Trapping of an Heterometallic Unsaturated Hydride: Structure and Properties of the Ammonia Complex [MoMnCp(μ-H)(μ-PPh2)(CO)5(NH3)]



Abstract
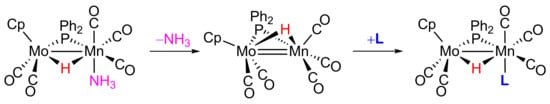
:
1. Introduction
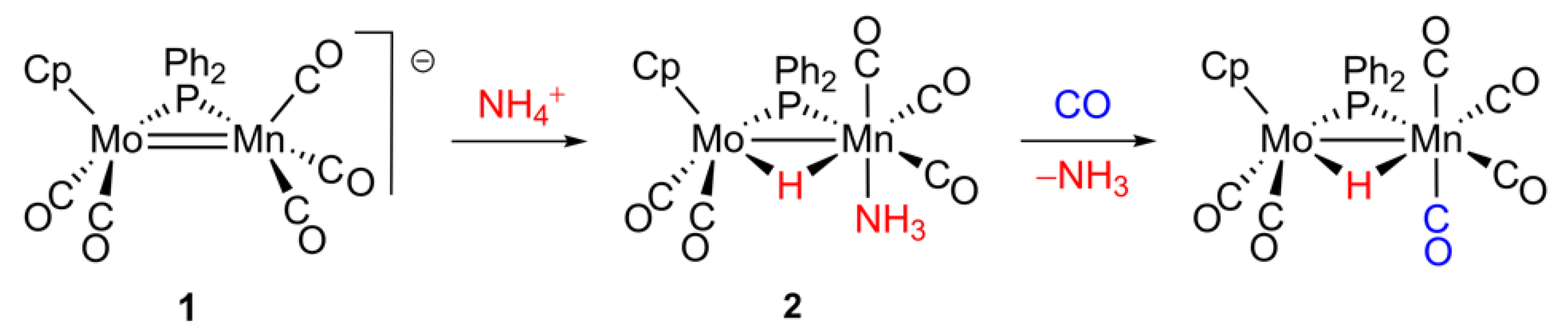
2. Results and Discussion
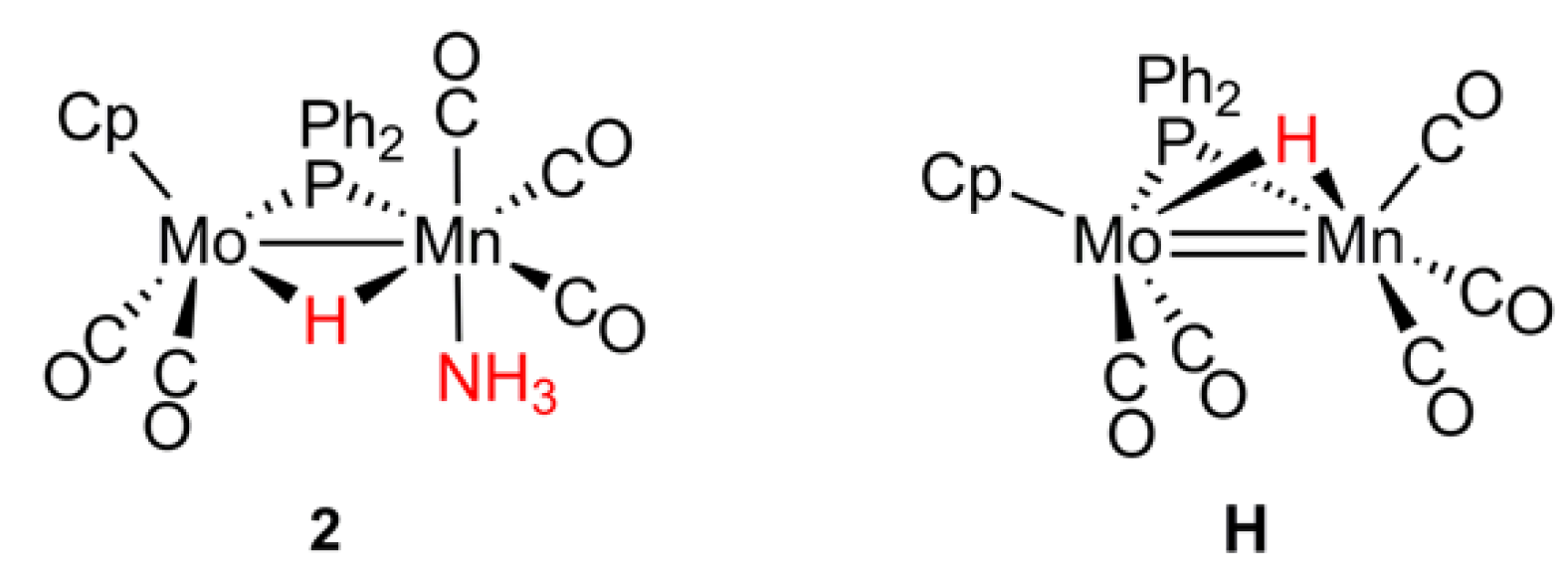
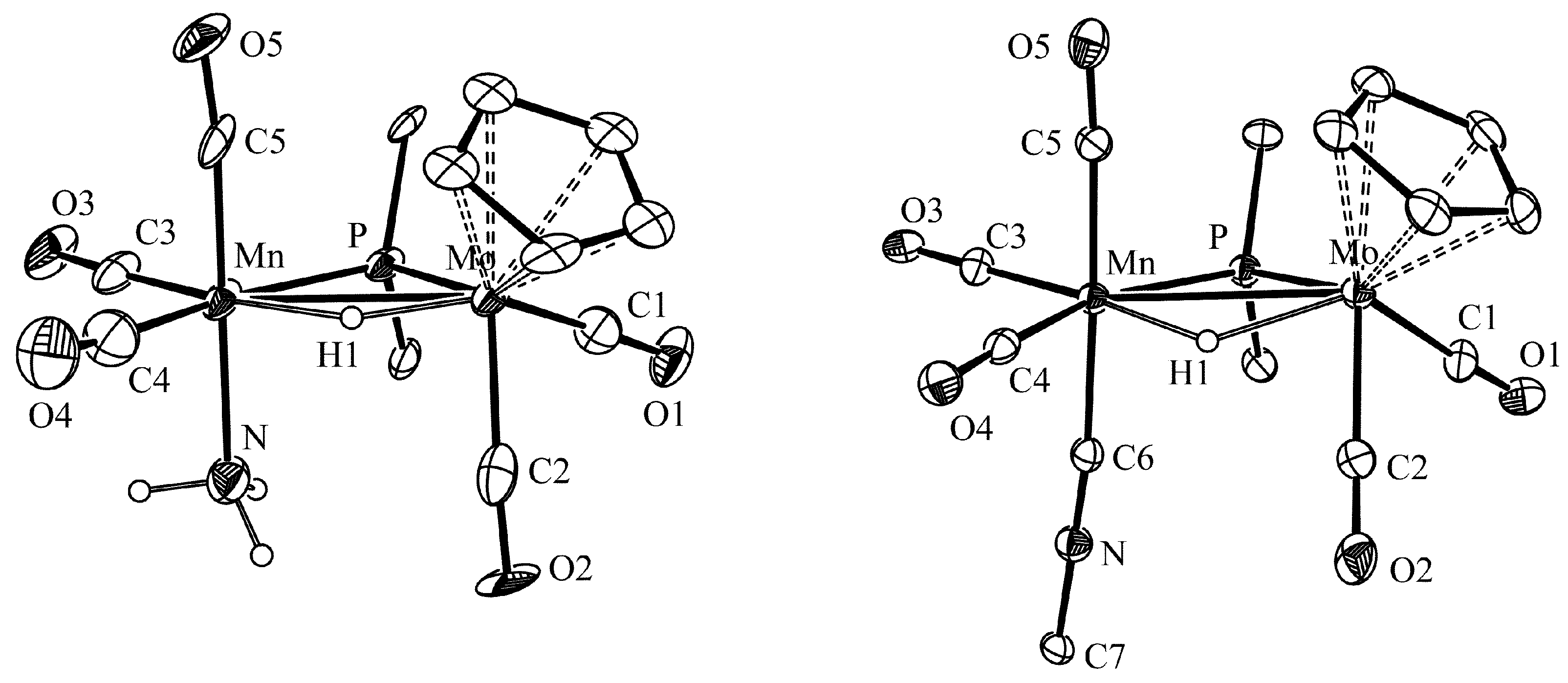
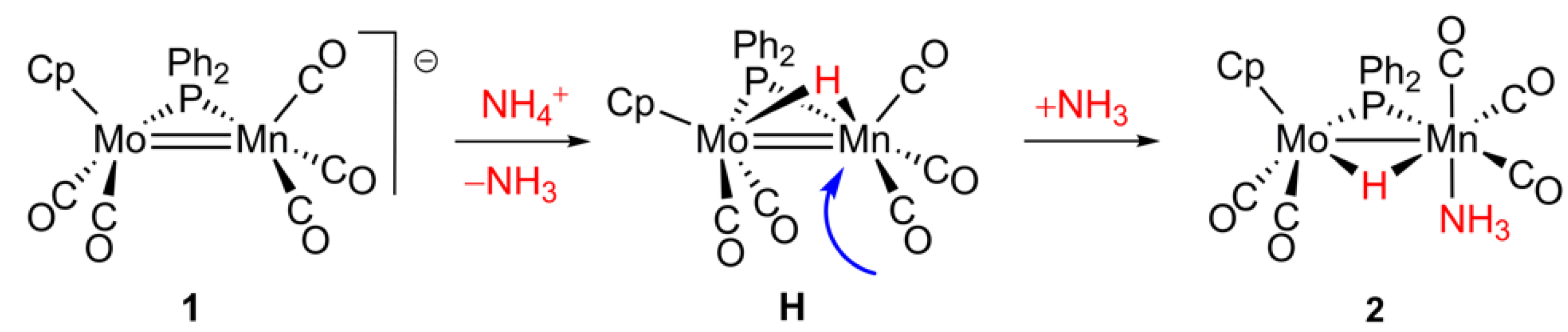
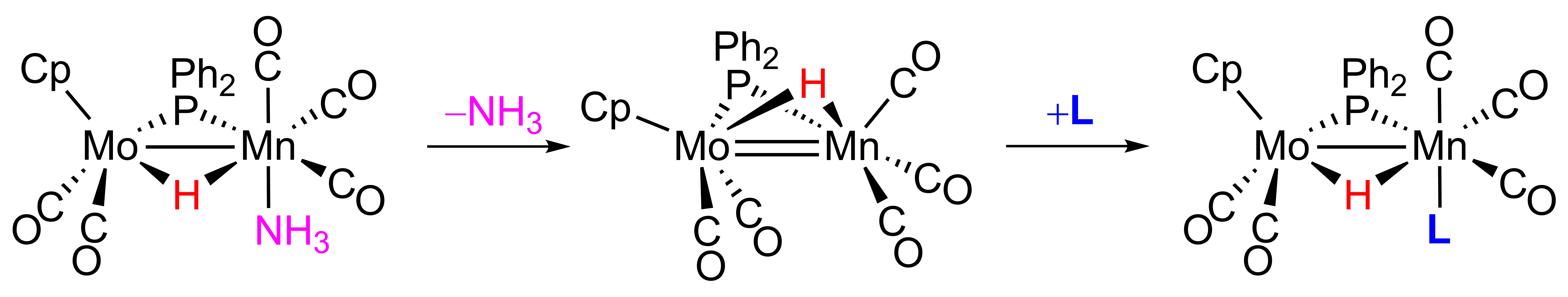
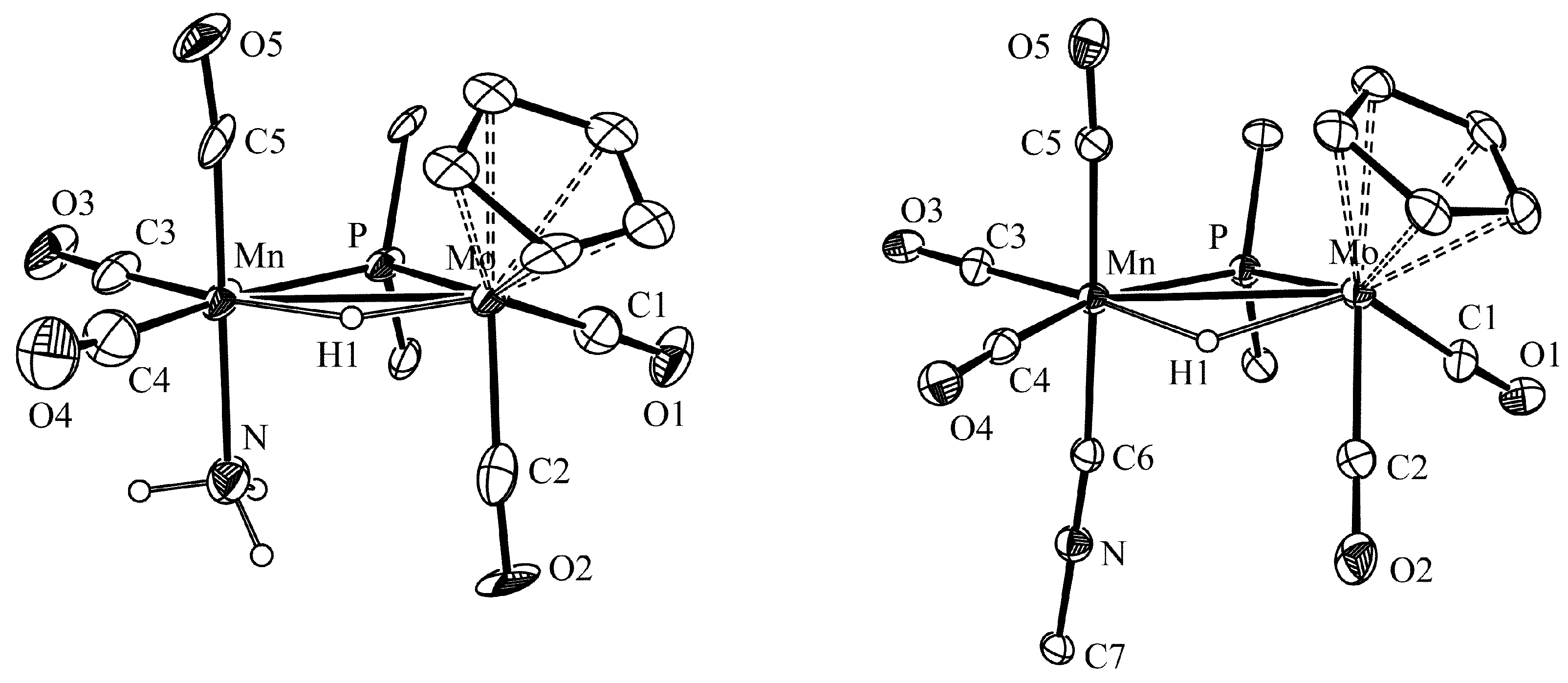
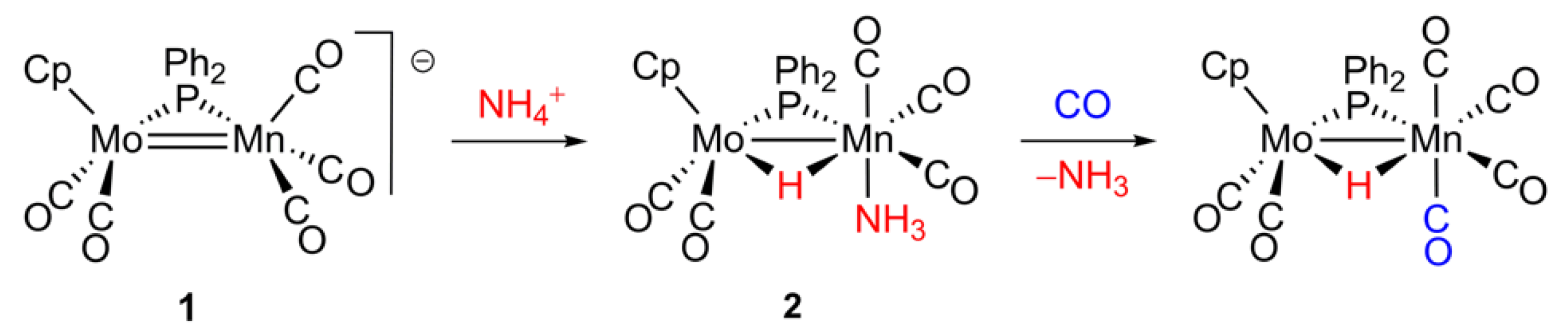
2.1. Protonation of Anion 1. Structural Characterization of the Ammonia Complex 2
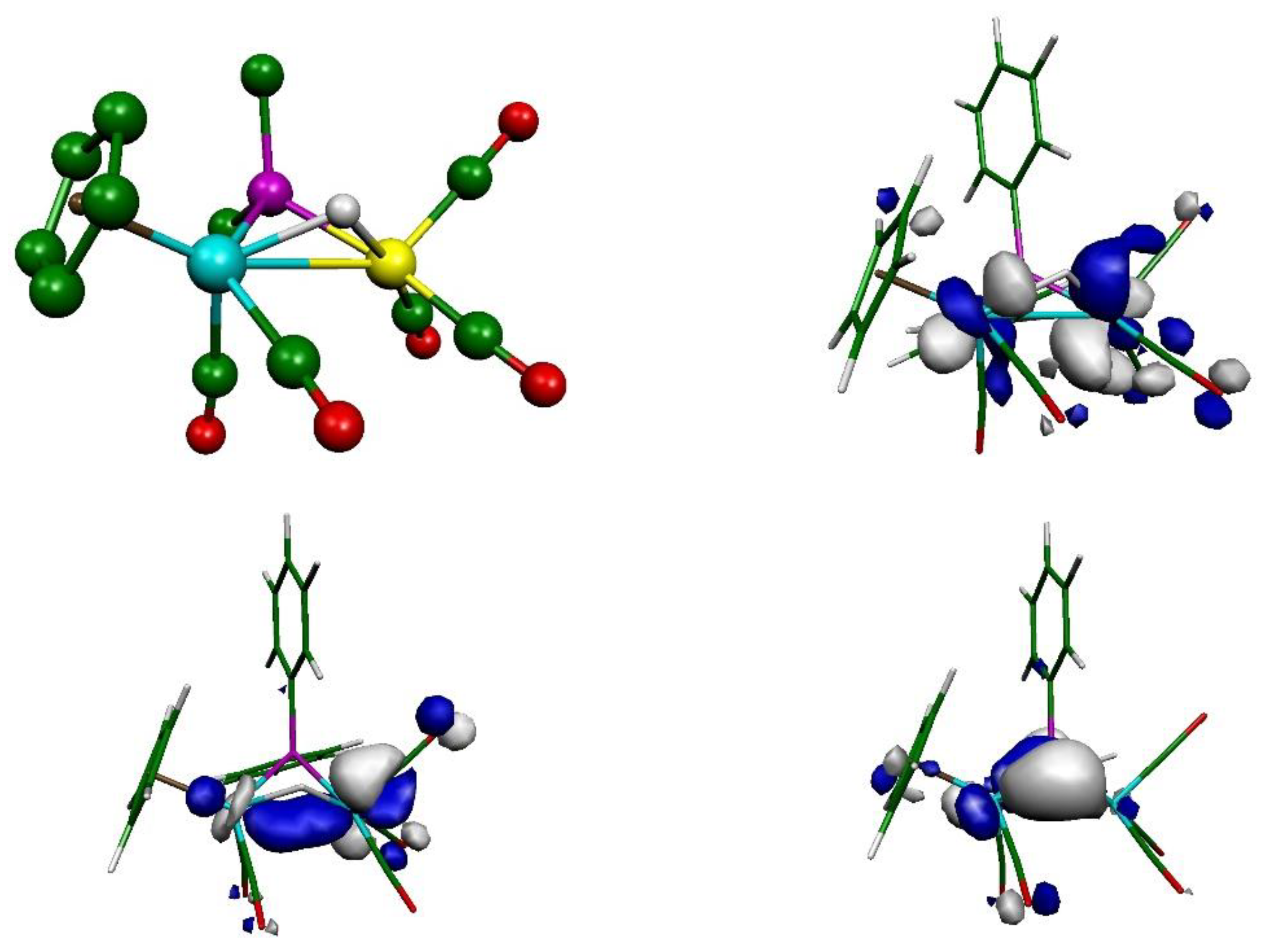
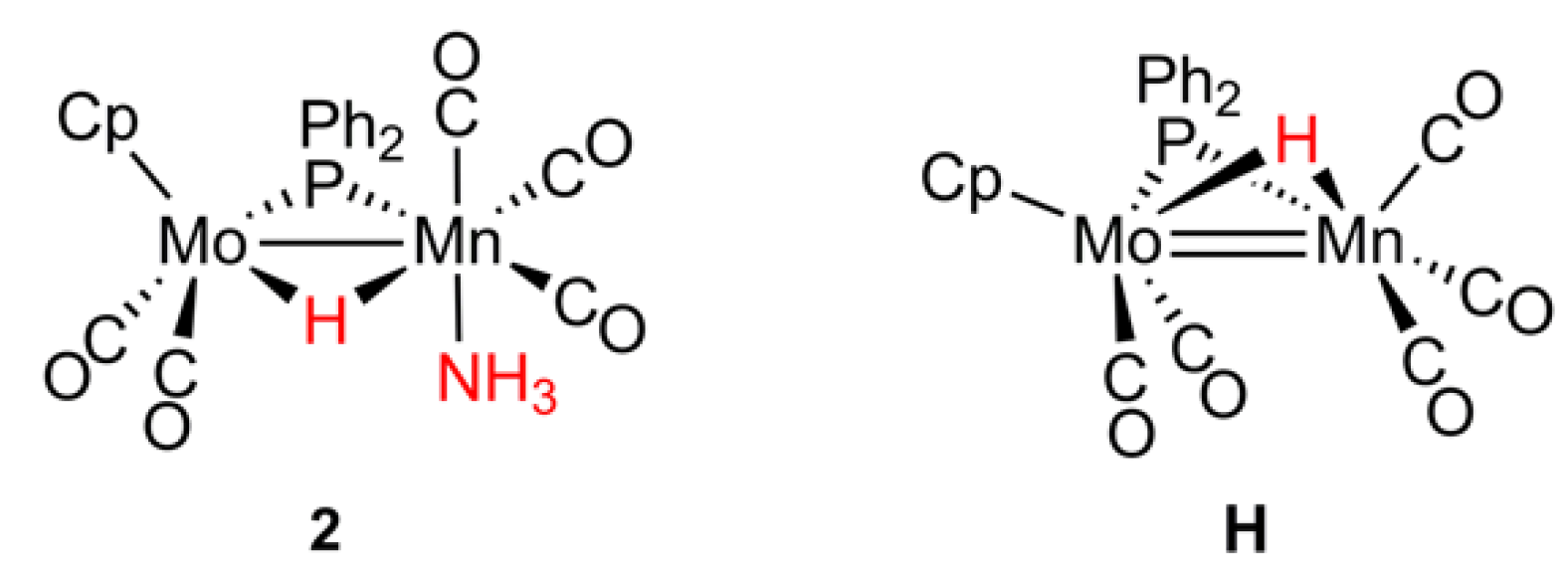
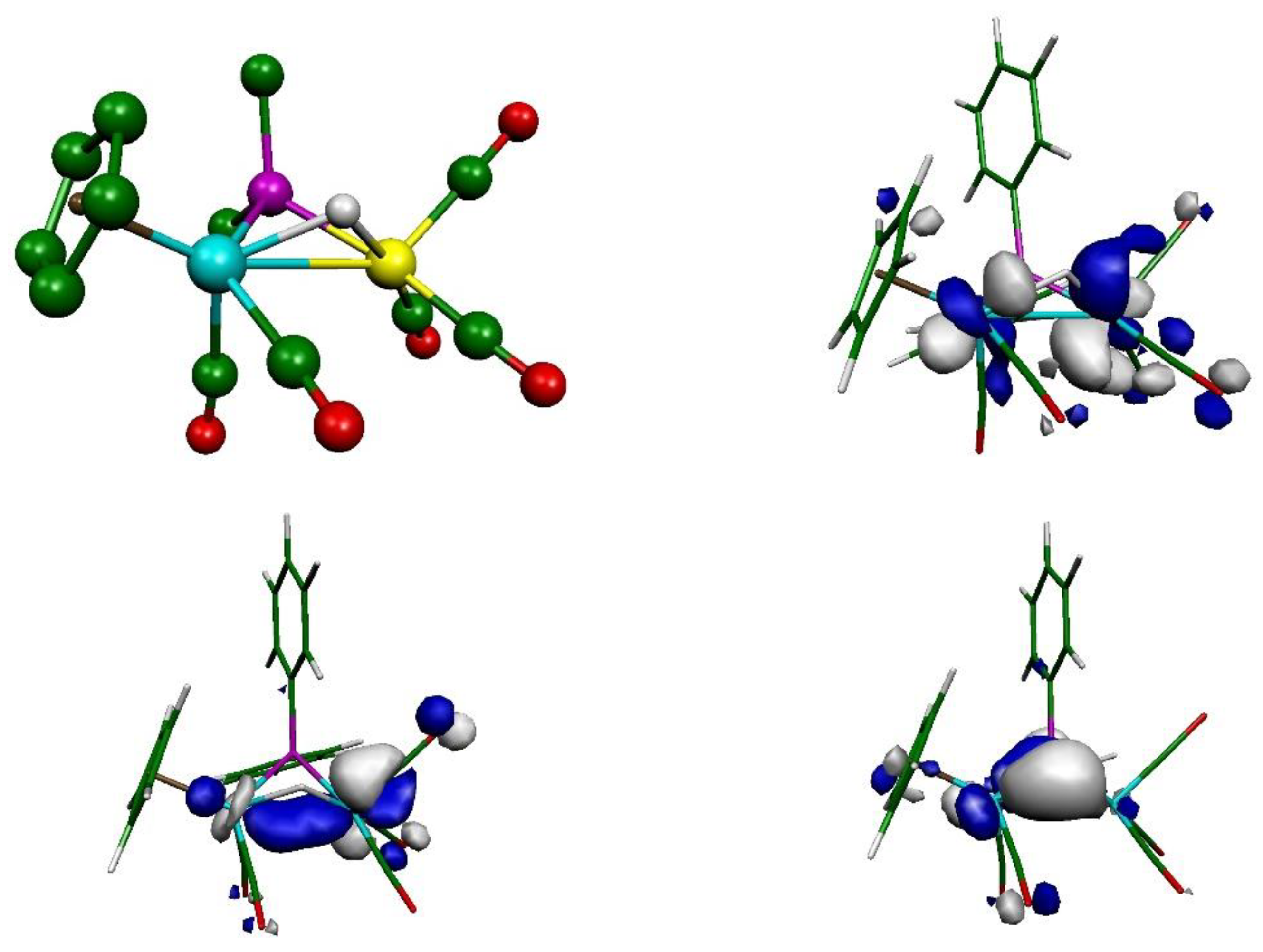
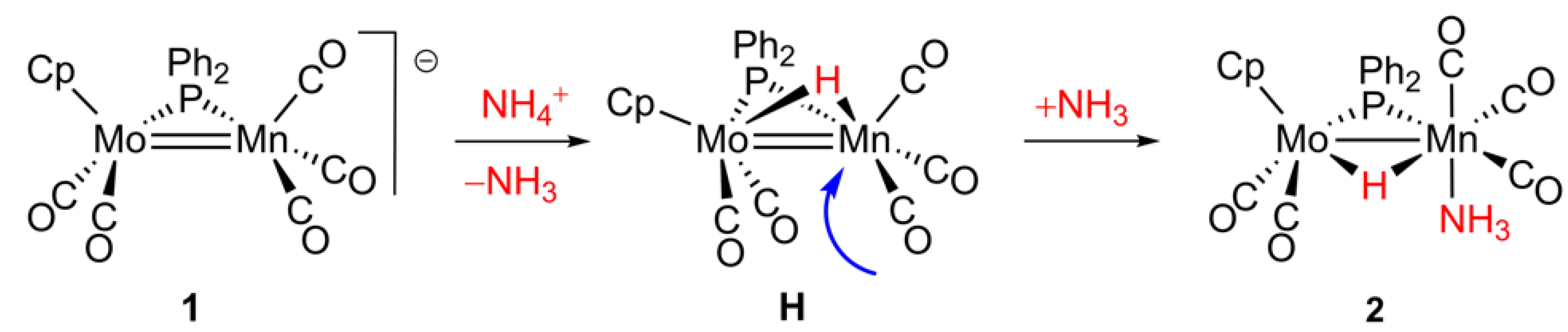
2.2. Mechanism of Formation of 2. DFT Calculations on the Putative Hydride Intermediate H
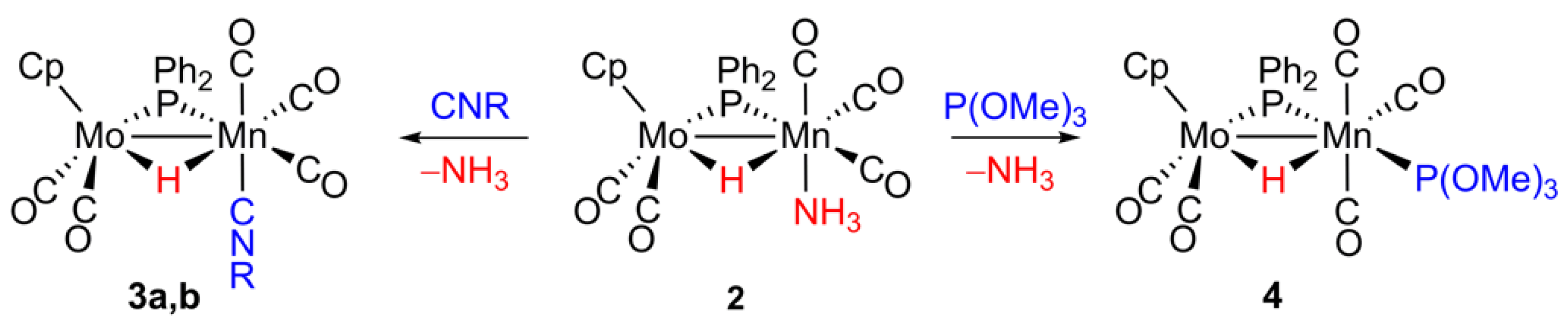
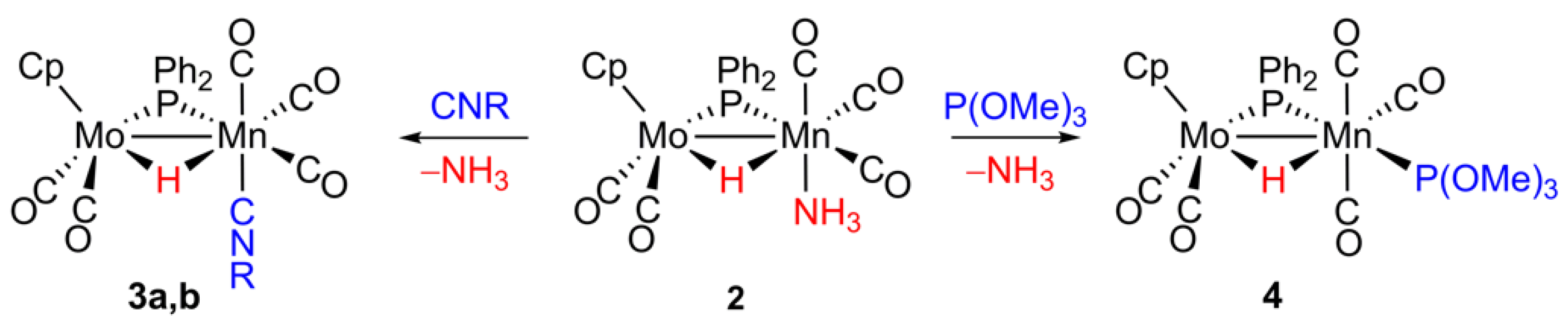
2.3. Reaction of 2 with Isocyanides
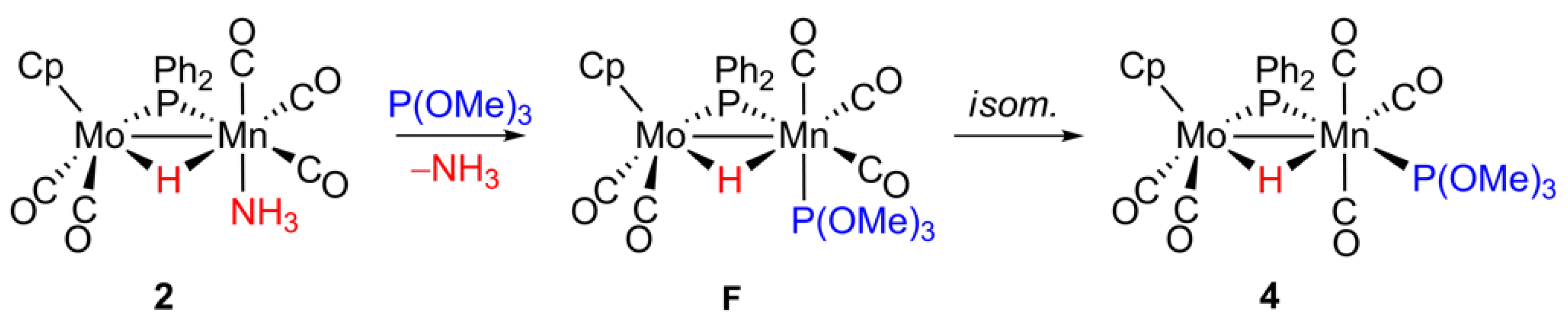
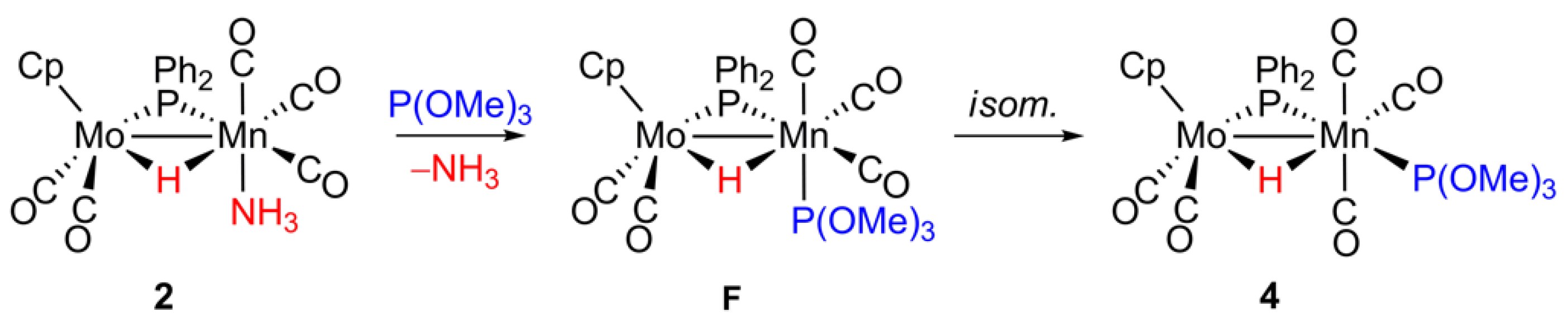
2.4. Reaction of 2 with P(OMe)3
3. Materials and Methods
3.1. General Procedures and Starting Materials
3.2. Preparation of [MoMnCp(μ-H)(μ-PPh2)(CO)5(NH3)] (2)
3.3. Preparation of [MoMnCp(μ-H)(μ-PPh2)(CO)5(CNXyl)] (3a)
