
Development and Validation of a Sensitive HPLC Assay for Determination of Sparfloxacin According to the European Medicines Agency Guideline

 , ,
, ,  , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
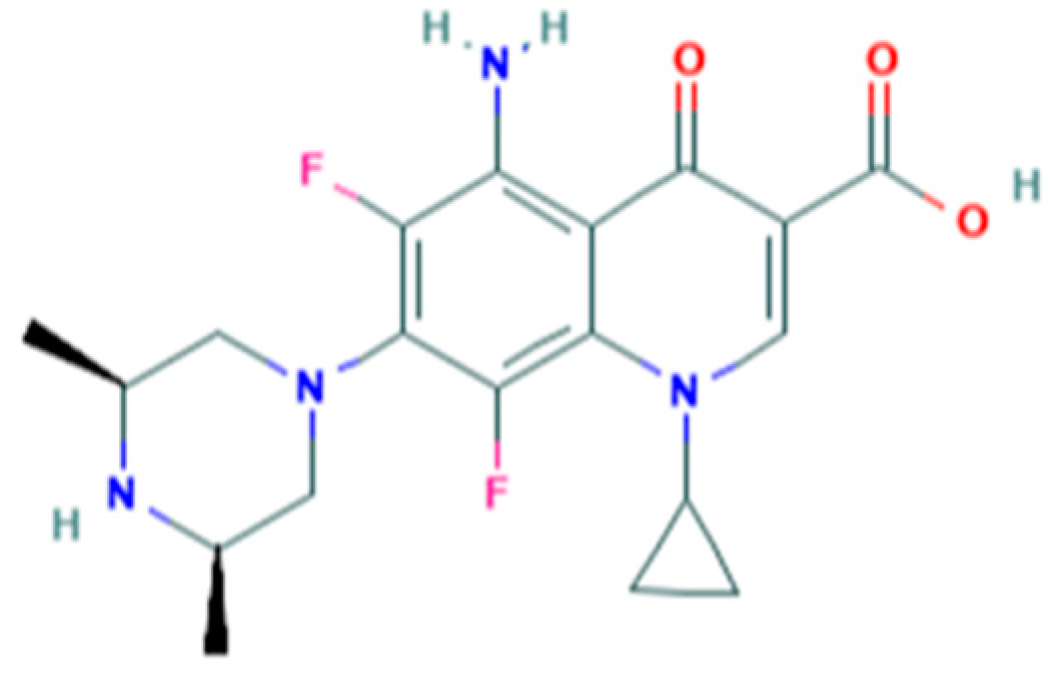
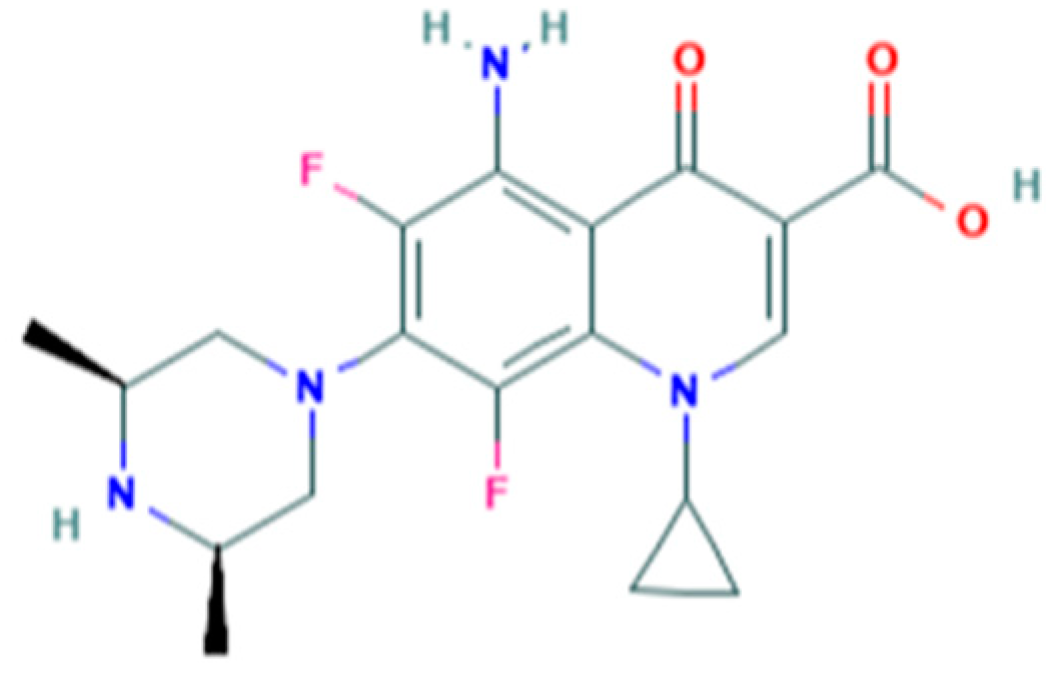
2.1. Chemicals and Reagents
2.2. Animals and Experimental Procedures
2.3. Preparations of Stock, Calibration and Quality Working Solutions
- Stock solution (1 mg/mL) was prepared in HPLC-grade water, also adding 100 µL NaOH 1N. IS (1 mg/mL) was also dissolved in HPLC-grade water and 100 µL NaOH 1N.
