Proteogenomics in Aid of Host–Pathogen Interaction Studies: A Bacterial Perspective


{kind=link}
{kind=link}
{kind=link}
Abstract
1. General Introduction
2. Current Limitations for the Application of Omics in Host–Pathogen Interaction Studies
3. Transcriptome Analyses during Infection to Study Gene Expression Profiles of Host and Pathogen Simultaneously: RNA-Seq Supplanting Microarrays
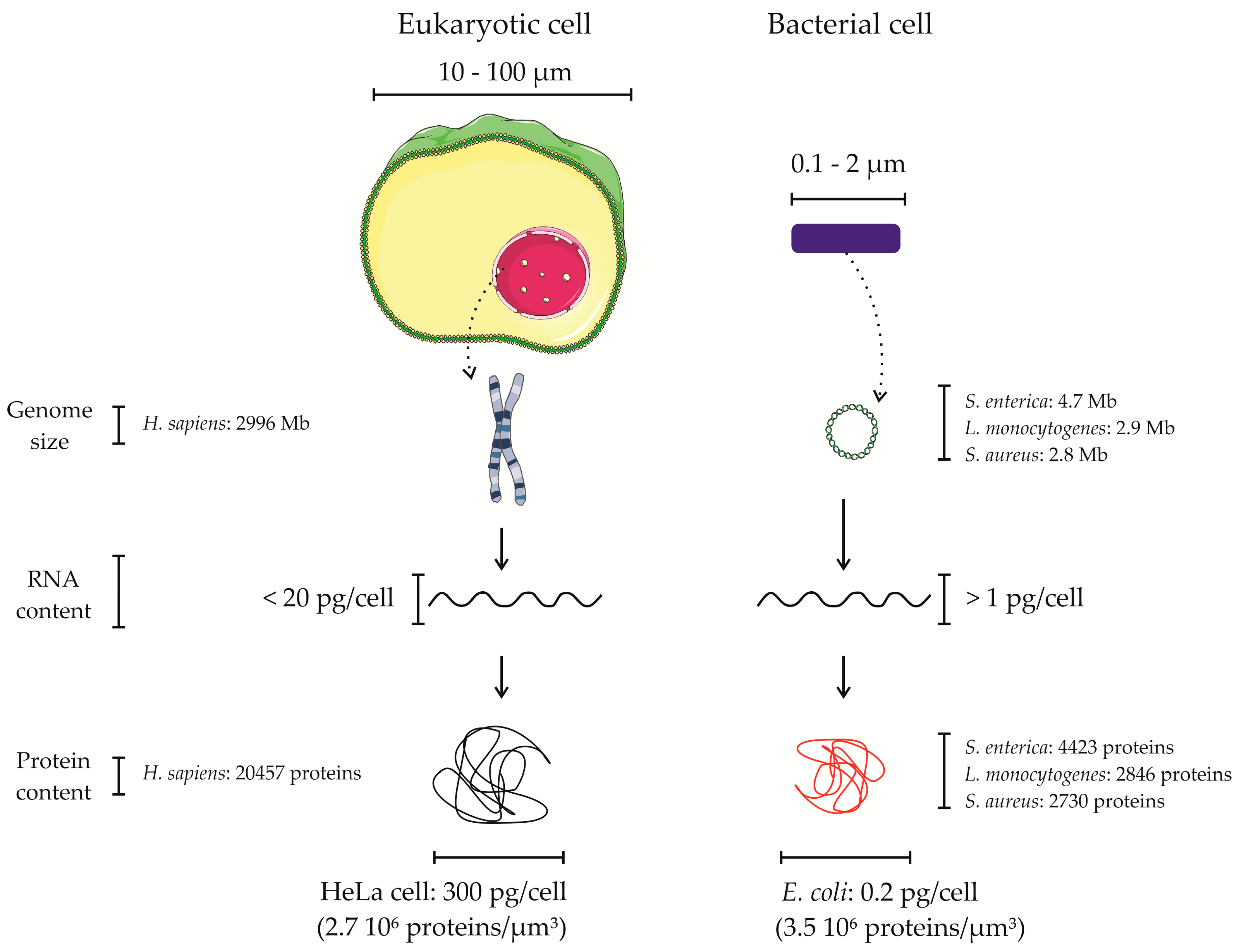
4. Proteomics of Bacterial Pathogens in the Context of the Host—Overcoming the Overwhelming Host Proteome Contribution
