Anticancer Activities of Meroterpenoids Isolated from the Brown Alga Cystoseira usneoides against the Human Colon Cancer Cells HT-29

 and
and 
Abstract
1. Introduction
2. Materials and Methods
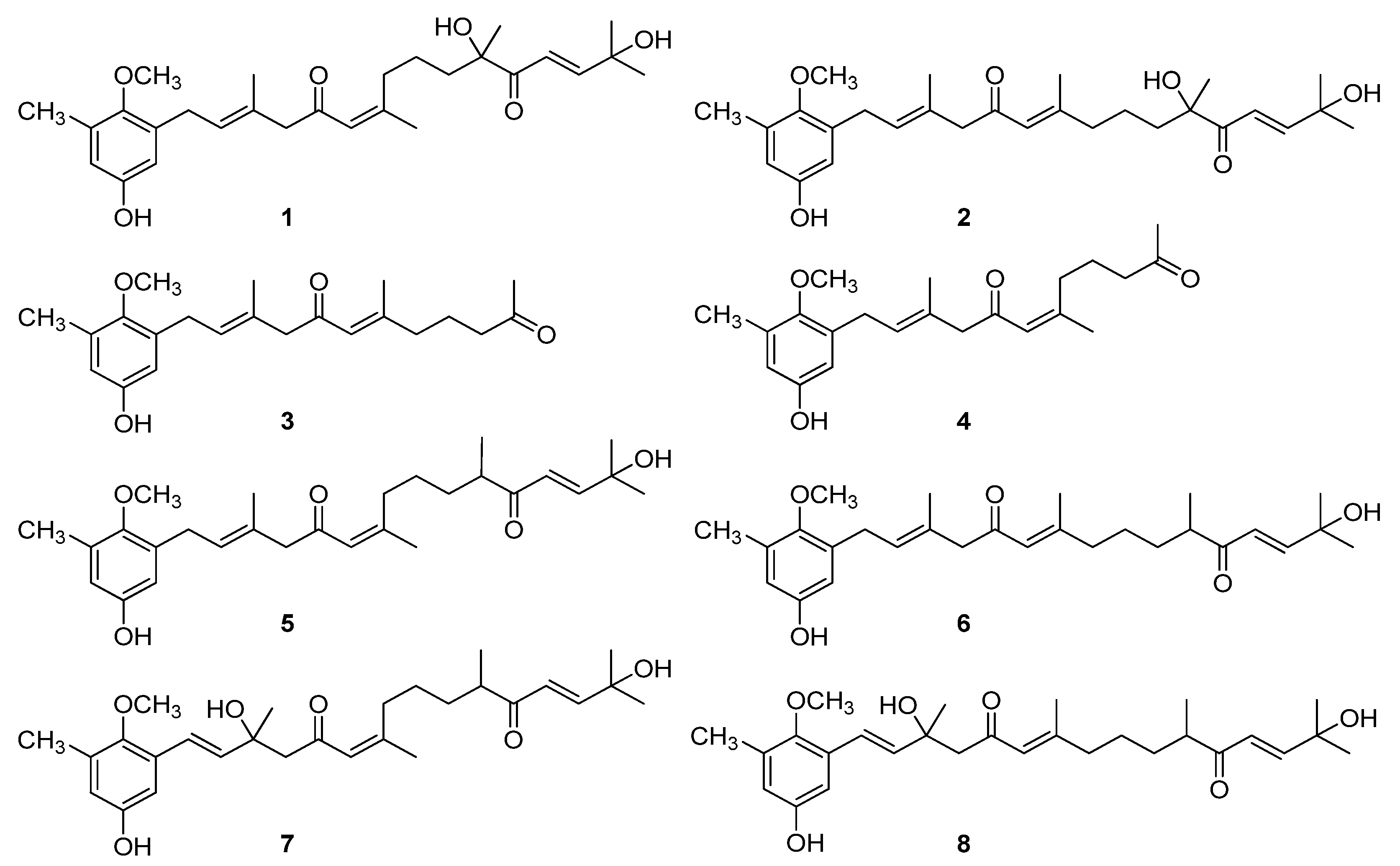
2.1. Isolation and Identification of the Meroterpenoids 1-8 from the Alga C. usneoides
2.2. Reagents for Bioactivity Assays
2.3. Cell Culture
2.4. Cell Viability Assay
2.5. Flow Cytometry Analysis for Apoptosis Induction Assay
2.6. Cell Cycle Analysis by Flow Cytometry
2.7. Wound Migration Assay
2.8. Cell Invasion Assay
2.9. Preparation of Cell Lysates
2.10. Western Blot Analysis
2.11. Statistical Analysis
3. Results
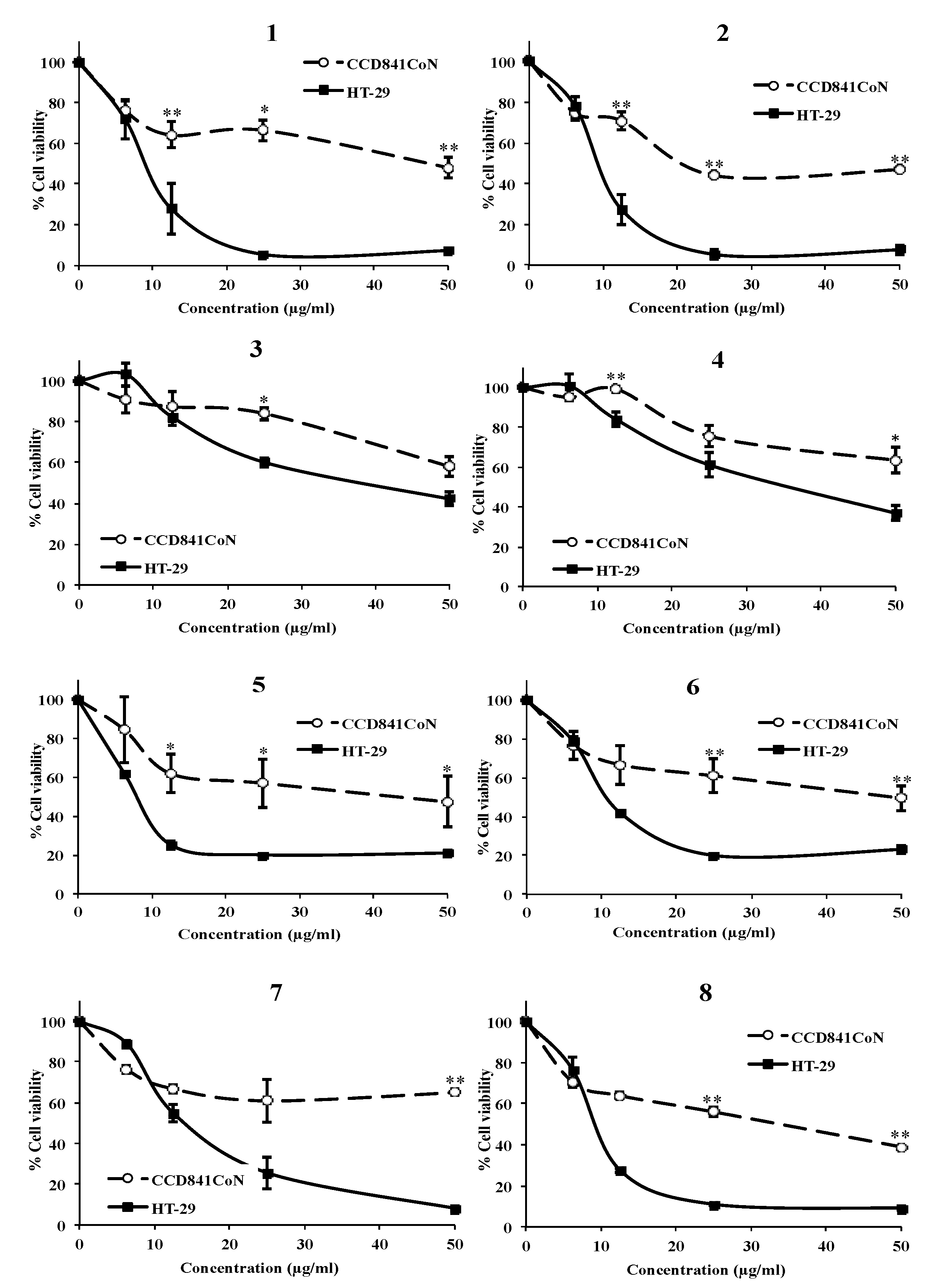
3.1. The AMTs 1-8 Inhibit Cell Proliferation in Human Colon Adenocarcinoma Cells HT-29
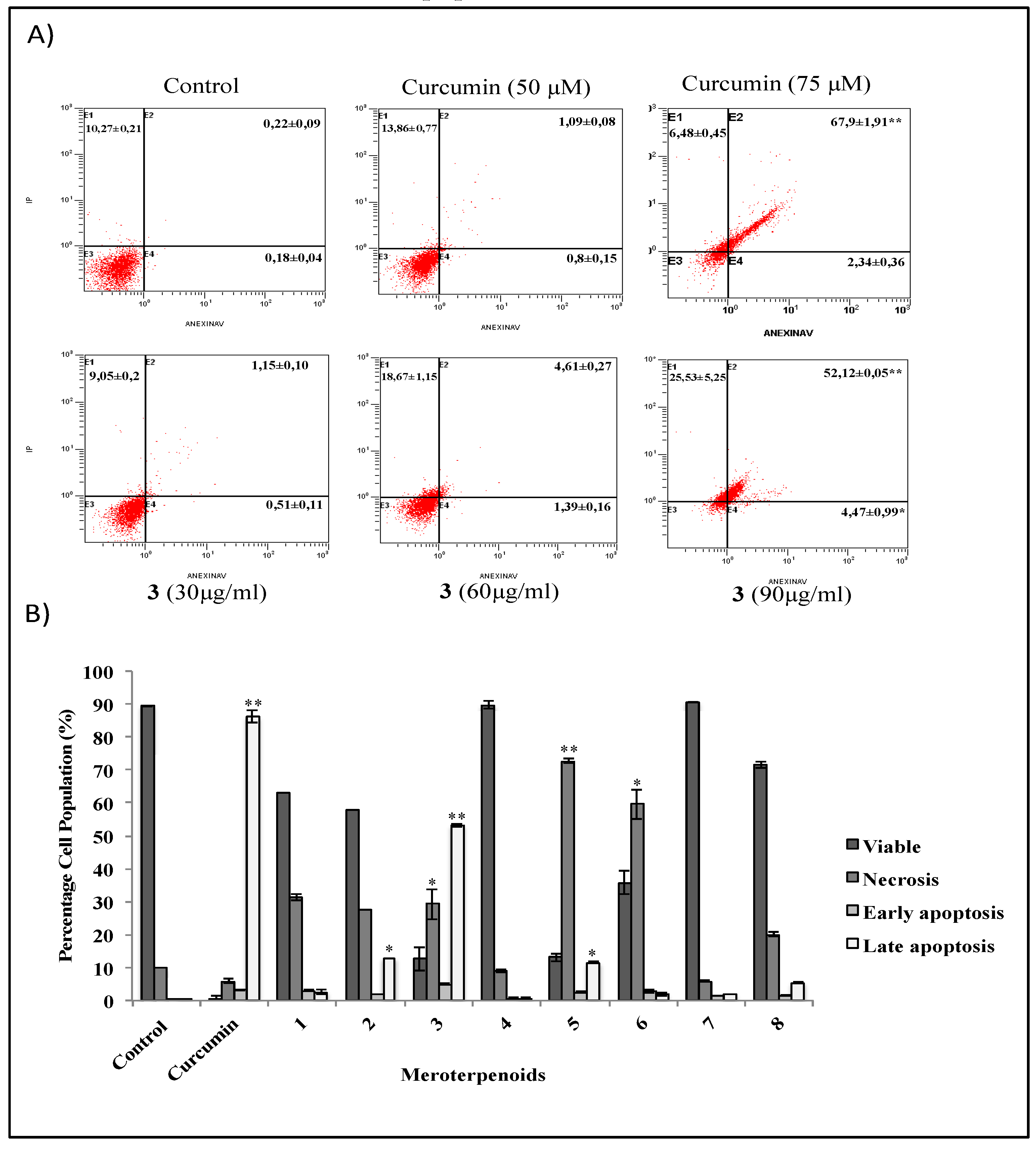
3.2. The AMTs 11-hydroxy-1′-O-methylamentadione (2), cystomexicone B (3), and 6-cis-amentadione-1′-methyl ether (5) Induce Apoptosis of HT-29 Colon Cancer Cells
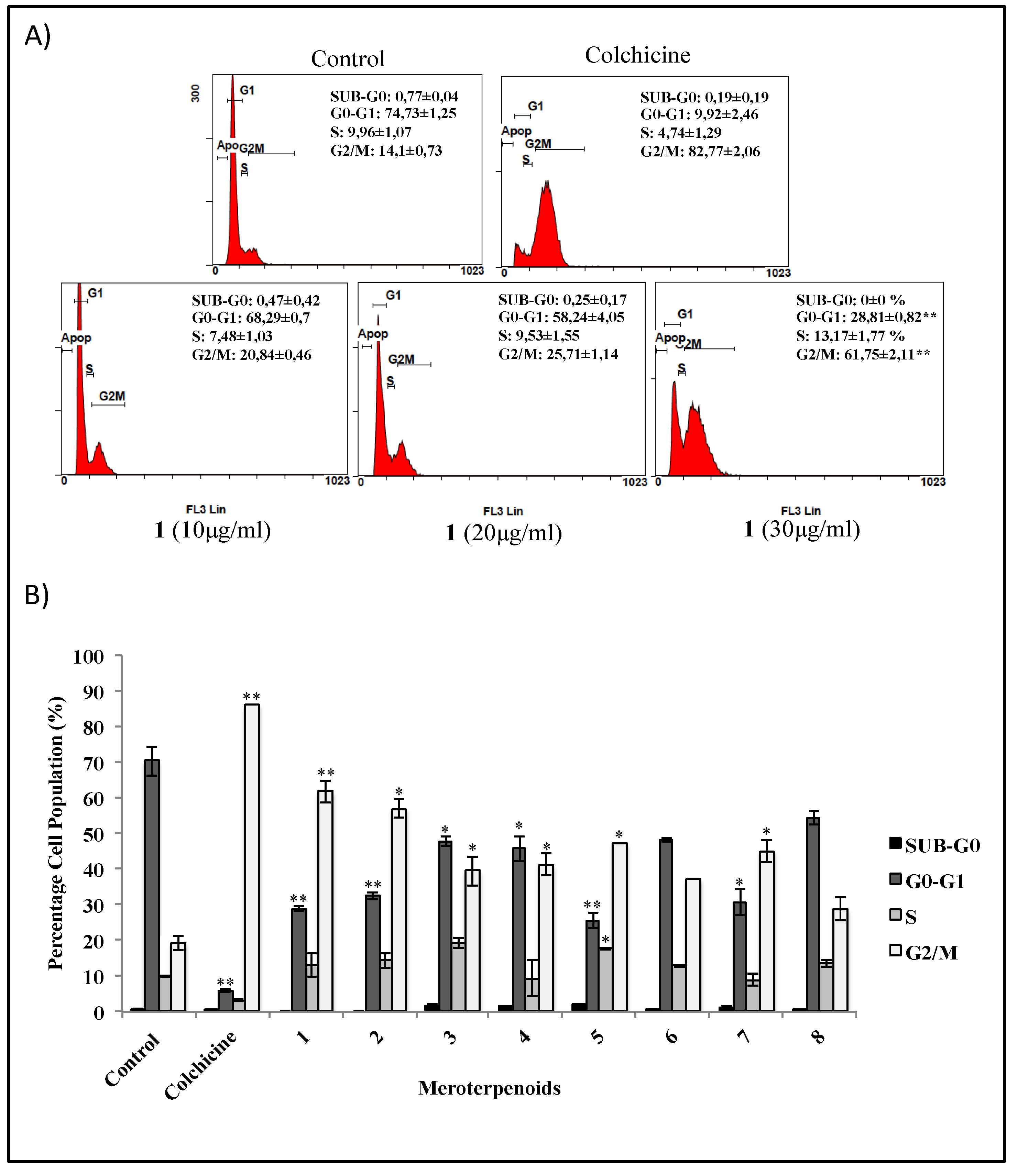
3.3. Effects of the AMTs 1-8 on Cell Cycle Arrest in HT-29 Cells
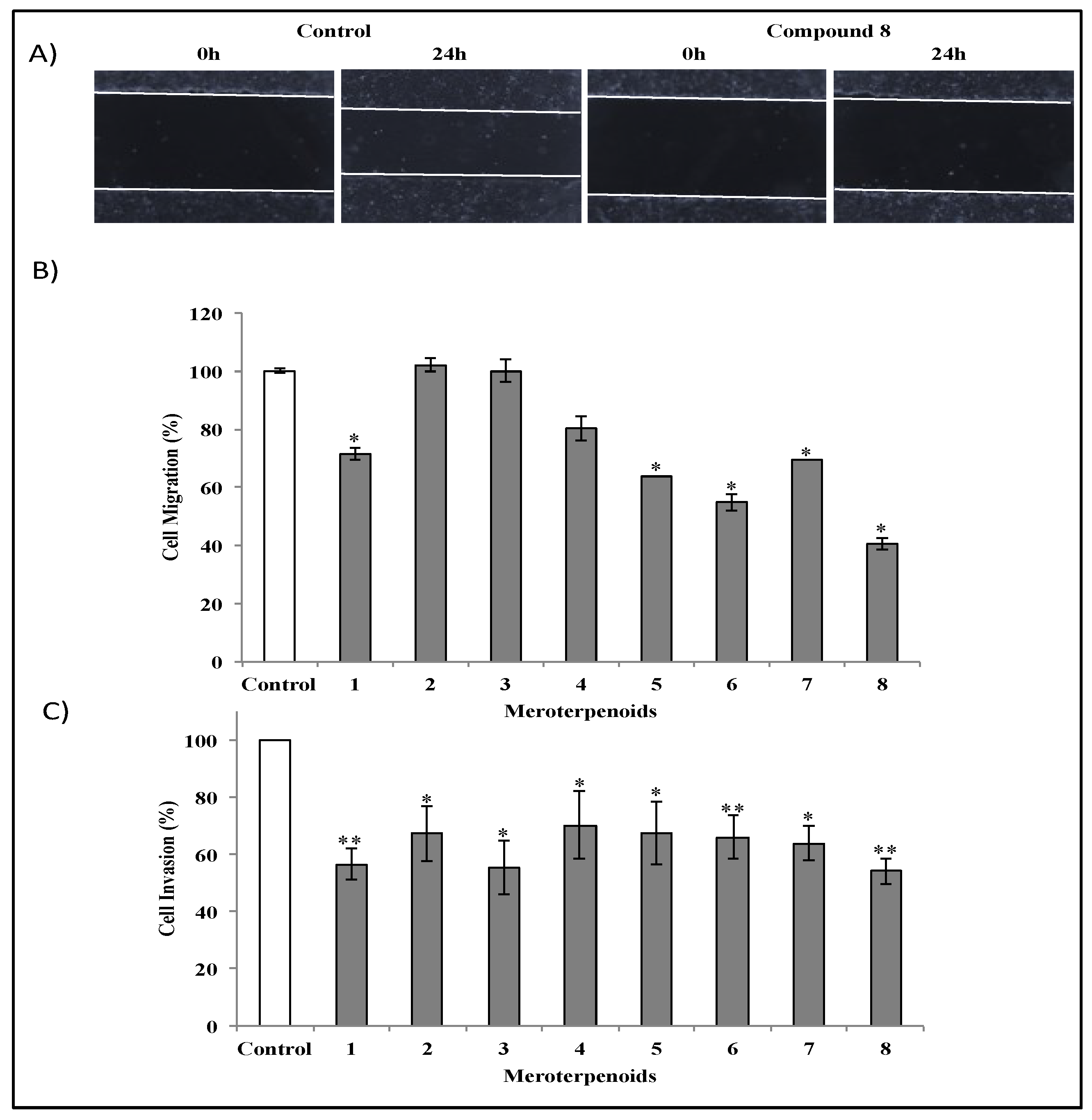
3.4. Effects of the AMTs 1-8 on the Migration and Invasion of HT-29 Cells
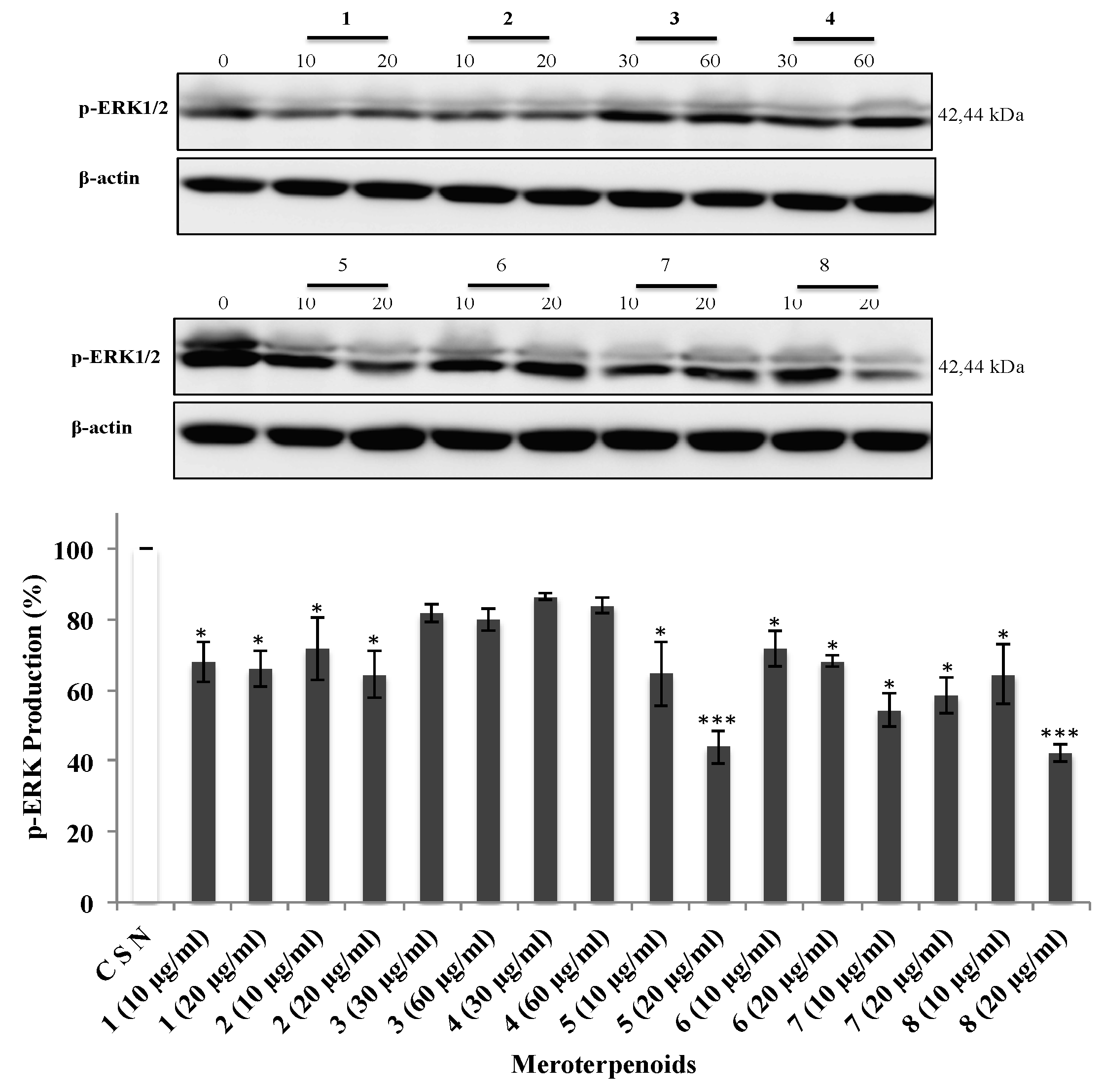
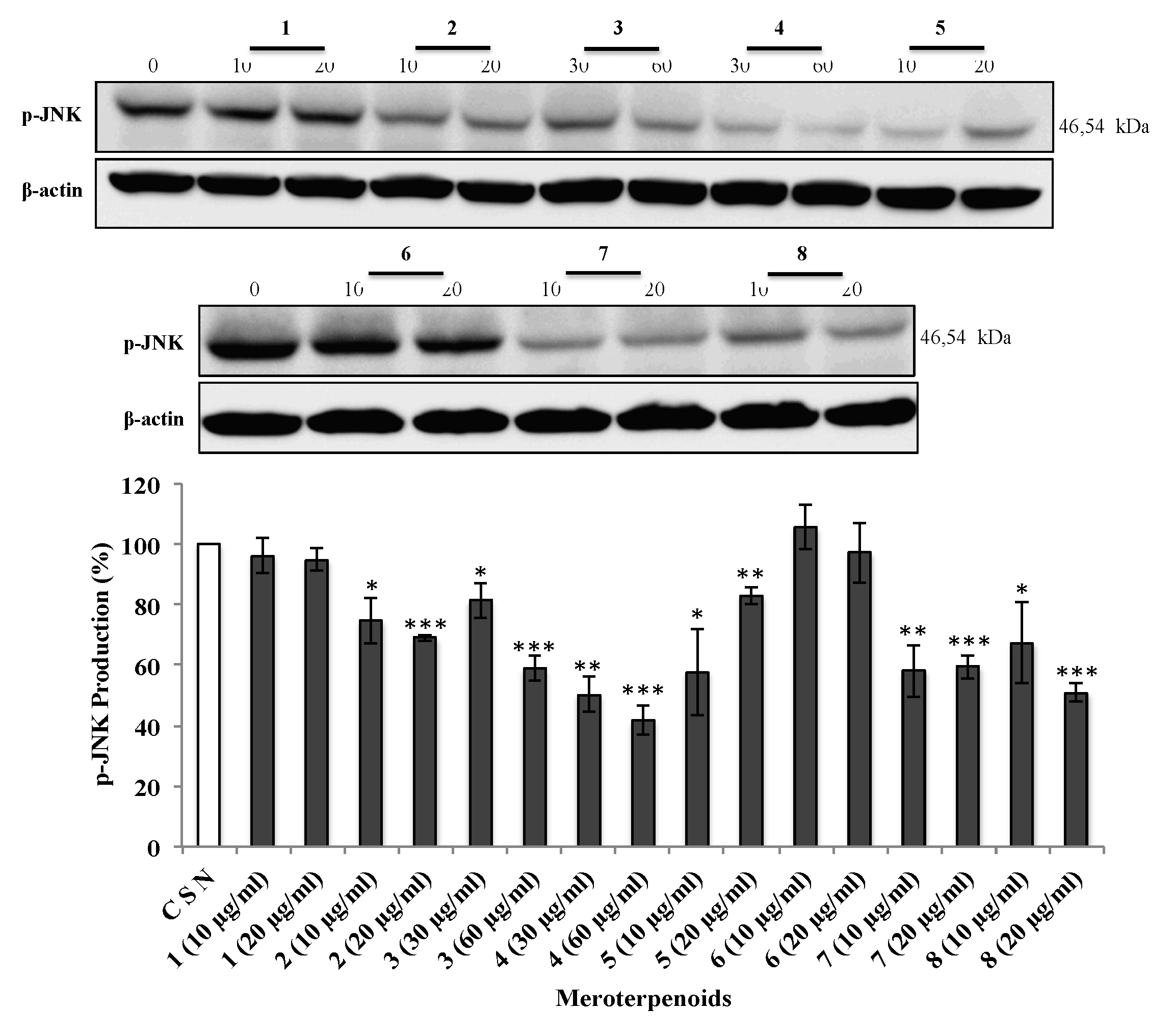
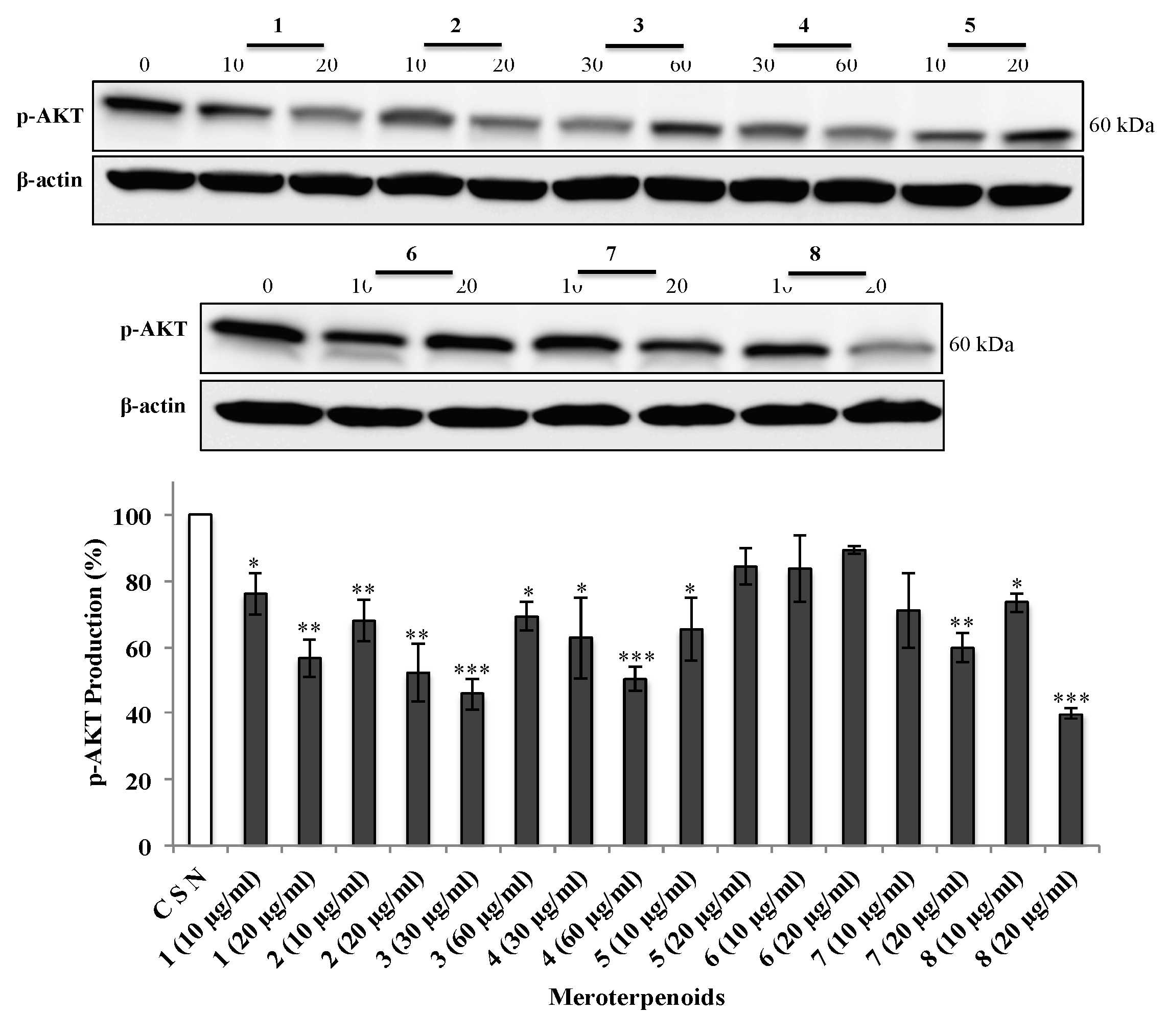
3.5. The AMTs 1-8 Inhibit Phosphorylation of ERK, JNK, and AKT
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, 359–386. [Google Scholar] [CrossRef] [PubMed]
- Kavousipour, S.; Khademi, F.; Zamani, M.; Vakili, B.; Mokarram, P. Novel biotechnology approaches in colorectal cancer diagnosis and therapy. Biotechnol. Lett. 2017, 39, 785–803. [Google Scholar] [CrossRef] [PubMed]
- Mayer, R.J. Targeted therapy for advanced colorectal cancer–more is not always better. N. Engl. J. Med. 2009, 360, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta 2013, 1830, 3670–3695. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Torres, V.; Encinar, J.A.; Herranz-López, M.; Pérez-Sánchez, A.; Galiano, V.; Barrajón-Catalán, E.; Micol, V. An updated review on marine anticancer compounds: The use of virtual screening for the discovery of small-molecule cancer drugs. Molecules 2017, 22, 1037. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Marine-sourced anti-cancer and cancer pain control agents in clinical and late preclinical development. Mar. Drugs 2014, 12, 255–278. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef]
- Leal, M.C.; Munro, M.H.G.; Blunt, J.W.; Puga, J.; Jesus, B.; Calado, R.; Rosa, R.; Madeira, C. Biogeography and biodiscovery hotspots of macroalgal marine natural products. Nat. Prod. Rep. 2013, 30, 1380–1390. [Google Scholar] [CrossRef]
- Huang, M.; Lu, J.J.; Huang, M.Q.; Bao, J.L.; Chen, X.P.; Wang, Y.T. Terpenoids: Natural products for cancer therapy. Expert Opin. Investig. Drugs 2012, 21, 1801–1818. [Google Scholar] [CrossRef]
- Rocha, D.H.A.; Seca, A.M.L.; Pinto, D.C.G.A. Seaweed secondary metabolites in vitro and in vivo anticancer activity. Mar. Drugs 2018, 16, 410. [Google Scholar] [CrossRef] [PubMed]
- Lisiak, N.; Paszel-Jaworska, A.; Totoń, E.; Rubiś, B.; Pakuła, M.; Bednarczyk-Cwynar, B.; Zaprutko, L.; Rybczyńska, M. Semisynthetic oleanane triterpenoids inhibit migration and invasion of human breast cancer cells through downregulated expression of the ITGB1/PTK2/PXN pathway. Chem. Biol. Interact. 2017, 268, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Sunasee, S.N.; Davies-Coleman, M.T. Cytotoxic and antioxidant marine prenylated quinones and hydroquinones. Nat. Prod. Rep. 2012, 29, 513–535. [Google Scholar] [CrossRef] [PubMed]
- Gouveia, V.; Seca, A.M.L.; Barreto, M.C.; Pinto, D.C.G.A. Di- and sesquiterpenoids from Cystoseira genus: Structure, intra-molecular transformations and biological activity. Mini Rev. Med. Chem. 2013, 13, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- Amico, V. Marine brown algae of family Cystoseiraceae: Chemistry and chemotaxonomy. Phytochemistry 1995, 39, 1257–1279. [Google Scholar] [CrossRef]
- Valls, R.; Piovetti, L. The chemistry of the Cystoseiraceae (Fucales: Pheophyceae): Chemotaxonomic relationships. Biochem. Syst. Ecol. 1995, 23, 723–745. [Google Scholar] [CrossRef]