3.4. Preparation of [MoMnCp(μ-H)(μ-PPh2)(CO)5{CN(p-C6H4OMe)}] (3b)
3.5. Preparation of [MoMnCp(μ-H)(μ-PPh2)(CO)5{P(OMe)3}] (4)
3.6. X-ray Structure Determination of Compounds 2 and 3a
3.7. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Knorr, M.; Jourdain, I. Activation of alkynes by diphosphine- and μ-phosphido-spanned heterobimetallic complexes. Coord. Chem. Rev. 2017, 350, 217–247. [Google Scholar] [CrossRef]
- Mankad, N.P. Selectivity Effects in Bimetallic Catalysis. Chem. Eur. J. 2016, 22, 5822–5829. [Google Scholar] [CrossRef] [PubMed]
- Buchwalter, P.; Rosé, J.; Braunstein, P. Multimetallic Catalysis Based on Heterometallic Complexes and Clusters. Chem. Rev. 2015, 115, 28–126. [Google Scholar] [CrossRef] [PubMed]
- Eisenhart, R.J.; Clouston, L.J.; Lu, C.C. Configuring Bonds between First-Row Transition Metals. Acc. Chem. Res. 2015, 48, 2885–2894. [Google Scholar] [CrossRef] [PubMed]
- Krogman, J.P.; Thomas, C.M. Metal-metal multiple bonding in C3-symmetric bimetallic complexes of the first row transition metals. Chem. Commun. 2014, 540, 5115–5127. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.M. Metal-Metal Multiple Bonds in Early/Late Heterobimetallic Complexes: Applications Toward Small Molecule Activation and Catalysis. Comments Inorg. Chem. 2011, 32, 14–38. [Google Scholar] [CrossRef]