- Calibration working solutions containing both SPAR (2, 5, 10, 20, 50 and 100 µg/mL) and IS (20 µg/mL) were then prepared by diluting an appropriate volume of the stock solution in 10 mL of HPLC-grade water.
- Quality control working solutions (QC) were obtained by adding an aliquot of each stock solution to obtain final concentrations of 0.2 µg/mL (QC1: LLOQ, lower limit of quantitation), 0.6 µg/mL (QC2: LOW, three times the LLOQ), 3 µg/mL (QC3: MED, between 30 and 50% of the calibration curve range) and 8 µg/mL (QC4: HIGH, 75% of the upper calibration curve range).
2.4. Preparation of Analysis Samples
- Blank samples: biological matrix without SPAR and IS (1 mL).
- Zero samples: biological matrix (0.9 mL) with 0.1 mL IS (20 µg/mL).
- Calibration standards: 0.9 mL plasma was spiked with 100 µL of each calibration working solution to obtain calibration curves. Thus, concentrations of calibration samples were 0.2, 0.5, 1, 2, 5 and 10 µg/mL for SPAR and 2 µg/mL for IS.
- Quality control samples were also prepared in plasma (0.9 mL) at concentrations of 0.2, 0.6, 3 and 8 µg/mL for SPAR (0.1 mL) and 2 µg/mL for IS (0.1 mL).
- All samples were fully thawed at room temperature.
2.5. Extraction Method
2.6. HPLC System and Conditions
2.7. Method Validation Procedure
2.8. Method Application
2.9. Data Analysis
3. Results and Discussion
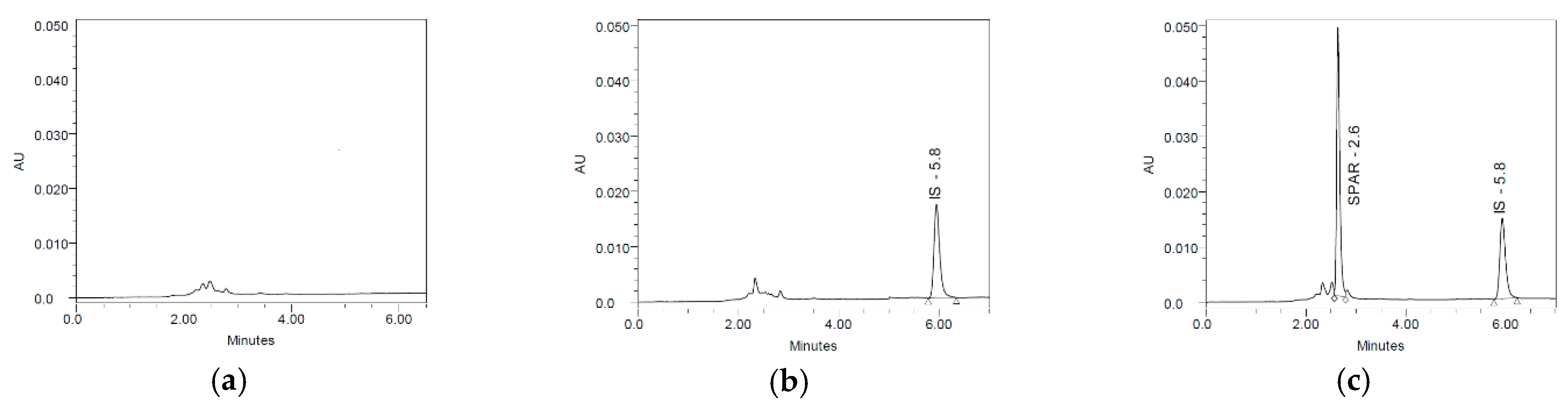
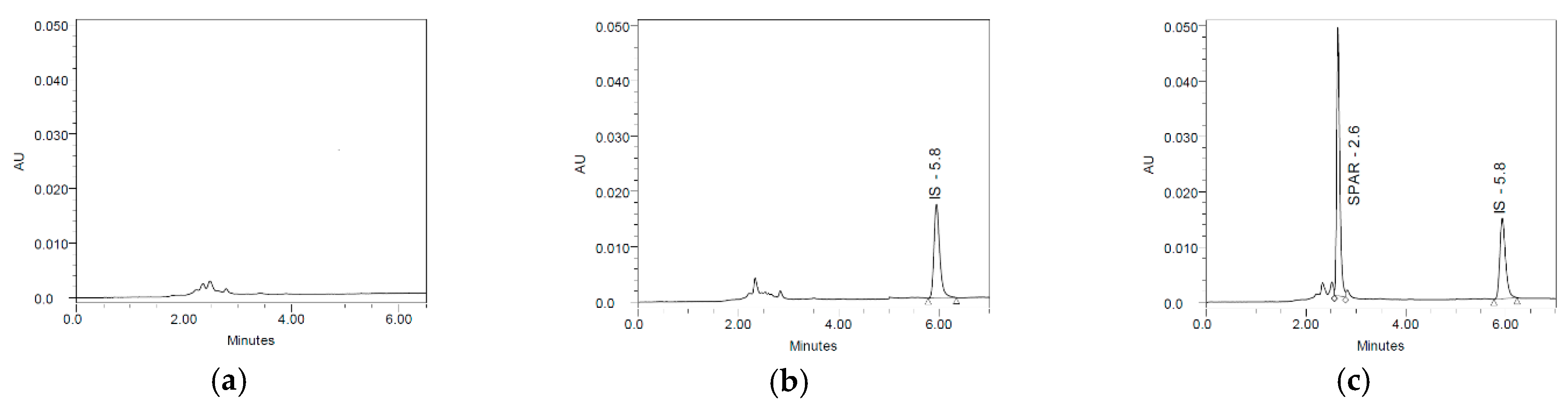
3.1. Selectivity
3.2. Carry-Over
3.3. Lower Limit of Quantification (LLOQ)
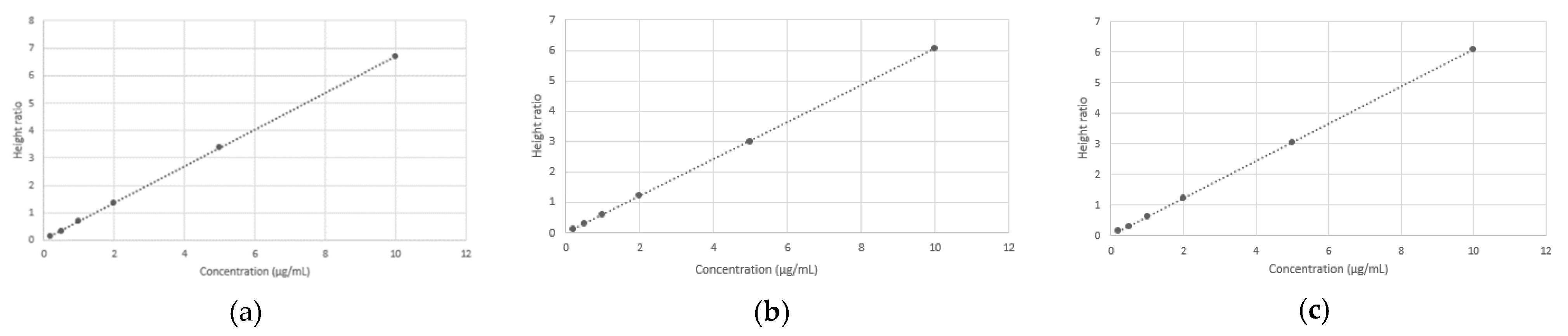