- Gouveia, V.L.M.; Seca, A.M.L.; Barreto, M.C.; Neto, A.I.; Kijjoa, A.; Silva, A.M.S. Cytotoxic meroterpenoids from the macroalga Cystoseira abies-marina. Phytochem. Lett. 2013, 6, 593–597. [Google Scholar] [CrossRef]
- Vizetto-Duarte, C.; Custódio, L.; Acosta, G.; Lago, J.H.G.; Morais, T.R.; Bruno de Sousa, C.; Gangadhar, K.N.; Rodrigues, M.J.; Pereira, H.; Lima, R.T.; et al. Can macroalgae provide promising anti-tumoral compounds? A closer look at Cystoseira tamariscifolia as a source for antioxidant and anti-hepatocarcinoma compounds. Peer J. 2016, 4, 1704. [Google Scholar] [CrossRef]
- De los Reyes, C.; Zbakh, H.; Motilva, V.; Zubía, E. Antioxidant and anti-inflammatory meroterpenoids from the brown alga Cystoseira usneoides. J. Nat. Prod. 2013, 76, 621–629. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R.J. New colorimetric cytotoxicity assay for anticancer-drug screening. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutellingsperger, C. A novel assay for apoptosis flow cytometric detection of phosphatidyl serine expression on early apoptotic cells using fluorescein labeled annexin V. J. Immunol. Methods 1995, 184, 39–51. [Google Scholar] [CrossRef]
- Nicoletti, I.; Migliorati, G.; Pagliacci, M.; Grignani, F.; Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods. 1991, 139, 271–279. [Google Scholar] [CrossRef]
- Moghadamtousi, S.Z.; Karimian, H.; Rouhollahi, E.; Paydar, A.; Fadaeinasab, M.; AbdulKadi, H. Annona muricata leaves induce G1 cell cycle arrest and apoptosis through mitochondria-mediated pathway in human HCT-116 and HT-29 colon cancer cells. J. Ethnopharmacol. 2014, 156, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Bézivin, C.; Tomasi, F.; Lohézie-Le, D.; Boustie, J. Cytotoxic activity of some lichen extracts on murine and human cancer cell lines. Phytomedicine 2003, 10, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Suffness, M.; Pezzuto, J.M. Assays related to cancer drug discovery. In Methods in Plant Biochemistry: Assays for Bioactivity; Hostettmann, K., Ed.; Academic Press: London, UK, 1990; pp. 71–133. [Google Scholar]