- Ritleng, V.; Chetcuti, M.J. Hydrocarbyl Ligand Transformations on Heterobimetallic Complexes. Chem. Rev. 2007, 107, 797–858. [Google Scholar] [CrossRef] [PubMed]
- Collman, J.P.; Boulatov, R. Heterodinuclear Transition-Metal Complexes with Multiple Metal–Metal Bonds. Angew. Chem. Int. Ed. 2002, 41, 3948–3961. [Google Scholar] [CrossRef]
- Clapham, S.; Braunstein, P.; Boag, N.M.; Welter, R.; Chetcuti, M.J. Carbon−Phosphorus Bond Cleavage in Allyldiphenylphosphine by a Heterobimetallic Ni=Mo Complex and Formation of an Electronically Unsaturated NiMo2 μ-Diphenylphosphido Cluster. Organometallics 2008, 27, 1758–1764. [Google Scholar] [CrossRef]
- Oishi, M.; Oshima, M.; Suzuki, H. A Study on Zr–Ir Multiple Bonding Active for C–H Bond Cleavage. Inorg. Chem. 2014, 53, 6634–6654. [Google Scholar] [CrossRef] [PubMed]
- Oishi, M.; Kino, M.; Saso, M.; Oshima, M.; Suzuki, H. Early–Late Heterobimetallic Complexes with a Ta–Ir Multiple Bond: Bimetallic Oxidative Additions of C–H, N–H, and O–H Bonds. Organometallics 2012, 31, 4658–4661. [Google Scholar] [CrossRef]