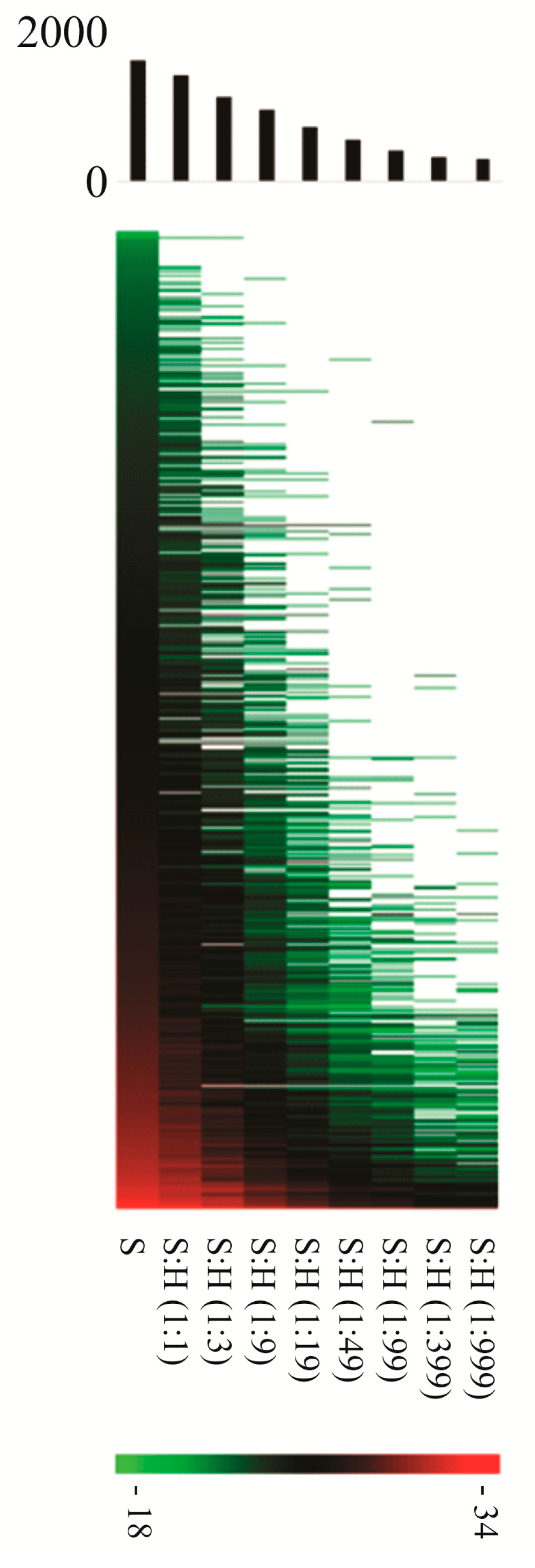
4.1. Physical Separation Based Approaches for Isolating Intact Bacterial Pathogens upon Infection
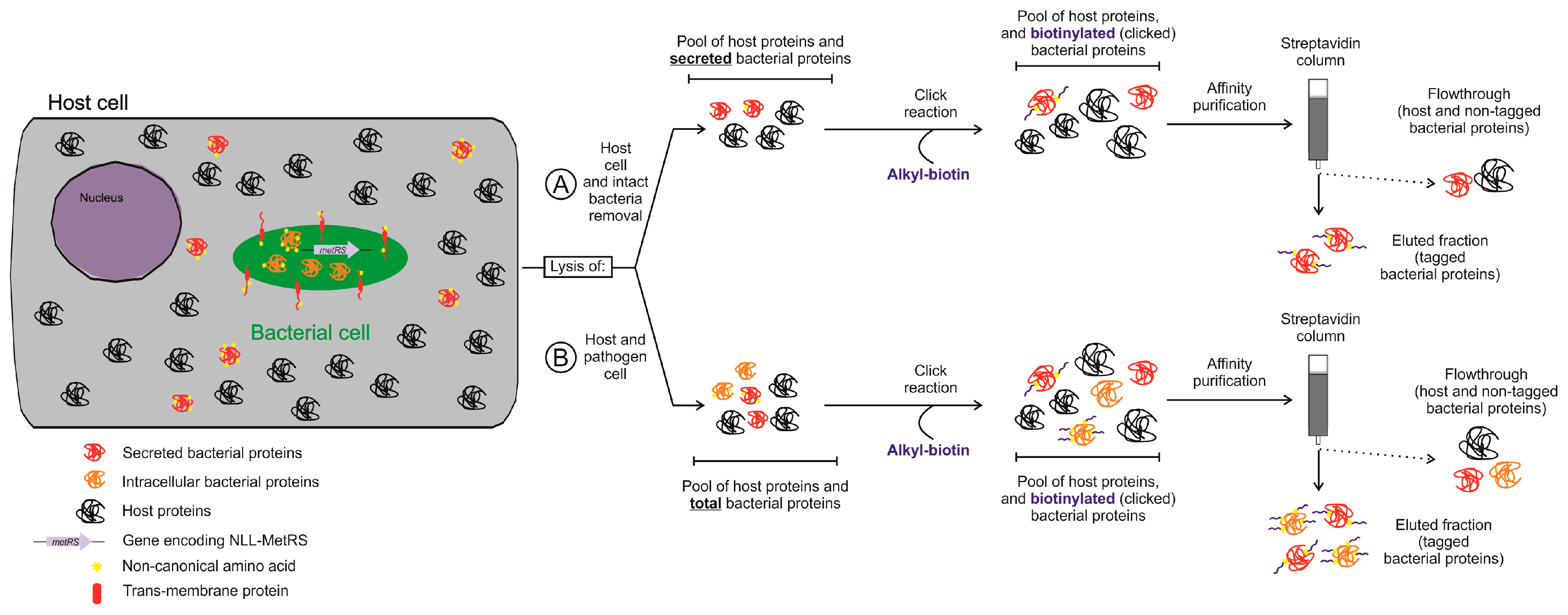
4.2. Protein Labeling-Based Isolation of the Bacterial Proteome in Infection Models
5. Secretomics for Identifying Bacterial Extracellular Effectors Involved in Infection
6. Ribosome Profiling as the Next Big Step of Future Endeavors for Studying Bacterial Translatomes upon Host Cell Infection?
7. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Methot, P.O.; Alizon, S. What is a pathogen? Toward a process view of host-parasite interactions. Virulence 2014, 5, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Falkow, S. Molecular Koch’s postulates applied to bacterial pathogenicity—A personal recollection 15 years later. Nat. Rev. Microbiol. 2004, 2, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, A.; Pirofski, L.-A. The damage-response framework of microbial pathogenesis. Nat. Rev. Microbiol. 2003, 1, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Jean Beltran, P.M.; Federspiel, J.D.; Sheng, X.; Cristea, I.M. Proteomics and integrative omic approaches for understanding host–pathogen interactions and infectious diseases. Mol. Syst. Biol. 2017, 13, 922. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Hu, M.; Fu, J.; Liu, Y.; Wu, M.; Yu, K.; Liu, X. Quantitative proteomic analysis of host epithelial cells infected by Salmonella enterica serovar Typhimurium. Proteomics 2017, 17, 1700092. [Google Scholar] [CrossRef] [PubMed]
- Chromy, B.A.; Perkins, J.; Heidbrink, J.L.; Gonzales, A.D.; Murphy, G.A.; Fitch, J.P.; McCutchen-Maloney, S.L. Proteomic characterization of host response to Yersinia pestis and near neighbors. Biochem. Biophys. Res. Commun. 2004, 320, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, R.; Dong, W.; Hu, L.; Zong, B.; Zhang, Y.; Wang, X.; Guo, A.; Zhang, A.; Xiang, Y.; et al. Comparative proteomics analysis of human macrophages infected with virulent mycobacterium bovis. Front. Cell. Infect. Microbiol. 2017, 7, 65. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhao, G.; Stein, R.; Zheng, X.; Hu, W.; Shang, N.; Bu, X.; Liu, X.; Wang, J.; Feng, E.; et al. The proteome of Shigella flexneri 2a 2457T grown at 30 and 37 degrees C. Mol. Cell. Proteom. MCP 2010, 9, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Stancik, L.M.; Stancik, D.M.; Schmidt, B.; Barnhart, D.M.; Yoncheva, Y.N.; Slonczewski, J.L. pH-dependent expression of periplasmic proteins and amino acid catabolism in Escherichia coli. J. Bacteriol. 2002, 184, 4246–4258. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Kogl, S.A.; Jung, K. Time-dependent proteome alterations under osmotic stress during aerobic and anaerobic growth in Escherichia coli. J. Bacteriol. 2006, 188, 7165–7175. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Yang, E.; Vu, G.-P.; Gong, H.; Su, J.; Liu, F.; Lu, S. Mass spectrometry-based quantitative proteomic analysis of Salmonella enterica serovar Enteritidis protein expression upon exposure to hydrogen peroxide. BMC Microbiol. 2010, 10, 166. [Google Scholar] [CrossRef] [PubMed]
- Grossman, N.T.; Casadevall, A. Physiological differences in Cryptococcus neoformans strains in vitro versus in vivo and their effects on antifungal susceptibility. Antimicrob. Agents Chemother. 2017, 61, e02108-16. [Google Scholar] [CrossRef] [PubMed]
- Zeigerer, A.; Wuttke, A.; Marsico, G.; Seifert, S.; Kalaidzidis, Y.; Zerial, M. Functional properties of hepatocytes in vitro are correlated with cell polarity maintenance. Exp. Cell Res. 2017, 350, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Disson, O.; Cossart, P.; Lecuit, M. The Issue of Species Specificity of Bacterial Infection, How to Address it Experimentally. In Bacterial Virulence; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 279–310. [Google Scholar] [CrossRef]
- Nolte, O.; Rickert, A.; Ehrhard, I.; Ledig, S.; Sonntag, H.-G. A modified ex vivo human whole blood model of infection for studying the pathogenesis of Neisseria meningitidis during septicemia. FEMS Immunol. Med. Microbiol. 2002, 32, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Shelburne, S.A., 3rd; Granville, C.; Tokuyama, M.; Sitkiewicz, I.; Patel, P.; Musser, J.M. Growth characteristics of and virulence factor production by group A Streptococcus during cultivation in human saliva. Infect. Immun. 2005, 73, 4723–4731. [Google Scholar] [CrossRef] [PubMed]
- Lecuit, M.; Nelson, D.M.; Smith, S.D.; Khun, H.; Huerre, M.; Vacher-Lavenu, M.C.; Gordon, J.I.; Cossart, P. Targeting and crossing of the human maternofetal barrier by Listeria monocytogenes: Role of internalin interaction with trophoblast E-cadherin. Proc. Natl. Acad. Sci. USA 2004, 101, 6152–6157. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B. Molecular Biology of the Cell; Garland Science: New York, NY, USA, 2015. [Google Scholar]
- Karlin, S.; Brocchieri, L.; Trent, J.; Blaisdell, B.E.; Mrázek, J. Heterogeneity of genome and proteome content in bacteria, archaea, and eukaryotes. Theor. Popul. Biol. 2002, 61, 367–390. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Weiss, M.; Simonovic, M.; Haertinger, G.; Schrimpf, S.P.; Hengartner, M.O.; von Mering, C. PaxDb, a database of protein abundance averages across all three domains of life. Mol. Cell. Proteom. MCP 2012, 11, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Kelleher, N.L. Proteoform: A single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Plasman, K.; Gevaert, K.; Impens, F.; Montoye, T. Perfecting Progress; Samedan Ltd. Pharmaceutical Publishers: London, UK, 2017; pp. 70–72. [Google Scholar]