- Chen, P.N.; Hsieh, Y.S.; Chiou, H.L.; Chu, S.C. Silibinin inhibits cell invasion through inactivation of both PI3K-Akt and MAPK signaling pathways. Chem. Biol. Interact. 2005, 156, 141–150. [Google Scholar] [CrossRef]
- Turner, N.A.; Aley, P.K.; Hall, K.T.; Warburton, P.; Galloway, S.; Midgley, L.; O’regan, D.J.; Wood, I.C.; Ball, S.G.; Porter, K.E. Simvastatin inhibits TNF-α induced invasion of human cardiac myofibroblasts via both MMP-9-dependent and -independent mechanisms. J. Mol. Cell Cardio. 2007, 43, 168–176. [Google Scholar] [CrossRef]
- Urones, J.G.; Basabe, P.; Marcos, I.S.; Pineda, J.; Lithgow, A.M.; Moro, R.F.; Brito Palma, F.M.S.; Araujo, M.E.M.; Gravalos, M.D.G. Meroterpenes from Cystoseira usneoides. Phytochemistry 1992, 31, 179–182. [Google Scholar] [CrossRef]
- Lee, J.I.; Park, B.J.; Kim, H.; Seo, Y. Isolation of two new meroterpenoids from Sargassum siliquastrum. Bull. Korean Chem. Soc. 2014, 35, 2867–2869. [Google Scholar] [CrossRef]
- Penicooke, N.; Walford, K.; Badal, S.; Delgoda, R.; Williams, L.A.D.; Joseph-Nathan, P.; Gordillo-Román, B.; Gallimore, W. Antiproliferative activity and absolute configuration of zonaquinone acetate from the Jamaican alga Stypopodium zonale. Phytochemistry 2013, 87, 96–101. [Google Scholar] [CrossRef]
- Choi, Y.K.; Kim, J.; Lee, K.M.; Choi, Y.J.; Ye, B.R.; Kim, M.S.; Ko, S.G.; Lee, S.H.; Kang, D.H.; Heo, S.J. Tuberatolide B suppresses cancer progression by promoting ROS-mediated inhibition of STAT3 signaling. Mar. Drugs 2017, 15, 55. [Google Scholar] [CrossRef]
- Heo, S.J.; Kim, K.N.; Yoon, W.J.; Oh, C.; Choi, Y.U.; Affan, A.; Lee, Y.J.; Lee, H.S.; Kang, D.H. Chromene induces apoptosis via caspase-3 activation in human leukemia HL-60 cells. Food Chem. Toxicol. 2011, 49, 1998–2004. [Google Scholar] [CrossRef]
- Tarhouni-Jabberi, S.; Zakraoui, O.; Ioannou, E.; Riahi-Chebbi, I.; Haoues, M.; Roussis, V.; Kharrat, R.; Essafi-Benkhadir, K. Mertensene, a halogenated monoterpene, induces G2/M cell cycle arrest and caspase dependent apoptosis of human colon adenocarcinoma HT29 cell line through the modulation of ERK-1/-2, AKT and NF-κB signaling. Mar. Drugs 2017, 15, 221. [Google Scholar] [CrossRef]