- Kameo, H.; Nakajima, Y.; Suzuki, H. Drastic Acceleration of Phosphine/Phosphite Incorporation into a Tetrahydrido Ruthenium/Osmium Complex, and One-way Ruthenium to Osmium Migration of a Phosphorus Ligand. Angew. Chem. Int. Ed. 2008, 47, 10159–10162. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.A.; García, M.E.; García-Vivó, D.; Huergo, E.; Ruiz, M.A. Synthesis of the Unsaturated [MMoCp(µ-PR2)(CO)5]– Anions (M = Mn, R = Ph; M = Re, R = Cy): Versatile Precursors of Heterometallic Complexes. Eur. J. Inorg. Chem. 2017, 9, 1280–1283. [Google Scholar] [CrossRef]
- Alvarez, M.A.; García, M.E.; García-Vivó, D.; Huergo, E.; Ruiz, M.A. Acetonitrile Adduct [MoReCp(μ-H)(μ-PCy2)(CO)5(NCMe)]: A Surrogate of an Unsaturated Heterometallic Hydride Complex. Inorg. Chem. 2018, 57, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Green, M.L.H.; Parkin, G. The Covalent Bond Classification Method and its Application to Compounds That Feature 3-Center 2-Electron Bonds. Struct. Bond. 2017, 117, 79–140. [Google Scholar]
- García-Vivó, D.; Ruiz, M.A. Reply to the Comment on “Hydride, gold(I) and related derivatives of the unsaturated ditungsten anion [W2Cp2(μ-PCy2)(μ-CO)2]”. Dalton Trans. 2018, 47, 6630–6631. [Google Scholar] [CrossRef] [PubMed]
- Horton, A.D.; Mays, M.J.; Raithby, P.R. Synthesis and substitution reactions of a heterodimetallic molybdenum–manganese complex, [MoMn(µ-H)(µ-PPh2)(η5-C5H5)(CO)6]; X-ray crystal structure of [MoMn(µ-H)(µ-PPh2)(η5-C5H5)(CO)4(dppm-PP′)]. J. Chem. Soc. Dalton Trans. 1987, 6, 1557–1563. [Google Scholar] [CrossRef]