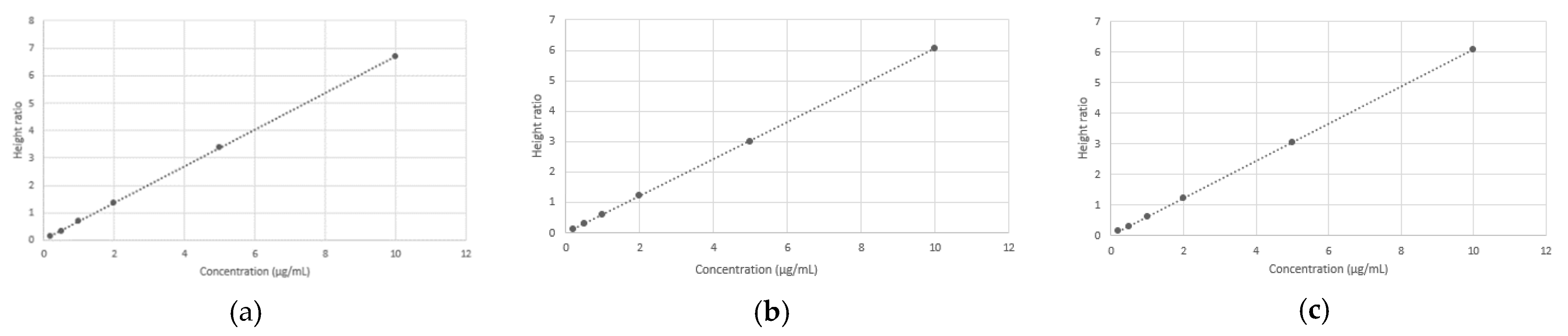
3.4. Calibration Curve
3.5. Accuracy and Precision
3.6. Stability
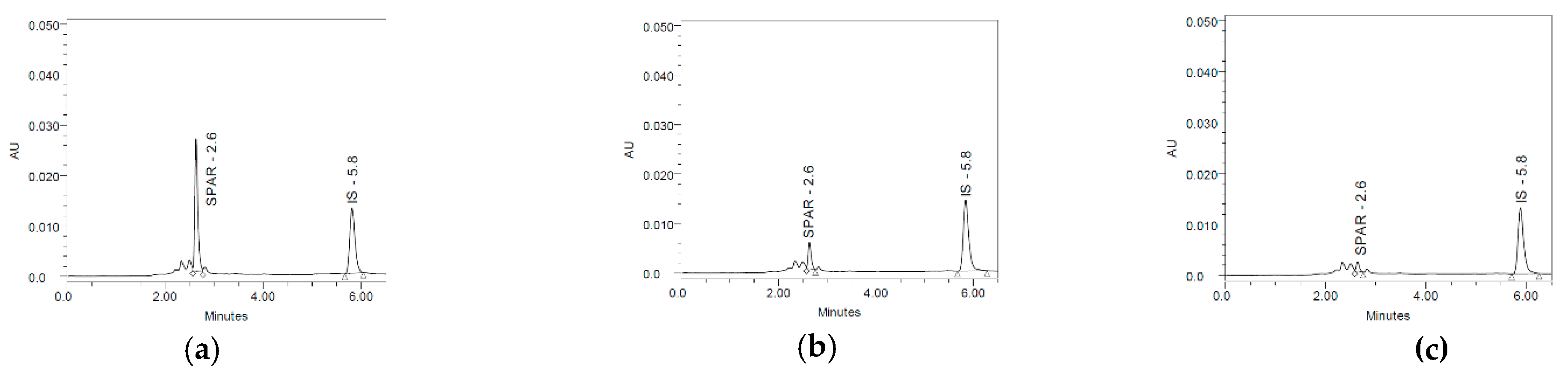
3.7. Method Application
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- O´Neil, M. The Merck Index, 15th ed.; Royal Society Chemistry: Whitehouse Station, NJ, USA, 2013; ISBN 9781849736701. [Google Scholar]
- Ravisankar, P.; Rao, G.D.; Rao, M.N.; Devadasu, C.A. Validated RP-HPLC method for the determination of sparfloxacin in bulk and pharmaceutical dosage form. Asian J. Pharm. Clin. Res. 2014, 7, 135–140. [Google Scholar]
- Bhar, M.K.; Khargharia, S.; Chakraborty, A.K.; Mandal, T.K. Disposition kinetics of sparfloxacin in healthy, hepatopathic, and nephropathic conditions in chicken after single intravenous administration. Indian J. Pharmacol. 2009, 41, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Shimada, J.; Nogita, T.; Ishibashi, Y. Clinical Pharmacokinetics of Sparfloxacin. Clin. Pharmacokinet. 1993, 25, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Alós, J. Quinolonas. Enferm. Infecc. Microbiol. Clin. 2009, 27, 290–297. [Google Scholar] [CrossRef]
- Oliphant, C.; Green, G. Quinolones: A comprehensive review. Am. Fam. Physician 2002, 65, 455–464. [Google Scholar] [PubMed]
- Sriranjani, D.; Kalaiselvi, L.; Ramesh, S.; Mathuram, L.N.; Sriram, P. Pharmacokinetics of sparfloxacin in broiler chicken. Br. Poult. Sci. 2006, 47, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Suraj, S.S.; Suresh, N.N.; Sanis, J.; Sanjay, B.M.; Saranya, C.; Asaf, M.; Shijin, M.S.; Nisha, A.R.; Anoopraj, R. Single dose oral pharmacokinetics of sparfloxacin in rats. Pharma Innov. J. 2019, 8, 317–322. [Google Scholar]