- Cen-Pacheco, F.; Villa-Pulgarin, J.A.; Mollinedo, F.; Martín, M.N.; Fernández, J.J.; Daranas, A.H. New polyether triterpenoids from Laurencia viridis and their biological evaluation. Mar. Drugs 2011, 9, 2220–2235. [Google Scholar] [CrossRef]
- Velatooru, L.R.; Baggu, C.B.; Janapala, V.R. Spatane diterpinoid from the brown algae, Stoechospermum marginatum induces apoptosis via ROS induced mitochondrial mediated caspase dependent pathway in murine B16F10 melanoma cells. Mol. Carcinog. 2016, 55, 2222–2235. [Google Scholar] [CrossRef]
- Campos, A.; Souza, C.B.; Lhullier, C.; Falkenberg, M.; Schenkel, E.P.; Ribeiro-do-Valle, R.M.; Siqueira, J.M. Anti-tumour effects of elatol, a marine derivative compound obtained from red algae Laurencia microcladia. J. Pharm. Pharmacol. 2012, 64, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi, P.T.; Vajravijayan, S.; Moumita, M.; Sakthivel, N.; Gunasekaran, K.; Krishna, R. A novel guaiane sesquiterpene derivative, guai-2-en-10α-ol, from Ulva fasciata Delile inhibits EGFR/PI3K/Akt signaling and induces cytotoxicity in triple-negative breast cancer cells. Mol. Cell Biochem. 2018, 438, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.H.; Stoeber, K. The cell cycle and cancer. J. Pathol. 2012, 226, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Brauer, C.T.; Thu, K.L.; Mason, J.M.; Blaser, H.; Bray, M.R.; Mak, T.W. Targeting mitosis in cancer: Emerging strategies. Mol. Cell. 2015, 60, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Palmer, N.; Kaldis, P. Regulation of the embryonic cell cycle during mammalian preimplantation development. Curr. Top. Dev. Biol. 2016, 120, 1–53. [Google Scholar] [CrossRef] [PubMed]
- Yang, V.W. The Cell Cycle. Physiology of the Gastrointestinal Tract, 5th ed.; Academic Press: Boston, MA, USA, 2012; chapter 15; pp. 451–471. [Google Scholar]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell death by mitotic catastrophe: A molecular definition. Oncogene 2004, 23, 2825–2837. [Google Scholar] [CrossRef]
