
Two New HPLC Methods, Assessed by GAPI, for Simultaneous Determination of Four Antipsychotics in Pharmaceutical Formulations: A Comparative Study

 , and
, and
Abstract
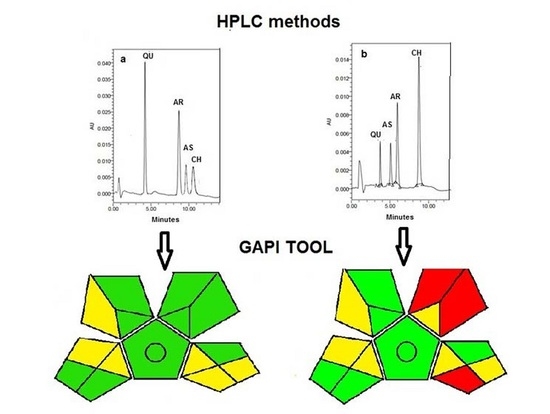
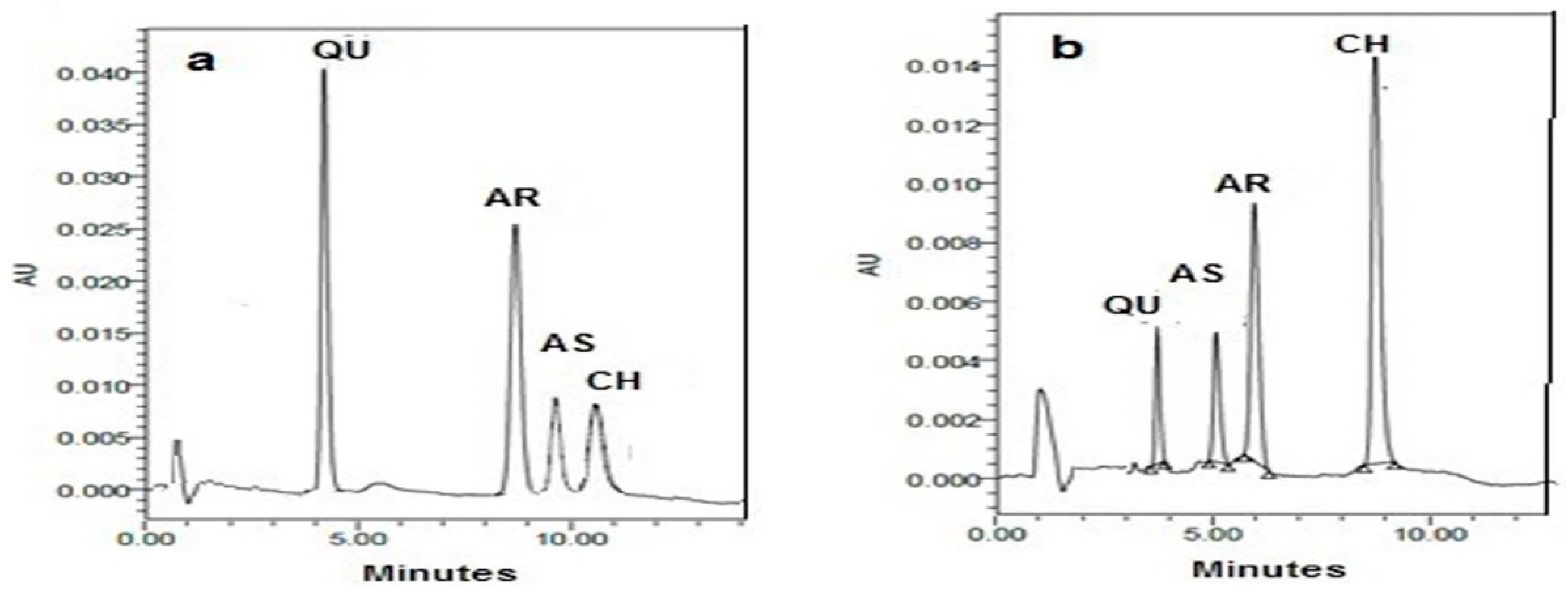
:
1. Introduction
2. Experimental
2.1. Instrumentation
2.2. Chemicals and Reagents
2.3. Pharmaceutical Samples
2.4. Chromatographic Conditions
2.4.1. Ecofriendly Green Method
2.4.2. Conventional Method
2.5. Standard Solutions
2.6. Sample Preparation
3. Results and Discussion
3.1. Method Optimization
3.2. The Validation of the Methods
3.2.1. Linearity
3.2.2. Precision
3.2.3. Range
3.2.4. Detection and Quantitation Limits
3.2.5. Selectivity
3.2.6. Accuracy
3.2.7. Stability
3.2.8. Analysis of Pharmaceutical Formulations
3.3. Evaluation of the Greenness of the Proposed Analytical Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ketter, T.A. Monotherapy versus combined treatment with second-generation antipsychotics in bipolar disorder. J. Clin. Psychiatry 2008, 69, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, A. Comparative efficacy and acceptability of antimanic drugs in acute mania: A multiple-treatments meta-analysis. Lancet 2011, 378, 1306–1315. [Google Scholar] [CrossRef]
- Roberts, R.; Neasham, A.; Lambrinudi, C. A quantitative analysis of antipsychotic prescribing trends for the treatment of schizophrenia in England and Wales. JRSM Open 2018, 9, 2054270418758570. [Google Scholar] [CrossRef] [PubMed]
- Bjornestad, J.; Lavik, K.O.; Davidson, L.; Hjeltnes, A. Antipsychotic treatment—A systematic literature review and meta-analysis of qualitative studies. J. Ment. Health 2020, 29, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Dev, V.; Raniwalla, J. Quetiapine: A review of its safety in the management of schizophrenia. Drug Saf. 2000, 23, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Sangeetha, D.; Goyal, R.; Reddy, P. A validated stability-indicating RP-LC method for the estimation of process-related impurities and degradation products of quetiapine fumarate in solid oral dosage form. Acta Chromatogr. 2013, 25, 393–409. [Google Scholar] [CrossRef]
- Venkata, K.B.; Battula, S.R.; Dubey, S. Validation of quetiapine fumarate in pharmaceutical dosage by reverse-phase HPLC with internal standard method. J. Chem. 2013, 2013, 578537. [Google Scholar] [CrossRef]
- Youssef, R.M.; Abdine, H.H.; Barary, M.A.; Wagih, M.M. Selective RP-HPLC method for determination of quetiapine in presence of coadministered drugs: Application for long-term stability study of quetiapine in whole blood. Acta Chromatogr. 2016, 28, 263–279. [Google Scholar] [CrossRef]
- Soponar, F.; Sandru, M.; David, V. Quantitative evaluation of Aripiprazole and its five related chemical impurities from pharmaceuticals using a HPLC-DAD method. Rev. Roum. Chim. 2014, 59, 1037–1046. [Google Scholar]
- Kalaichelvi, R.; Thangabalan, B.; Srinivasa Rao, D. Validated RP-HPLC method for analysis of Aripiprazole in a formulation. E-J. Chem. 2010, 7, 827–832. [Google Scholar] [CrossRef]