- Matsunaga, Y.; Miyazaki, H.; Oh-e, Y.; Nambu, K.; Furukawa, H.; Yoshida, K.; Hashimoto, M. Disposition and metabolism of [14C] sparfloxacin in the rat. Arzneimittelforschung 1991, 41, 747–759. [Google Scholar] [PubMed]
- Montay, G. Pharmacokinetics of sparfloxacin in healthy volunteers and patiensts: A review. J. Antimicrob. Chemother. 1996, 37, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Montay, G.; Bruno, R.; Vergniol, J.C.; Ebmeier, M.; Roux, Y.L.; Guimart, C.; Frydman, A.; Chassard, D.; Thebault, J.J. Pharmacokinetics of Sparfioxacin in Humans After Single Oral Administration at Doses of 200, 400, 600, and 800 mg. J. Clin. Pharmacol. 1994, 34, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Mody, V.D.; Pandya, K.K.; Satia, M.C.; Modi, I.A.; Modi, R.I.; Gandhi, T.P. High performance thin-layer chromatographic method for the determination of sparfloxacin in human plasma and its use in pharmacokinetic studies. J. Pharm. Biomed. Anal. 1998, 16, 1289–1294. [Google Scholar] [CrossRef]
- Cho, H.-Y.; Park, S.-A.; Lee, Y.-B. Improvement and validation of an HPLC method for examining the effects of the MDR1 gene polymorphism on sparfloxacin pharmacokinetics. J. Chromatogr. B 2006, 834, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Prakash, K.; Sireesha, K. HPLC-UV method for simultaneous determination of sparfloxacin and dexamethasone sodium phosphate in eye drops. Pak. J. Pharm. Sci. 2019, 32, 1057–1061. [Google Scholar] [PubMed]