- Horton, A.D.; Mays, M.J.; Raithby, P.R. Formation of complexes with C···H···Mn interactions from reactions of dienes or acetylene with a µ-hydrido heterometallic dimer; X-ray crystal structures of [(η5-C5H5)MoMn(µ-H)(µ-PPh2)(CO)6], [(η5-C5H5)MoMn{µ–σ:η3-CH2C(Me)CHMe}(µ-PPh2)(CO)4], and [(η5-C5H5)MoMn{µ–σ: η4-CHCHCH2CHPPh2}(CO)4]. J. Chem. Soc. Chem. Commun. 1985, 5, 247–250. [Google Scholar] [CrossRef]
- Cordero, B.; Gómez, V.; Platero-Prats, A.E.; Revés, M.; Echevarría, J.; Cremades, E.; Barragán, F.; Alvarez, S. Covalent Radii Revisited. Dalton Trans. 2008, 21, 2832–2838. [Google Scholar] [CrossRef] [PubMed]
- Herberhold, M.; Wehrmann, F.; Neugebauer, D.; Huttner, G. Die photo-induzierte reaktion von dekacarbonyl-dimangan mit ammoniak, kristall- und molekülstrusktur des produkts [fac-Mn(CO)3(NH3)3][Mn(CO)5]. J. Organomet. Chem. 1978, 152, 329–336. [Google Scholar] [CrossRef]
- Ni, C.; Lei, H.; Power, P.P. Reaction of M(II) Diaryls (M = Mn or Fe) with Ammonia to Afford Parent Amido Complexes. Organometallics 2010, 29, 1988–1991. [Google Scholar] [CrossRef]