- Bekker-Jensen, D.B.; Kelstrup, C.D.; Batth, T.S.; Larsen, S.C.; Haldrup, C.; Bramsen, J.B.; Sorensen, K.D.; Hoyer, S.; Orntoft, T.F.; Andersen, C.L.; et al. An optimized shotgun strategy for the rapid generation of comprehensive human proteomes. Cell Syst. 2017, 4, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. MCP 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed]
- Buchrieser, C.; Glaser, P.; Rusniok, C.; Nedjari, H.; D’Hauteville, H.; Kunst, F.; Sansonetti, P.; Parsot, C. The virulence plasmid pWR100 and the repertoire of proteins secreted by the type III secretion apparatus of Shigella flexneri. Mol. Microbiol. 2000, 38, 760–771. [Google Scholar] [CrossRef] [PubMed]
- Westermann, A.J.; Barquist, L. Resolving host–pathogen interactions by dual RNA-seq. PLoS Pathog. 2017, 13, e1006033. [Google Scholar] [CrossRef] [PubMed]
- Milo, R. What is the total number of protein molecules per cell volume? A call to rethink some published values. BioEssays 2013, 35, 1050–1055. [Google Scholar] [CrossRef] [PubMed]
- Mastroeni, P.; Grant, A.; Restif, O.; Maskell, D. A dynamic view of the spread and intracellular distribution of Salmonella enterica. Nat. Rev. Microbiol. 2009, 7, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Emoto, Y.; Emoto, M. A simple, reproducible, inexpensive, yet old-fashioned method for determining phagocytic and bactericidal activities of macrophages. Yonsei Med. J. 2016, 57, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Malik-Kale, P.; Winfree, S.; Steele-Mortimer, O. The bimodal lifestyle of intracellular Salmonella in epithelial cells: Replication in the cytosol obscures defects in vacuolar replication. PLoS ONE 2012, 7, e38732. [Google Scholar] [CrossRef]
- Raybourne, R.B.; Roth, G.; Deuster, P.A.; Sternberg, E.M.; Singh, A. Uptake and killing of Listeria monocytogenes by normal human peripheral blood granulocytes and monocytes as measured by flow cytometry and cell sorting. FEMS Immunol. Med. Microbiol. 2001, 31, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Chalfie, M.; Tu, Y.; Euskirchen, G.; Ward, W.W.; Prasher, D.C. Green fluorescent protein as a marker for gene expression. Science 1994, 263, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Surmann, K.; Michalik, S.; Hildebrandt, P.; Gierok, P.; Depke, M.; Brinkmann, L.; Bernhardt, J.; Salazar, M.G.; Sun, Z.; Shteynberg, D.; et al. Comparative proteome analysis reveals conserved and specific adaptation patterns of Staphylococcus aureus after internalization by different types of human non-professional phagocytic host cells. Front. Microbiol. 2014, 5, 392. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.; Scharf, S.S.; Hildebrandt, P.; Burian, M.; Bernhardt, J.; Dhople, V.; Kalinka, J.; Gutjahr, M.; Hammer, E.; Volker, U. Time-resolved quantitative proteome profiling of host–pathogen interactions: The response of Staphylococcus aureus RN1HG to internalisation by human airway epithelial cells. Proteomics 2010, 10, 2801–2811. [Google Scholar] [CrossRef] [PubMed]
- Burton, N.A.; Schurmann, N.; Casse, O.; Steeb, A.K.; Claudi, B.; Zankl, J.; Schmidt, A.; Bumann, D. Disparate impact of oxidative host defenses determines the fate of Salmonella during systemic infection in mice. Cell Host Microbe 2014, 15, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Knodler, L.A.; Bestor, A.; Ma, C.; Hansen-Wester, I.; Hensel, M.; Vallance, B.A.; Steele-Mortimer, O. Cloning vectors and fluorescent proteins can significantly inhibit Salmonella enterica virulence in both epithelial cells and macrophages: Implications for bacterial pathogenesis studies. Infect. Immun. 2005, 73, 7027–7031. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.; Martinez-Argudo, I.; Humphrey, T.J.; Jepson, M.A. GFP plasmid-induced defects in Salmonella invasion depend on plasmid architecture, not protein expression. Microbiology 2009, 155, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Westermann, A.J.; Förstner, K.U.; Amman, F.; Barquist, L.; Chao, Y.; Schulte, L.N.; Müller, L.; Reinhardt, R.; Stadler, P.F.; Vogel, J. Dual RNA-seq unveils noncoding RNA functions in host–pathogen interactions. Nature 2016, 529, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Wang, X.; Han, Y.; Guo, Z.; Yang, R. Yersinia pestis versus Yersinia pseudotuberculosis: Effects on host macrophages. Scand. J. Immunol. 2012, 76, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Arnett, E.; Vadia, S.; Nackerman, C.C.; Oghumu, S.; Satoskar, A.R.; McLeish, K.R.; Uriarte, S.M.; Seveau, S. The pore-forming toxin listeriolysin O is degraded by neutrophil metalloproteinase-8 and fails to mediate Listeria monocytogenes intracellular survival in neutrophils. J. Immunol. 2014, 192, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Oviedo-Boyso, J.; Barriga-Rivera, J.G.; Valdez-Alarcon, J.J.; Bravo-Patino, A.; Carabez-Trejo, A.; Cajero-Juarez, M.; Baizabal-Aguirre, V.M. Internalization of Staphylococcus aureus by bovine endothelial cells is associated with the activity state of NF-kappaB and modulated by the pro-inflammatory cytokines TNF-alpha and IL-1beta. Scand. J. Immunol. 2008, 67, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Bettencourt, P.; Carmo, N.; Pires, D.; Timóteo, P.; Anes, E. Mycobacterial Infection of Macrophages: The Effect of the Multiplicity of Infection. In Antimicrobial Research: Novel Bioknowledge and Educational Programs; Méndez-Vilas, A., Ed.; Formatex Research Center: Badajoz, Spain, 2017; pp. 651–664. [Google Scholar]