- Srinivas, N.; Narasu, L.; Shankar, B.; Mullangi, R. Development and validation of a HPLC method for simultaneous quantitation of gatifloxacin, sparfloxacin and moxifloxacin using levofloxacin as internal standard in human plasma: Application to a clinical pharmacokinetic study. Biomed. Chromatogr. 2008, 22, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.; Jan, M.; Khan, I.; Khan, M. Quantification of sparfloxacin in pharmaceutical dosages and biological samples. Pak. J. Pharm. Sci. 2012, 25, 823–829. [Google Scholar] [PubMed]
- Hn, O.; Cj, M. Spectrophotometric determination of sparfloxacin in bulk and dosage forms. Int. J. Curr. Res. Rev. 2010, 2, 27–33. [Google Scholar]
- Stahlmann, R.; Zippel, U.T.A.; Fo, C.; Schwabe, R.; Shakibaei, M.; Merker, H.; Borner, K. Chondrotoxicity and Toxicokinetics of Sparfloxacin in Juvenile Rats. Antimicrob. Agents Chemother. 1998, 42, 1470–1475. [Google Scholar] [CrossRef] [PubMed]
- López, C.; Díez, R.; Rodríguez, J.; Sierra, M.; García, J.; Fernández, N.; Diez, M.; Sahagún, A. Determination of Menbutone: Development and Validation of a Sensitive HPLC Assay according to the European Medicines Agency Guideline. Separations 2022, 9, 84. [Google Scholar] [CrossRef]
- European Medicines Agency (EMA). Guideline on Bioanalytical Method Validation (EMEA/CHMP/EWP/192217/2009). Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/includes/document/document_detail.jsp?webContentId=WC500109686%26mid=WC0b01ac058009a3dc (accessed on 22 March 2022).