- Djordjević Filijović, N.; Pavlović, A.; Nikolić, K.; Agbaba, D. Validation of an HPLC method for determination of Aripiprazole and its impurities in pharmaceuticals. Acta Chromatogr. 2014, 26, 13–28. [Google Scholar] [CrossRef]
- Srinivas, K.S.V.; Buchireddy, R.; Madhusudhan, G. Stress degradation studies on Aripiprazole and development of a validated stability indicating LC method. Chromatographia 2008, 68, 635–640. [Google Scholar] [CrossRef]
- Shimokawa, Y. High performance liquid chromatographic methods for the determination of aripiprazole with ultraviolet detection in rat plasma and brain: Application to the pharmacokinetic study. J. Chromatogr. B 2005, 821, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Shahid, M.; Walker, G.B.; Zorn, S.H. Asenapine: A novel psychopharmacologic agent with a unique human receptor signature. J. Psychopharmacol. 2009, 23, 65–73. [Google Scholar] [CrossRef]
- Chhalotiya, U.K.; Bhatt, K.K.; Shah, D.A.; Patel, J.R. Stability-indicating liquid chromatographic method for the quantification of the new antipsychotic agent Asenapine in bulk and in pharmaceutical formulation. Sci. Pharm. 2012, 80, 407–417. [Google Scholar] [CrossRef]
- Managuli, R.S. Development and validation of a stability-indicating RP-HPLC method by a statistical optimization process for the quantification of asenapine maleate in lipidic nanoformulations. J. Chromatogr. Sci. 2016, 54, 1290–1300. [Google Scholar] [CrossRef]
- Govindarajan, N.R.; Koulagari, S.; Methuku, A.; Podhuturi, S. Method development and validation of RP-HPLC method for determination of new antipsychotic agent Asenapine maleate in bulk and pharmaceutical formulation. Eur. J. Anal. Chem. 2014, 9, 58–65. [Google Scholar]
- Karaca, S.A.; Uǧur, D.Y. A stability indicating ion-pair lc method for the determination of Asenapine in pharmaceuticals. J. Chil. Chem. Soc. 2017, 62, 3325–3329. [Google Scholar] [CrossRef]
- Fadhel, S.R.; Khalil, S.I. Simultaneous estimation of chlorpromazine hydrochloride and carvedilol in bulk and pharmaceutical dosage forms using HPLC. J. Biochem. Technol. 2018, 9, 5–9. [Google Scholar]
- Mahadik, K.R.; Aggarwal, H.; Kaul, N. Development and validation of HPLC method for simultaneous estimation of trihexyphenidyl hydrochloride and chlorpromazine hydrochloride from tablet dosage form. Indian Drugs 2002, 39, 441–445. [Google Scholar]
- Rani, N.U.; Divya, K.; Sahithi, G. New validated RP-HPLC method for simultaneous estimation of chlorpromazine and Trihexyphenidyl HCl in tablets. Int. J. Adv. Pharm. Anal. 2014, 4, 134–137. [Google Scholar]
- Midha, K. High performance liquid chromatographic assay for nanogram determination of chlorpromazine and its comparison with a radioimmunoassay. J. Pharm. Sci. 1981, 70, 1043–1046. [Google Scholar] [CrossRef] [PubMed]
- Khelfi, A.; Azzouz, M.; Abtroun, R.; Reggabi, M.; Alamir, B. Determination of chlorpromazine, haloperidol, levomepromazine, olanzapine, risperidone, and sulpiride in human plasma by liquid chromatography/tandem mass spectrometry (LC-MS/MS). Int. J. Anal. Chem. 2018, 2018, 5807218. [Google Scholar] [CrossRef] [PubMed]