- Gog, J.R.; Murcia, A.; Osterman, N.; Restif, O.; McKinley, T.J.; Sheppard, M.; Achouri, S.; Wei, B.; Mastroeni, P.; Wood, J.L.N.; et al. Dynamics of Salmonella infection of macrophages at the single cell level. J. R. Soc. Interface 2012, 9, 0163. [Google Scholar] [CrossRef] [PubMed]
- Jantsch, J.; Cheminay, C.; Chakravortty, D.; Lindig, T.; Hein, J.; Hensel, M. Intracellular activities of Salmonella enterica in murine dendritic cells. Cell. Microbiol. 2003, 5, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, J.A.; Knodler, L.A.; Sturdevant, D.E.; Virtaneva, K.; Carmody, A.B.; Fischer, E.R.; Porcella, S.F.; Steele-Mortimer, O. Induction of Salmonella pathogenicity island 1 under different growth conditions can affect Salmonella–host cell interactions in vitro. Microbiology 2010, 156, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement system part II: Role in immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Phagocytosis: An immunobiologic process. Immunity 2016, 44, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Geier, H.; Celli, J. Phagocytic receptors dictate phagosomal escape and intracellular proliferation of Francisella tularensis. Infec. Immun. 2011, 79, 2204–2214. [Google Scholar] [CrossRef] [PubMed]
- Kolb-Maurer, A.; Gentschev, I.; Fries, H.W.; Fiedler, F.; Brocker, E.B.; Kampgen, E.; Goebel, W. Listeria monocytogenes-infected human dendritic cells: Uptake and host cell response. Infect. Immun. 2000, 68, 3680–3688. [Google Scholar] [CrossRef] [PubMed]
- Saliba, A.E.; Li, L. Single-cell RNA-seq ties macrophage polarization to growth rate of intracellular Salmonella. Nat. Microbiol. 2016, 2, 16206. [Google Scholar] [CrossRef] [PubMed]
- Fodor, S.P.; Rava, R.P.; Huang, X.C.; Pease, A.C.; Holmes, C.P.; Adams, C.L. Multiplexed biochemical assays with biological chips. Nature 1993, 364, 555–556. [Google Scholar] [CrossRef] [PubMed]
- Veres, G.; Gibbs, R.A.; Scherer, S.E.; Caskey, C.T. The molecular basis of the sparse fur mouse mutation. Science 1987, 237, 415–417. [Google Scholar] [CrossRef] [PubMed]
- Rotter, A.; Hren, M.; Baebler, Š.; Blejec, A.; Gruden, K. Finding differentially expressed genes in two-channel DNA microarray datasets: How to increase reliability of data preprocessing. OMICS J. Integr. Biol. 2008, 12, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Hossain, H.; Tchatalbachev, S.; Chakraborty, T. Host gene expression profiling in pathogen-host interactions. Curr. Opin. Immunol. 2006, 18, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Westermann, A.J.; Gorski, S.A.; Vogel, J. Dual RNA-seq of pathogen and host. Nat. Rev. Microbiol. 2012, 10, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Fung-Leung, W.-P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.S.; Khan, Z.; Kruglyak, L.; Singh, M.; Caudy, A.A. Measuring differential gene expression by short read sequencing: Quantitative comparison to 2-channel gene expression microarrays. BMC Genom. 2009, 10, 221. [Google Scholar] [CrossRef] [PubMed]
- Szafranska, A.K.; Oxley, A.P.; Chaves-Moreno, D.; Horst, S.A.; Rosslenbroich, S.; Peters, G.; Goldmann, O.; Rohde, M.; Sinha, B.; Pieper, D.H.; et al. High-resolution transcriptomic analysis of the adaptive response of Staphylococcus aureus during acute and chronic phases of osteomyelitis. MBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Mavromatis, C.H.; Bokil, N.J.; Totsika, M.; Kakkanat, A.; Schaale, K.; Cannistraci, C.V.; Ryu, T.; Beatson, S.A.; Ulett, G.C.; Schembri, M.A.; et al. The co-transcriptome of uropathogenic Escherichia coli-infected mouse macrophages reveals new insights into host–pathogen interactions. Cell. Microbiol. 2015, 17, 730–746. [Google Scholar] [CrossRef] [PubMed]
- Humphrys, M.S.; Creasy, T.; Sun, Y.; Shetty, A.C.; Chibucos, M.C.; Drabek, E.F.; Fraser, C.M.; Farooq, U.; Sengamalay, N.; Ott, S.; et al. Simultaneous transcriptional profiling of bacteria and their host cells. PLoS ONE 2013, 8, e80597. [Google Scholar] [CrossRef] [PubMed]
- Rienksma, R.A.; Suarez-Diez, M.; Mollenkopf, H.J.; Dolganov, G.M.; Dorhoi, A.; Schoolnik, G.K.; Martins Dos Santos, V.A.; Kaufmann, S.H.; Schaap, P.J.; Gengenbacher, M. Comprehensive insights into transcriptional adaptation of intracellular mycobacteria by microbe-enriched dual RNA sequencing. BMC Genom. 2015, 16, 34. [Google Scholar] [CrossRef] [PubMed]
- Baddal, B.; Muzzi, A.; Censini, S.; Calogero, R.A.; Torricelli, G.; Guidotti, S.; Taddei, A.R.; Covacci, A.; Pizza, M.; Rappuoli, R.; et al. Dual RNA-seq of nontypeable haemophilus influenzae and host cell transcriptomes reveals novel insights into host-pathogen cross talk. MBio 2015, 6, e01765-15. [Google Scholar] [CrossRef] [PubMed]