- Spanish Agency of Medicines and Medical Devices (AEMPS). List of Laboratories Certified for Good Laboratory Practice Compliance [Listado de Laboratorios Certificados para el Cumplimiento de Buenas Prácticas de Laboratorio]. Available online: https://www.aemps.gob.es/industria-farmaceutica/buenas-practicas-de-laboratorio/listadolab-bpl/ (accessed on 18 April 2022).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intra-Day | CV (%) | Accuracy (%) |
|---|---|---|
| Batch 1 | 19.7 | 103.1 |
| Batch 2 | 3.7 | 112.3 |
| Batch 3 | 15.4 | 105.0 |
| Inter-Day | 12.9 | 106.8 |
| Equation of the Line | R2 | p | |
|---|---|---|---|
| Calibration curve 1 | y = 0.6694x + 0.0226 | 0.999 | <0.001 |
| Calibration curve 2 | y = 0.6068x + 0.0076 | 0.999 | |
| Calibration curve 3 | y = 0.6081x + 0.0126 | 0.999 |
| Nominal Concentration (µg/mL) | |||||||
|---|---|---|---|---|---|---|---|
| 0.2 | 0.5 | 1 | 2 | 5 | 10 | ||
| Accuracy (%) | Calibration curve 1 | 103.1 | 93.5 | 101.4 | 100.1 | 100.4 | 99.9 |
| Calibration curve 2 | 112.3 | 101.4 | 99.0 | 100.3 | 99.0 | 100.3 | |
| Calibration curve 3 | 105.0 | 98.6 | 98.7 | 100.2 | 100.2 | 100.0 | |
| LLOQ | LOW | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Nominal Concentration (µg/mL) | 0.2 | 0.6 | ||||||||
| Sample Number | 1 | 2 | 3 | 4 | 5 | 1 | 2 | 3 | 4 | 5 |
| Intra-Day | ||||||||||
| Batch 1 | 0.00 | 1.16 | 0.18 | 0.13 | 0.65 | 0.35 | 0.81 | 0.22 | 0.33 | 0.82 |
| Batch 2 | 0.40 | 0.43 | 0.88 | 0.71 | 0.24 | 0.87 | 0.10 | 0.18 | 0.63 | 1.03 |
| Batch 3 | 0.08 | 0.04 | 0.42 | 0.82 | 0.17 | 0.63 | 0.23 | 2.16 | 0.58 | 2.51 |
| Inter-Day | 0.42 | 0.77 | ||||||||
| MED | HIGH | |||||||||
| Nominal Concentration (µg/mL) | 3 | 8 | ||||||||
| Sample Number | 1 | 2 | 3 | 4 | 5 | 1 | 2 | 3 | 4 | 5 |
| Intra-Day | ||||||||||
| Batch 1 | 0.12 | 0.21 | 0.21 | 0.04 | 0.10 | 0.16 | 0.08 | 0.05 | 0.33 | 0.22 |
| Batch 2 | 0.25 | 0.32 | 0.32 | 0.50 | 0.47 | 0.01 | 0.22 | 0.15 | 0.45 | 0.16 |
| Batch 3 | 0.07 | 0.47 | 0.78 | 0.28 | 0.76 | 0.63 | 0.40 | 0.20 | 0.05 | 0.45 |
| Inter-Day | 0.33 | 0.24 | ||||||||
| LLOQ | LOW | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Nominal Concentration (µg/mL) | 0.2 | 0.6 | ||||||||
| Sample Number | 1 | 2 | 3 | 4 | 5 | 1 | 2 | 3 | 4 | 5 |
| Intra-Day | ||||||||||
| Batch 1 | 90.3 | 92.8 | 96.3 | 95.3 | 97.7 | 87.0 | 87.6 | 88.5 | 87.8 | 87.2 |
| Batch 2 | 113.0 | 115.6 | 118.0 | 110.4 | 105.1 | 101.9 | 103.0 | 104.0 | 106.3 | 104.2 |
| Batch 3 | 96.4 | 99.2 | 101.9 | 104.8 | 112.0 | 93.6 | 97.0 | 100.1 | 98.1 | 99.9 |
| Inter-Day | 103.2 ± 8.8 | 96.4 ± 7.1 | ||||||||