- Braterman, P.S. Metal Carbonyl Spectra; Academic Press: London, UK, 1975. [Google Scholar]
- Carty, A.J.; MacLaughlin, S.A.; Nucciarone, D. Phosphorus-31 NMR Spectroscopy in Stereochemical Analysis; Verkade, J.G., Quin, L.D., Eds.; VCH: Deerfield Beach, FL, USA, 1987; Chapter 16. [Google Scholar]
- Jameson, C.J. Phosphorus-31 NMR Spectroscopy in Stereochemical Analysis; Verkade, J.G., Quin, L.D., Eds.; VCH: Deerfield Beach, FL, USA, 1987; Chapter 6. [Google Scholar]
- Cramer, C.J. Essentials of Computational Chemistry, 2nd ed.; Wiley: Chichester, UK, 2004. [Google Scholar]
- Koch, W.; Holthausen, M.C. A Chemist’s Guide to Density Functional Theory, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Alvarez, M.A.; García, M.E.; García-Vivó, D.; Ruiz, M.A.; Toyos, A. The Doubly-Bonded Ditungsten Anion [W2Cp2(μ-PPh2)(NO)2]: An Entry to the Chemistry of Unsaturated Nitrosyl Complexes. Dalton Trans. 2016, 45, 13300–13303. [Google Scholar] [CrossRef] [PubMed]
- García, M.E.; Ramos, A.; Ruiz, M.A.; Lanfranchi, M.; Marchiò, L. Structure and Bonding in the Unsaturated Hydride- and Hydrocarbyl-Bridged Complexes [Mo2(η5-C5H5)2(μ-X)(μ-PCy2)(CO)2] (X = H, CH3, CH2Ph, Ph). Evidence for the presence of α-Agostic and π-Bonding Interactions. Organometallics 2007, 26, 6197–6212. [Google Scholar] [CrossRef]