- Sistik, P.; Urinovska, R.; Brozmanova, H. Fast simultaneous LC/MS/MS determination of 10 active compounds in human serum for therapeutic drug monitoring in psychiatric medication. Biomed. Chromatogr. 2016, 30, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Wróblewski, K. Optimization of chromatographic systems for analysis of selected psychotropic drugs and their metabolites in serum and saliva by HPLC in order to monitor therapeutic drugs. Open Chem. 2019, 17, 1361–1373. [Google Scholar] [CrossRef]
- Kannaiah, K.P.; Sugumaran, A.; Chanduluru, H.K. Environmental impact of greeness assessment tools in liquid chromatography, A review. Microchem. J. 2021, 170, 106685. [Google Scholar] [CrossRef]
- Anastas, P.T.; Kirchhoff, M.M. Origins, current status, and future challenges of green chemistry. Acc. Chem. Res. 2002, 35, 686–694. [Google Scholar] [CrossRef]
- Gałuszka, A.; Migaszewski, Z.; Namieśnik, J. The 12 principles of green analytical chemistry and the Significance mnemonic of green analytical practices. TrAC-Trends Anal. Chem. 2013, 50, 78–84. [Google Scholar] [CrossRef]
- Yabré, M. Green analytical methods of antimalarial artemether-lumefantrine analysis for falsification detection using a low-cost handled NIR spectrometer with DD-SIMCA and drug quantification by HPLC. Molecules 2020, 25, 3397. [Google Scholar] [CrossRef]
- ICH, Q8(R2): Pharmaceutical development. In Proceedings of the International Conference on Harmonization, August 2009; Available online: https://www.ema.europa.eu/en/ich-q8-r2-pharmaceutical-development (accessed on 25 July 2022).
- Juan, J.B.N.; Carmen, G.C.; Maria, J.V.L. Enantiomeric determination, validation and robustness studies of racemic citalopram in pharmaceutical formulations by capillary electrophoresis. J. Chromatogr. A 2005, 1072, 249–257. [Google Scholar]
- Sprent, P.; Draper, N.R.; Smith, H. Applied regression analysis. Biometrics 1981, 37, 863. [Google Scholar] [CrossRef]
- Płotka-Wasylka, J. A new tool for the evaluation of the analytical procedure: Green Analytical Procedure Index. Talanta 2018, 181, 204–209. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Method | Compound | Retention Time (min) a | %RSD b of Retention Time | Capacity Factor (K) | Selectivity (α) c | Resolution (Rs) c | Tailing Factor | Plate Count |
|---|---|---|---|---|---|---|---|---|
| Green | QU | 4.21 | 1.24 | 3.63 | 2.36 (a1) | 10.59 (b1) | 1.24 | 3799.56 |
| AR | 8.71 | 1.11 | 8.57 | 1.11 (a2) | 1.69 (b2) | 1.11 | 6111.079 | |
| AS | 9.57 | 1.29 | 9.52 | 1.10 (a3) | 3.41 (b3) | 1.24 | 5976.887 | |
| CH | 10.44 | 1.37 | 10.47 | 1.37 | 4273.030 | |||
| Conventional | QU | 3.70 | 1.44 | 3.35 | 1.36 (a4) | 4.54 (b4) | 1.44 | 5925.405 |
| AS | 5.05 | 1.29 | 4.55 | 1.21 (a5) | 1.75 (b5) | 1.29 | 5477.001 | |
| AR | 5.94 | 1.18 | 5.53 | 1.55 (a6) | 8.21 (b6) | 1.18 | 4868.921 | |
| CH | 8.78 | 1.37 | 8.59 | 1.37 | 10,244.90 |
| Method | Green | |||
| Parameters | QU | AR | AS | CH |
| Calibration range (µg mL−1) | 0.1–20 | 0.1–20 | 0.1–20 | 0.1–20 |
| Detection limit (µg mL−1) | 0.03 | 0.02 | 0.03 | 0.02 |
