| Cisplatin + Carvedilol | 5.949 ± 5.97 | Additive interaction with synergistic tendencies at lower concentrations of both drugs and antagonism at higher concentrations. | 1.72 μM of cisplatin and 11.49 μM of carvedilol |
| Cisplatin + Metoprolol | 4.098 ± 5.46 | Additive interaction with synergistic tendencies at lower concentrations of cisplatin | 1.72 μM of cisplatin and 375 μM of metoprolol |
| Propranolol + Carvedilol | 27.633 ± 7.45 | Synergistic interaction at higher concentrations of both drugs, with some antagonistic tendencies at lower concentrations. | Most of high concentrations combinations |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rama, L.; Almeida, M.; Jose, J.; Pereira, M.d.L.; Oliveira, M. Beta-Blockers as Potential Adjuvants in Melanoma Treatment. Toxics 2025, 13, 981. https://doi.org/10.3390/toxics13110981
Rama L, Almeida M, Jose J, Pereira MdL, Oliveira M. Beta-Blockers as Potential Adjuvants in Melanoma Treatment. Toxics. 2025; 13(11):981. https://doi.org/10.3390/toxics13110981
Chicago/Turabian StyleRama, Laura, Mónica Almeida, Jiya Jose, Maria de Lourdes Pereira, and Miguel Oliveira. 2025. "Beta-Blockers as Potential Adjuvants in Melanoma Treatment" Toxics 13, no. 11: 981. https://doi.org/10.3390/toxics13110981
APA StyleRama, L., Almeida, M., Jose, J., Pereira, M. d. L., & Oliveira, M. (2025). Beta-Blockers as Potential Adjuvants in Melanoma Treatment. Toxics, 13(11), 981. https://doi.org/10.3390/toxics13110981