- Alvarez, M.A.; García, M.E.; García-Vivó, D.; Ruiz, M.A.; Vega, M.F. Synthesis and reactivity of the triply bonded binuclear anion [W2(η5-C5H5)2(μ-PCy2)(μ-CO)2]: Tungsten makes a difference. Organometallics 2010, 29, 512–515. [Google Scholar] [CrossRef]
- Alvarez, M.A.; García, M.E.; García-Vivó, D.; Ruiz, M.A.; Vega, M.F. Hydride, Gold(I) and Related Derivatives of the Unsaturated Ditungsten Anion [W2Cp2(μ-PCy2)(μ-CO)2]. Dalton Trans. 2014, 43, 16044–16055. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules—A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Doyle, M.J.; Duckworth, T.J.; Manojlovic-Muir, L.; Mays, M.J.; Raithby, P.R.; Robertson, F.J. Substitution Reactions of a Heterodimetallic Molybdenum-Manganese Complex. J. Chem. Soc. Dalton Trans. 1982, 18, 2703–2714. [Google Scholar] [CrossRef]
- Wrackmeyer, B.; Alt, H.G.; Maisel, H.E. Ein- und zwei-dimensionale Multikern NMR-Spektroskopie an den isomeren Halbsandwich-Komplexen cis- und trans-[(η5-C5H5)W(CO)2(H)PMe3]. J. Organomet. Chem. 1990, 399, 125–130. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C. Purification of Laboratory Chemicals, 7th ed.; Butterworth-Heinemann: Oxford, UK, 2012. [Google Scholar]
- CrysAlis Pro; Oxford Diffraction Limited, Ltd.: Oxford, UK, 2006.