| Quantitation limit (µg mL−1) | 0.10 | 0.07 | 0.09 | 0.07 |
| Regression equation (Y) a: Slope (b) | 43,961.73 | 43,627.12 | 8968.81 | 7272.07 |
| Standard deviation of the slope (Sb) | 802.48 | 387.87 | 99.49 | 62.08 |
| Relative standard deviation of slope (%) | 1.8 | 0.89 | 1.1 | 0.85 |
| Intercept (a) | 2161.03 | 116.73 | 60.17 | 61.59 |
| Correlation coefficient (r) | 0.9998 | 0.9999 | 0.9999 | 0.9999 |
| Method | Conventional | |||
| Parameters | QU | AS | AR | CH |
| Calibration range (µg mL−1) | 0.1–20 | 0.1–20 | 0.1–20 | 0.1–20 |
| Detection limit (µg mL−1) | 0.03 | 0.03 | 0.04 | 0.03 |
| Quantitation limit (µg mL−1) | 0.11 | 0.11 | 0.12 | 0.11 |
| Regression equation (Y) a: Slope (b) | 26,543.81 | 4002.5 | 25,633.3 | 13,094.44 |
| Standard deviation of the slope (Sb) | 380.95 | 57.96 | 418.84 | 190.77 |
| Relative standard deviation slope (%) | 1.44 | 1.45 | 1.63 | 1.46 |
| Intercept (a) | 1745.39 | 189.48 | 2234.69 | 1104.77 |
| Correlation coefficient (r) | 0.9999 | 0.9999 | 0.9998 | 0.9998 |
| Green HPLC Method | Conventional HPLC Method | ||||
|---|---|---|---|---|---|
| Comp. | % Recovery | Comp. | % Recovery | ||
| QU | Mean a | 100.39 | QU | Mean a | 99.03 |
| ± S.D. a | 1.89 | ± S.D. a | 1.86 | ||
| t (2.18) b | 0.17 | ||||
| F (4.28) b | 1.00 | ||||
| AR | Mean a | 99.59 | AR | Mean a | 98.93 |
| ± S.D. a | 1.40 | ± S.D. a | 1.39 | ||
| t (2.18) b | 1.74 | ||||
| F (4.28) b | 0.98 | ||||
| AS | Meana | 98.16 | AS | Mean a | 98.14 |
| ± S.D.a | 2.00 | ± S.D. a | 2.09 | ||
| t (2.18) b | 1.74 | ||||
| F (4.28) b | 0.97 | ||||
| CH | Meana | 99.57 | CH | Mean a | 99.56 |
| ± S.D.a | 2.23 | ± S.D. a | 2.19 | ||
| t (2.18) b | 0.09 | ||||
| F (4.28) b | 1.00 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdel Hameed, E.A.; Abd El-Naby, Z.A.; El Gindy, A.; Zaitone, S.A.; Alshaman, R.; Saraya, R.E.; Khairy, G.M. Two New HPLC Methods, Assessed by GAPI, for Simultaneous Determination of Four Antipsychotics in Pharmaceutical Formulations: A Comparative Study. Separations 2022, 9, 220. https://doi.org/10.3390/separations9080220
Abdel Hameed EA, Abd El-Naby ZA, El Gindy A, Zaitone SA, Alshaman R, Saraya RE, Khairy GM. Two New HPLC Methods, Assessed by GAPI, for Simultaneous Determination of Four Antipsychotics in Pharmaceutical Formulations: A Comparative Study. Separations. 2022; 9(8):220. https://doi.org/10.3390/separations9080220
Chicago/Turabian StyleAbdel Hameed, Eman A., Zaitona A. Abd El-Naby, Alaa El Gindy, Sawsan A. Zaitone, Reem Alshaman, Roshdy E. Saraya, and Gasser M. Khairy. 2022. "Two New HPLC Methods, Assessed by GAPI, for Simultaneous Determination of Four Antipsychotics in Pharmaceutical Formulations: A Comparative Study" Separations 9, no. 8: 220. https://doi.org/10.3390/separations9080220
APA StyleAbdel Hameed, E. A., Abd El-Naby, Z. A., El Gindy, A., Zaitone, S. A., Alshaman, R., Saraya, R. E., & Khairy, G. M. (2022). Two New HPLC Methods, Assessed by GAPI, for Simultaneous Determination of Four Antipsychotics in Pharmaceutical Formulations: A Comparative Study. Separations, 9(8), 220. https://doi.org/10.3390/separations9080220