- Damron, F.H.; Oglesby-Sherrouse, A.G.; Wilks, A.; Barbier, M. Dual-seq transcriptomics reveals the battle for iron during Pseudomonas aeruginosa acute murine pneumonia. Sci. Rep. 2016, 6, 39172. [Google Scholar] [CrossRef] [PubMed]
- Nuss, A.M.; Beckstette, M.; Pimenova, M.; Schmühl, C.; Opitz, W.; Pisano, F.; Heroven, A.K.; Dersch, P. Tissue dual RNA-seq allows fast discovery of infection-specific functions and riboregulators shaping host–pathogen transcriptomes. Proc. Natl. Acad. Sci. USA 2017, 114, E791–E800. [Google Scholar] [CrossRef] [PubMed]
- Silva, M. Classical labeling of bacterial pathogens according to their lifestyle in the host: Inconsistencies and alternatives. Front. Microbiol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Q.; Hu, M.; Yu, K.; Fu, J.; Zhou, F.; Liu, X. Proteomic analyses of intracellular Salmonella enterica serovar typhimurium reveal extensive bacterial adaptations to infected host epithelial cells. Infect. Immun. 2015, 83, 2897–2906. [Google Scholar] [CrossRef] [PubMed]
- Pieper, R.; Zhang, Q.; Parmar, P.P.; Huang, S.T.; Clark, D.J.; Alami, H.; Donohue-Rolfe, A.; Fleischmann, R.D.; Peterson, S.N.; Tzipori, S. The Shigella dysenteriae serotype 1 proteome, profiled in the host intestinal environment, reveals major metabolic modifications and increased expression of invasive proteins. Proteomics 2009, 9, 5029–5045. [Google Scholar] [CrossRef] [PubMed]
- Pieper, R.; Fisher, C.R.; Suh, M.J.; Huang, S.T.; Parmar, P.; Payne, S.M. Analysis of the proteome of intracellular Shigella flexneri reveals pathways important for intracellular growth. Infect. Immun. 2013, 81, 4635–4648. [Google Scholar] [CrossRef] [PubMed]
- Van de Velde, S.; Delaive, E.; Dieu, M.; Carryn, S.; Van Bambeke, F.; Devreese, B.; Raes, M.; Tulkens, P.M. Isolation and 2-D-DIGE proteomic analysis of intracellular and extracellular forms of Listeria monocytogenes. Proteomics 2009, 9, 5484–5496. [Google Scholar] [CrossRef] [PubMed]
- Garcia-del Portillo, F.; Calvo, E.; D’Orazio, V.; Pucciarelli, M.G. Association of ActA to peptidoglycan revealed by cell wall proteomics of intracellular Listeria monocytogenes. J. Biol. Chem. 2011, 286, 34675–34689. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, J.R.; Nanduri, B.; Pittman, J.R.; Givaruangsawat, S.; Burgess, S.C.; Lawrence, M.L. Proteomic expression profiles of virulent and avirulent strains of Listeria monocytogenes isolated from macrophages. J. Proteom. 2011, 74, 1906–1917. [Google Scholar] [CrossRef] [PubMed]
- Ponnusamy, D.; Hartson, S.D.; Clinkenbeard, K.D. Intracellular Yersinia pestis expresses general stress response and tellurite resistance proteins in mouse macrophages. Vet. Microbiol. 2011, 150, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Al Dahouk, S.; Jubier-Maurin, V.; Neubauer, H.; Köhler, S. Quantitative analysis of the Brucella suis proteome reveals metabolic adaptation to long-term nutrient starvation. BMC Microbiol. 2013, 13, 199. [Google Scholar] [CrossRef] [PubMed]
- Steele-Mortimer, O. The Salmonella-containing vacuole—Moving with the times. Curr. Opin. Microbiol. 2008, 11, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.C.; Duchateau, M.; Fredlund, J.; Weiner, A.; Mallet, A.; Schmitt, C.; Matondo, M.; Hourdel, V.; Chamot-Rooke, J.; Enninga, J. The COPII complex and lysosomal VAMP7 determine intracellular Salmonella localization and growth. Cell. Microbiol. 2015, 17, 1699–1720. [Google Scholar] [CrossRef] [PubMed]
- Vorwerk, S.; Krieger, V.; Deiwick, J.; Hensel, M.; Hansmeier, N. Proteomes of host cell membranes modified by intracellular activities of Salmonella enterica. Mol. Cell. Proteom. MCP 2015, 14, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Knodler, L.A.; Vallance, B.A.; Celli, J.; Winfree, S.; Hansen, B.; Montero, M.; Steele-Mortimer, O. Dissemination of invasive Salmonella via bacterial-induced extrusion of mucosal epithelia. Proc. Natl. Acad. Sci. USA 2010, 107, 17733–17738. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.; Selbach, M.; Rollenhagen, C.; Ballmaier, M.; Meyer, T.F.; Mann, M.; Bumann, D. Robust Salmonella metabolism limits possibilities for new antimicrobials. Nature 2006, 440, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Twine, S.M.; Mykytczuk, N.C.; Petit, M.D.; Shen, H.; Sjostedt, A.; Wayne Conlan, J.; Kelly, J.F. In vivo proteomic analysis of the intracellular bacterial pathogen, Francisella tularensis, isolated from mouse spleen. Biochem. Biophys. Res. Commun. 2006, 345, 1621–1633. [Google Scholar] [CrossRef] [PubMed]
- Monk, I.R.; Foster, T.J. Genetic manipulation of Staphylococci—Breaking through the barrier. Front. Cell. Infect. Microbiol. 2012, 2, 49. [Google Scholar] [CrossRef] [PubMed]
- Depke, M.; Surmann, K.; Hildebrandt, P.; Jehmlich, N.; Michalik, S.; Stanca, S.E.; Fritzsche, W.; Volker, U.; Schmidt, F. Labeling of the pathogenic bacterium Staphylococcus aureus with gold or ferric oxide-core nanoparticles highlights new capabilities for investigation of host–pathogen interactions. Cytom. Part A J. Int. Soc. Anal. Cytol. 2014, 85, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Ngo, J.T.; Champion, J.A.; Mahdavi, A.; Tanrikulu, I.C.; Beatty, K.E.; Connor, R.E.; Yoo, T.H.; Dieterich, D.C.; Schuman, E.M.; Tirrell, D.A. Cell-selective metabolic labeling of proteins. Nat. Chem. Biol. 2009, 5, 715–717. [Google Scholar] [CrossRef] [PubMed]