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision B.02; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for potassium to gold including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. Influence of polarization functions on MO hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Petersson, G.A.; Al-Laham, M.A. A complete basis set model chemistry. II. Open-shell systems and the total energies of the first-row atoms. J. Chem. Phys. 1991, 94, 6081–6090. [Google Scholar] [CrossRef]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A.; Mantzaris, J. A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elements. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Portmann, S.; Luthi, H.P. MOLEKEL: An Interactive Molecular Graphics Tool. CHIMIA 2000, 54, 766–770. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 2 | 3a | Parameter | 2 | 3a |
|---|---|---|---|---|---|
| Mo Mn | 3.087(3) | 3.0770(4) | P Mo C1 | 73.1(7) | 78.7(1) |
| Mo P | 2.446(4) | 2.4405(6) | P Mo C2 | 105.3(6) | 111.1(1) |
| Mn P | 2.277(5) | 2.2816(7) | P Mn C3 | 98.1(7) | 101.1(1) |
| Mo H | 1.56(1) | 1.83(3) | P Mn C4 | 165.5(9) | 161.5(1) |
| Mn H | 1.82(1) | 1.74(3) | P Mn C5 | 89.0(7) | 91.7(1) |
| Mo C1 | 1.91(2) | 1.973(3) | P Mn N1/C6 | 90.5(4) | 88.0(1) |
| Mo C2 | 1.95(3) | 1.962(3) | Mo P Mn | 81.5(2) | 81.25(2) |
| Mn C3 | 1.78(2) | 1.793(3) | C6 N6 C7 | - | 179.6(3) |
| Mn C4 | 1.84(3) | 1.819(3) | Mo Mn N/C6 | 92.9(5) | 97.9(1) |
| Mn C5 | 1.80(2) | 1.827(3) | - | - | - |
| Mn N1/C6 | 2.10(2) | 1.919(3) | - | - | - |
| Compound | ν(CO) | δ(P) |
|---|---|---|
| Na[MoMnCp(μ-PPh2)(CO)5] (1-Na) 3 | 1968 (s), 1881 (vs) 1864 (s, sh), 1804 (w) | 131.3 |
| [MoMnCp(μ-H)(μ-PPh2)(CO)5(NH3)] (2) | 2001 (s), 1944 (vs), 1915 (s), 1893 (m), 1866 (m) 4 | 161.4 5 |
| [MoMnCp(μ-H)(μ-PPh2)(CO)5(CNXyl)] (3a) | 2009 (s), 1951 (vs) 1931 (m, sh), 1871 (w) | 160.9 6 |
| [MoMnCp(μ-H)(μ-PPh2)(CO)5{CN(p-C6H4OMe)}] (3b) | 2010 (s), 1949 (vs) 1933 (s, sh), 1870 (m) | 160.9 |
| [MoMnCp(μ-H)(μ-PPh2)(CO)5{P(OMe)3}] (4) | 2030 (w), 1951 (vs) 1925 (m, sh), 1869 (m) | 191.81 63.9 (μ-P) |
| Parameter | 2·C7H8 | 3a |
|---|---|---|
| mol formula | C29H27MnMoNO5P | C31H25MnMoNO5P |
| mol wt | 651.37 | 673.37 |
| cryst syst | monoclinic | triclinic |
| space group | P21/c | P 1 |
| radiation (λ, Å) | 1.54184 | 1.54184 |
| a, Å | 19.0906(14) | 10.9412(6) |
| b, Å | 8.5500(4) | 11.1757(6) |
| c, Å | 17.2528(11) | 11.9618(6) |
| α, deg | 90 | 84.979(4) |
| β, deg | 100.240(6) | 78.905(5) |
| γ, deg | 90 | 84.550(4) |
| V, Å3 | 2771.2(3) | 1425.22(13) |
| Z | 4 | 2 |
| calcd density, g·cm−3 | 1.561 | 1.569 |
| absorp coeff, mm−1 | 8.285 | 8.080 |
| temperature, K | 154(5) | 156(5) |
| θ range (deg) | 4.71–69.41 | 3.76–69.69 |
| index ranges (h; k; l) | −22, 21; −10, 8; −20, 15 | −12, 13; −13, 13; −14, 12 |
| no. of reflns collected | 12,356 | 12,082 |
| no. of indep reflns (Rint) | 5094(0.0912) | 5243(0.0320) |
| reflns with I > 2σ(I) | 4275 | 4743 |
| R indexes [data with I > 2σ(I)] a | R1 = 0.2410, wR2 = 0.5368 b | R1 = 0.0276, wR2 = 0.0721 c |
| R indexes (all data) a | R1 = 0.2510, wR2 = 0.5492 b | R1 = 0.0316, wR2 = 0.0752 c |
| GOF | 2.390 | 1.058 |
| no. of restraints/params | 24/305 | 0/367 |
| ∆ρ(max./min.), eÅ−3 | 10.949/−3.325 | 0.480/−0.662 |
| CCDC deposition no | 1871692 | 1871693 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alvarez, M.A.; García-Vivó, D.; Huergo, E.; Ruiz, M.A. Trapping of an Heterometallic Unsaturated Hydride: Structure and Properties of the Ammonia Complex [MoMnCp(μ-H)(μ-PPh2)(CO)5(NH3)]. Inorganics 2018, 6, 125. https://doi.org/10.3390/inorganics6040125
Alvarez MA, García-Vivó D, Huergo E, Ruiz MA. Trapping of an Heterometallic Unsaturated Hydride: Structure and Properties of the Ammonia Complex [MoMnCp(μ-H)(μ-PPh2)(CO)5(NH3)]. Inorganics. 2018; 6(4):125. https://doi.org/10.3390/inorganics6040125
Chicago/Turabian StyleAlvarez, M. Angeles, Daniel García-Vivó, Estefanía Huergo, and Miguel A. Ruiz. 2018. "Trapping of an Heterometallic Unsaturated Hydride: Structure and Properties of the Ammonia Complex [MoMnCp(μ-H)(μ-PPh2)(CO)5(NH3)]" Inorganics 6, no. 4: 125. https://doi.org/10.3390/inorganics6040125
APA StyleAlvarez, M. A., García-Vivó, D., Huergo, E., & Ruiz, M. A. (2018). Trapping of an Heterometallic Unsaturated Hydride: Structure and Properties of the Ammonia Complex [MoMnCp(μ-H)(μ-PPh2)(CO)5(NH3)]. Inorganics, 6(4), 125. https://doi.org/10.3390/inorganics6040125