- Hood, J.D.; Cheresh, D.A. Role of integrins in cell invasion and migration. Nat. Rev. Cancer 2002, 2, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Kajanne, R.; Miettinen, P.; Mehlem, A.; Leivonen, S.K.; Birrer, M.; Foschi, M.; Kähäri, V.M.; Leppä, S. EGFR regulates MMP function in fibroblasts through MAPK and AP-1 pathways. J. Cell Physiol. 2007, 212, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Go, H.; Hwang, H.J.; Kim, I.H.; Nam, T.J. Anoikis induction by glycoprotein from Laminaria japonica in HT-29 cells. Prev. Med. 2011, 1, 49–61. [Google Scholar] [CrossRef][Green Version]
- Shan, J.Z.; Xuan, Y.Y.; Zheng, S.; Dong, Q.; Zhang, S.Z. Ursolic acid inhibits proliferation and induces apoptosis of HT-29 colon cancer cells by inhibiting the EGFR/MAPK pathway. J. Zhejiang Univ. Sci. B 2009, 10, 668–674. [Google Scholar] [CrossRef]
- Jiang, H.; Zhu, Y.; Zhou, Z.; Xu, J.; Jin, S.; Xu, K.; Zhang, H.; Sun, Q.; Wang, J.; Xu, J. PRMT5 promotes cell proliferation by inhibiting BTG2 expression via the ERK signaling pathway in hepatocellular carcinoma. Cancer Med. 2018, 7, 869–882. [Google Scholar] [CrossRef]
- Park, J.I. Growth arrest signaling of the Raf/MEK/ERK pathway in cancer. Front. Biol. 2014, 9, 95–103. [Google Scholar] [CrossRef]
- Cagnol, S.; Chambard, J.C. ERK and cell death: Mechanisms of ERK-induced cell death--apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Li, Z.; Li, J.; Mo, B.; Hu, C.; Liu, H.; Qi, H.; Wang, X.; Xu, J. Genistein induces G2/M cell cycle arrest via stable activation of ERK1/2 pathway in MDA-MB-231 breast cancer cells. Cell. Biol. Toxicol. 2008, 24, 401–409. [Google Scholar] [CrossRef]
- Abrieu, A.; Fisher, D.; Simon, M.N.; Doree, M.; Picard, A. MAPK inactivation is required for the G2 to M-phase transition of the first mitotic cell cycle. EMBO J. 1997, 16, 6407–6413. [Google Scholar] [CrossRef]
- Wu, W.S.; Wu, J.R.; Hu, C.T. Signal cross talks for sustained MAPK activation and cell migration: The potential role of reactive oxygen species. Cancer Metastasis Rev. 2008, 27, 303–314. [Google Scholar] [CrossRef]
- Kuphal, S.; Bauer, R.; Bosserhoff, A.K. Integrin signaling in malignant melanoma. Cancer Metastasis Rev. 2005, 24, 195–222. [Google Scholar] [CrossRef] [PubMed]