- Grammel, M.; Zhang, M.M.; Hang, H.C. Orthogonal alkynyl-amino acid reporter for selective labeling of bacterial proteomes during infection. Angew. Chem. (Int. Ed. Engl.) 2010, 49, 5970–5974. [Google Scholar] [CrossRef] [PubMed]
- Ngo, J.T.; Tirrell, D.A. Noncanonical amino acids in the interrogation of cellular protein synthesis. Acc. Chem. Res. 2011, 44, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, D.C.; Link, A.J.; Graumann, J.; Tirrell, D.A.; Schuman, E.M. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proc. Natl. Acad. Sci. USA 2006, 103, 9482–9487. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, D.C.; Lee, J.J.; Link, A.J.; Graumann, J.; Tirrell, D.A.; Schuman, E.M. Labeling, detection and identification of newly synthesized proteomes with bioorthogonal non-canonical amino-acid tagging. Nat. Protoc. 2007, 2, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Hong, V.; Presolski, S.I.; Ma, C.; Finn, M.G. Analysis and optimization of copper-catalyzed azide-alkyne cycloaddition for bioconjugation. Angew. Chem. (Int. Ed. Engl.) 2009, 48, 9879–9883. [Google Scholar] [CrossRef] [PubMed]
- Tanrikulu, I.C.; Schmitt, E.; Mechulam, Y.; Goddard, W.A., 3rd; Tirrell, D.A. Discovery of Escherichia coli methionyl-tRNA synthetase mutants for efficient labeling of proteins with azidonorleucine in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 15285–15290. [Google Scholar] [CrossRef] [PubMed]
- Wier, G.M.; McGreevy, E.M.; Brown, M.J.; Boyle, J.P. New method for the orthogonal labeling and purification of Toxoplasma gondii proteins while inside the host cell. MBio 2015, 6, e01628. [Google Scholar] [CrossRef] [PubMed]
- Tjalsma, H.; Bolhuis, A.; Jongbloed, J.D.H.; Bron, S.; van Dijl, J.M. Signal peptide-dependent protein transport in Bacillus subtilis: A genome-based survey of the secretome. Microbiol. Mol. Biol. Rev. 2000, 64, 515–547. [Google Scholar] [CrossRef] [PubMed]
- Antelmann, H.; Tjalsma, H.; Voigt, B.; Ohlmeier, S.; Bron, S.; Dijl, J.M.; Hecker, M. A proteomic view on genome-based signal peptide predictions. Genome Res. 2001, 11. [Google Scholar] [CrossRef] [PubMed]
- Tjalsma, H. Feature-based reappraisal of the Bacillus subtilis exoproteome. Proteomics 2007, 7, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Desvaux, M.; Hebraud, M.; Talon, R.; Henderson, I.R. Secretion and subcellular localizations of bacterial proteins: A semantic awareness issue. Trends Microbiol. 2009, 17, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Cornejo-Granados, F.; Zatarain-Barrón, Z.L.; Cantu-Robles, V.A.; Mendoza-Vargas, A.; Molina-Romero, C.; Sánchez, F.; Del Pozo-Yauner, L.; Hernández-Pando, R.; Ochoa-Leyva, A. Secretome prediction of two M. tuberculosis clinical isolates reveals their high antigenic density and potential drug targets. Front. Microbiol. 2017, 8, 128. [Google Scholar] [CrossRef] [PubMed]
- Samudrala, R.; Heffron, F.; McDermott, J.E. Accurate prediction of secreted substrates and identification of a conserved putative secretion signal for type III secretion systems. PLoS Pathog. 2009, 5, e1000375. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Ren, X.; Wei, C.; Yang, J.; Hu, Y.; Liu, L.; Xu, X.; Wang, J.; Jin, Q. Analysis of the secretome and identification of novel constituents from culture filtrate of bacillus Calmette-Guérin using high-resolution mass spectrometry. Mol. Cell. Proteom. MCP 2013, 12, 2081–2095. [Google Scholar] [CrossRef] [PubMed]
- Trost, M.; Wehmhoner, D.; Karst, U.; Dieterich, G.; Wehland, J.; Jansch, L. Comparative proteome analysis of secretory proteins from pathogenic and nonpathogenic Listeria species. Proteomics 2005, 5, 1544–1557. [Google Scholar] [CrossRef] [PubMed]
- Niemann, G.S.; Brown, R.N.; Gustin, J.K.; Stufkens, A.; Shaikh-Kidwai, A.S.; Li, J.; McDermott, J.E.; Brewer, H.M.; Schepmoes, A.; Smith, R.D.; et al. Discovery of novel secreted virulence factors from Salmonella enterica serovar typhimurium by proteomic analysis of culture supernatants. Infect. Immun. 2011, 79, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Yu, H.B.; de Hoog, C.L.; Stoynov, N.; Li, Y.; Foster, L.J.; Finlay, B.B. Quantitative proteomic analysis of type III secretome of enteropathogenic Escherichia coli reveals an expanded effector repertoire for attaching/effacing bacterial pathogens. Mol. Cell. Proteom. MCP 2012, 11, 692–709. [Google Scholar] [CrossRef] [PubMed]
- Chinison, J.J.; Danelishvili, L.; Gupta, R.; Rose, S.J.; Babrak, L.M.; Bermudez, L.E. Identification of Mycobacterium avium subsp. hominissuis secreted proteins using an in vitro system mimicking the phagosomal environment. BMC Microbiol. 2016, 16, 270. [Google Scholar] [CrossRef] [PubMed]