| MED | HIGH | |||||||||
| Nominal Concentration (µg/mL) | 0.2 | 0.6 | ||||||||
| Sample Number | 1 | 2 | 3 | 4 | 5 | 1 | 2 | 3 | 4 | 5 |
| Intra-Day | ||||||||||
| Batch 1 | 94.3 | 100.0 | 97.7 | 98.9 | 98.4 | 97.9 | 96.4 | 96.0 | 96.9 | 96.9 |
| Batch 2 | 101.5 | 100.0 | 103.1 | 114.2 | 100.8 | 97.8 | 98.3 | 99.3 | 100.8 | 99.4 |
| Batch 3 | 96.6 | 94.8 | 97.4 | 93.0 | 96.2 | 92.3 | 92.5 | 93.3 | 95.2 | 94.6 |
| Inter-Day | 99.1 ± 5.0 | 96.5 ± 2.5 | ||||||||
| Temperature (°C) | Time | QC2 | QC4 | ||
|---|---|---|---|---|---|
| CV (%) | Accuracy (%) | CV (%) | Accuracy (%) | ||
| −20 | 24 h | 2.34 | 107.3 | 1.88 | 107.1 |
| 48 h | 3.08 | 97.5 | 1.32 | 107.4 | |
| 72 h | 1.58 | 91.3 | 3.59 | 96.0 | |
| 4 | 24 h | 0.72 | 109.2 | 8.05 | 104.2 |
| 48 h | 4.39 | 99.0 | 3.39 | 108.4 | |
| 72 h | 3.31 | 90.4 | 2.15 | 102.7 | |
| 25 | 24 h | 0.86 | 102.7 | 0.40 | 110.7 |
| −20 | 7 days | 2.39 | 105.4 | 2.20 | 95.6 |
| 1 month | 0.87 | 107.0 | 1.24 | 94.8 | |
| 2 months | 2.58 | 105.6 | 2.50 | 110.1 | |
| 4 | 7 days | 1.11 | 109.0 | 0.60 | 103.2 |
| 4 | 24 h after extraction | 4.00 | 108.7 | 3.80 | 107.8 |
| 25 | 24 h after extraction | 0.80 | 102.8 | 6.99 | 104.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López, C.; Díez, R.; Rodríguez, J.M.; Sierra, M.; García, J.J.; Fernández, N.; Diez, M.J.; Sahagún, A.M. Development and Validation of a Sensitive HPLC Assay for Determination of Sparfloxacin According to the European Medicines Agency Guideline. Separations 2022, 9, 223. https://doi.org/10.3390/separations9080223
López C, Díez R, Rodríguez JM, Sierra M, García JJ, Fernández N, Diez MJ, Sahagún AM. Development and Validation of a Sensitive HPLC Assay for Determination of Sparfloxacin According to the European Medicines Agency Guideline. Separations. 2022; 9(8):223. https://doi.org/10.3390/separations9080223
Chicago/Turabian StyleLópez, Cristina, Raquel Díez, José M. Rodríguez, Matilde Sierra, Juan J. García, Nélida Fernández, M. José Diez, and Ana M. Sahagún. 2022. "Development and Validation of a Sensitive HPLC Assay for Determination of Sparfloxacin According to the European Medicines Agency Guideline" Separations 9, no. 8: 223. https://doi.org/10.3390/separations9080223
APA StyleLópez, C., Díez, R., Rodríguez, J. M., Sierra, M., García, J. J., Fernández, N., Diez, M. J., & Sahagún, A. M. (2022). Development and Validation of a Sensitive HPLC Assay for Determination of Sparfloxacin According to the European Medicines Agency Guideline. Separations, 9(8), 223. https://doi.org/10.3390/separations9080223