- Tournier, K. The 2 Faces of JNK signaling in cancer. Genes Cancer 2013, 4, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Mingo-Sion, A.M.; Marietta, P.M.; Koller, E.; Wolf, D.M.; Van Den Berg, C.L. Inhibition of JNK reduces G2/M transit independent of p53, leading to endoreduplication, decreased proliferation, and apoptosis in breast cancer cells. Oncogene 2004, 23, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.; Ryan, K.M.; Phillips, A.C.; Vousden, K.H. Cell cycle arrest and DNA endoreduplication following p21Waf1/Cip1 expression. Oncogene 1998, 17, 1691–1703. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. JNK-signaling: A multiplexing hub in programmed cell death. Genes Cancer 2017, 8, 682–694. [Google Scholar] [CrossRef]
- Cai, J.; Du, S.; Wang, H.; Xin, B.; Wang, J.; Shen, W.; Wei, W.; Guo, Z.; Shen, X. Tenascin-C induces migration and invasion through JNK/c-Jun signalling in pancreatic cancer. Oncotarget 2017, 8, 74406–74422. [Google Scholar] [CrossRef]
- Jung, S.K.; Jeong, C.H. Dehydroglyasperin D inhibits the proliferation of HT-29 human colorectal cancer cells through direct interaction with phosphatidylinositol 3-kinase. J. Cancer Prev. 2016, 21, 26–31. [Google Scholar] [CrossRef][Green Version]
- Hollborn, M.; Stathopoulos, C.; Steffen, A.; Wiedemann, P.; Kohen, L.; Bringmann, A. Positive feedback regulation between MMP-9 and VEGF in human RPE cells. Invest. Ophthalmol. Vis. Sci. 2007, 48, 4360–4367. [Google Scholar] [CrossRef]
- Chang, F.; Lee, J.T.; Navolanic, P.M.; Steelman, L.S.; Shelton, J.G.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: A target for cancer chemotherapy. Leukemia 2003, 17, 590–603. [Google Scholar] [CrossRef]
- Hiraoka, D.; Aono, R.; Hanada, S.; Okumura, E.; Kishimoto, T. Two new competing pathways establish the threshold for cyclin-B–Cdk1 activation at the meiotic G2/M transition. J. Cell. Sci. 2016, 129, 3153–3166. [Google Scholar] [CrossRef]