- Dreisbach, A.; Hempel, K.; Buist, G.; Hecker, M.; Becher, D.; van Dijl, J.M. Profiling the surfacome of Staphylococcus aureus. Proteomics 2010, 10, 3082–3096. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.T.; Wang, J.S.; Tsai, S.H.; Chuan, C.N.; Wu, J.J.; Liao, P.C. Label-free proteomic analysis of environmental acidification-influenced Streptococcus pyogenes secretome reveals a novel acid-induced protein histidine triad protein A (HtpA) involved in necrotizing fasciitis. J. Proteom. 2014, 109, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Maddi, A.; Haase, E.; Scannapieco, F. Mass spectrometric analysis of whole secretome and amylase-precipitated secretome proteins from Streptococcus gordonii. J. Proteom. Bioinform. 2014, 7, 287–295. [Google Scholar] [CrossRef]
- Gagic, D.; Ciric, M.; Wen, W.X.; Ng, F.; Rakonjac, J. Exploring the secretomes of microbes and microbial communities using filamentous phage display. Front. Microbiol. 2016, 7, 429. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, D.; Collett, M.A.; Lubbers, M.W.; Rakonjac, J. Direct selection and phage display of a Gram-positive secretome. Genome Biol. 2007, 8, R266. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, A.; Szychowski, J.; Ngo, J.T.; Sweredoski, M.J.; Graham, R.L.J.; Hess, S.; Schneewind, O.; Mazmanian, S.K.; Tirrell, D.A. Identification of secreted bacterial proteins by noncanonical amino acid tagging. Proc. Natl. Acad. Sci. USA 2014, 111, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Chande, A.G.; Siddiqui, Z.; Midha, M.K.; Sirohi, V.; Ravichandran, S.; Rao, K.V.S. Selective enrichment of mycobacterial proteins from infected host macrophages. Sci. Rep. 2015, 5, 13430. [Google Scholar] [CrossRef] [PubMed]
- Colston, M.J.; Cox, R.A. Mycobacterial Growth and Dormancy. In Mycobacteria; Blackwell Publishing Ltd.: Hoboken, NY, USA, 2009; pp. 198–219. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Ghaemmaghami, S.; Newman, J.R.; Weissman, J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 2009, 324, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Gawron, D.; Gevaert, K.; Van Damme, P. The proteome under translational control. Proteomics 2014, 14, 2647–2662. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.Y.; Lu, J.; Sun, X.; He, Q.Y. Application of subproteomics in the characterization of Gram-positive bacteria. J. Proteom. 2012, 75, 2803–2810. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Becker, A.H.; Sandikci, A.; Huber, D.; Chaba, R.; Gloge, F.; Nichols, R.J.; Typas, A.; Gross, C.A.; Kramer, G.; et al. Selective ribosome profiling reveals the co-translational chaperone action of trigger factor in vivo. Cell 2011, 147, 1295–1308. [Google Scholar] [CrossRef] [PubMed]
- Ndah, E.; Jonckheere, V.; Giess, A.; Valen, E.; Menschaert, G.; Van Damme, P. REPARATION: Ribosome profiling assisted (Re-)annotation of bacterial genomes. bioRxiv 2017, 113530. [Google Scholar] [CrossRef] [PubMed]
- Giess, A.; Ndah, E.; Jonckheere, V.; Van Damme, P.; Valen, E. A unique ribosome signature reveals bacterial translation initiation sites. bioRxiv 2016, 095893. [Google Scholar] [CrossRef]
- Baek, J.; Lee, J.; Yoon, K.; Lee, H. Identification of unannotated small genes in Salmonella. G3 2017, 7, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Stern-Ginossar, N.; Ingolia, N.T. Ribosome profiling as a tool to decipher viral complexity. Annu. Rev. Virol. 2015, 2, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Sanz, E.; Yang, L.; Su, T.; Morris, D.R.; McKnight, G.S.; Amieux, P.S. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc. Natl. Acad. Sci. USA 2009, 106, 13939–13944. [Google Scholar] [CrossRef] [PubMed]
- Heiman, M.; Schaefer, A.; Gong, S.; Peterson, J.D.; Day, M.; Ramsey, K.E.; Suarez-Farinas, M.; Schwarz, C.; Stephan, D.A.; Surmeier, D.J.; et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell 2008, 135, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Hornstein, N.; Torres, D.; Das Sharma, S.; Tang, G.; Canoll, P.; Sims, P.A. Ligation-free ribosome profiling of cell type-specific translation in the brain. Genome Biol. 2016, 17, 149. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wan, J.; Liu, B.; Ma, M. Quantitative profiling of initiating ribosomes in vivo. Nat. Methods 2015, 12, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Ederth, J.; Mandava, C.S.; Dasgupta, S.; Sanyal, S. A single-step method for purification of active His-tagged ribosomes from a genetically engineered Escherichia coli. Nucl. Acids Res. 2009, 37, e15. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fels, U.; Gevaert, K.; Van Damme, P. Proteogenomics in Aid of Host–Pathogen Interaction Studies: A Bacterial Perspective. Proteomes 2017, 5, 26. https://doi.org/10.3390/proteomes5040026
Fels U, Gevaert K, Van Damme P. Proteogenomics in Aid of Host–Pathogen Interaction Studies: A Bacterial Perspective. Proteomes. 2017; 5(4):26. https://doi.org/10.3390/proteomes5040026
Chicago/Turabian StyleFels, Ursula, Kris Gevaert, and Petra Van Damme. 2017. "Proteogenomics in Aid of Host–Pathogen Interaction Studies: A Bacterial Perspective" Proteomes 5, no. 4: 26. https://doi.org/10.3390/proteomes5040026
APA StyleFels, U., Gevaert, K., & Van Damme, P. (2017). Proteogenomics in Aid of Host–Pathogen Interaction Studies: A Bacterial Perspective. Proteomes, 5(4), 26. https://doi.org/10.3390/proteomes5040026