- Agarwal, E.; Brattain, M.G.; Chowdhury, S. Cell survival and metastasis regulation by akt signalling in colorectal cancer. Cell Signal. 2013, 25, 1711–1719. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Basson, M.D. AKT directly regulates focal adhesion kinase through association and serine phosphorylation: Implication for pressure-induced colon cancer metastasis. Am. J. Physiol. Cell Physiol. 2011, 300, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.M.; Mendis, E.; Kim, S.K. Laurencia okamurai extract containing laurinterol induces apoptosis in melanoma cells. J. Med. Chem. 2008, 11, 260–266. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | Selectivity Index (SI) | ||
|---|---|---|---|
| Compound | CCD841 CoN | HT-29 | |
| 1 | 46.41 ± 3.87 | 8.81 ± 1.55 | 5.26 |
| 2 | 21.41 ± 2.50 | 9.14 ± 0.23 | 2.34 |
| 3 | >50 | 34.34 ± 1.81 | >1.45 |
| 4 | 61.86 ± 2.37 | 36.86 ± 2.86 | 1.68 |
| 5 | 40.97 ± 9.80 | 7.83 ± 0.05 | 5.23 |
| 6 | 48.38 ± 3.84 | 10.72 ± 0.27 | 4.51 |
| 7 | >50 | 14.00 ± 1.33 | >3.57 |
| 8 | 31.88 ± 7.51 | 9.08 ± 1.04 | 3.51 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zbakh, H.; Zubía, E.; De Los Reyes, C.; Calderón-Montaño, J.M.; Motilva, V. Anticancer Activities of Meroterpenoids Isolated from the Brown Alga Cystoseira usneoides against the Human Colon Cancer Cells HT-29. Foods 2020, 9, 300. https://doi.org/10.3390/foods9030300
Zbakh H, Zubía E, De Los Reyes C, Calderón-Montaño JM, Motilva V. Anticancer Activities of Meroterpenoids Isolated from the Brown Alga Cystoseira usneoides against the Human Colon Cancer Cells HT-29. Foods. 2020; 9(3):300. https://doi.org/10.3390/foods9030300
Chicago/Turabian StyleZbakh, Hanaa, Eva Zubía, Carolina De Los Reyes, José M. Calderón-Montaño, and Virginia Motilva. 2020. "Anticancer Activities of Meroterpenoids Isolated from the Brown Alga Cystoseira usneoides against the Human Colon Cancer Cells HT-29" Foods 9, no. 3: 300. https://doi.org/10.3390/foods9030300
APA StyleZbakh, H., Zubía, E., De Los Reyes, C., Calderón-Montaño, J. M., & Motilva, V. (2020). Anticancer Activities of Meroterpenoids Isolated from the Brown Alga Cystoseira usneoides against the Human Colon Cancer Cells HT-29. Foods, 9(3), 300. https://doi.org/10.3390/foods9030300