Response of Downy Oak (Quercus pubescens Willd.) to Climate Change: Transcriptome Assembly, Differential Gene Analysis and Targeted Metabolomics


 , and
, and
Abstract
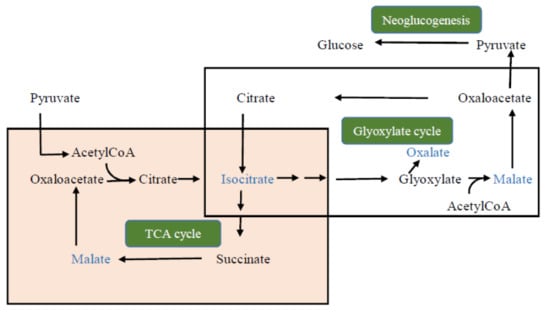
:
1. Introduction
2. Materials and Methods
2.1. Climate Change Simulation in Oak Forest
2.2. RNA Extraction
2.3. Library Preparation and Sequencing
2.4. Transcriptome Assembly and Completeness
2.5. Functional Annotation
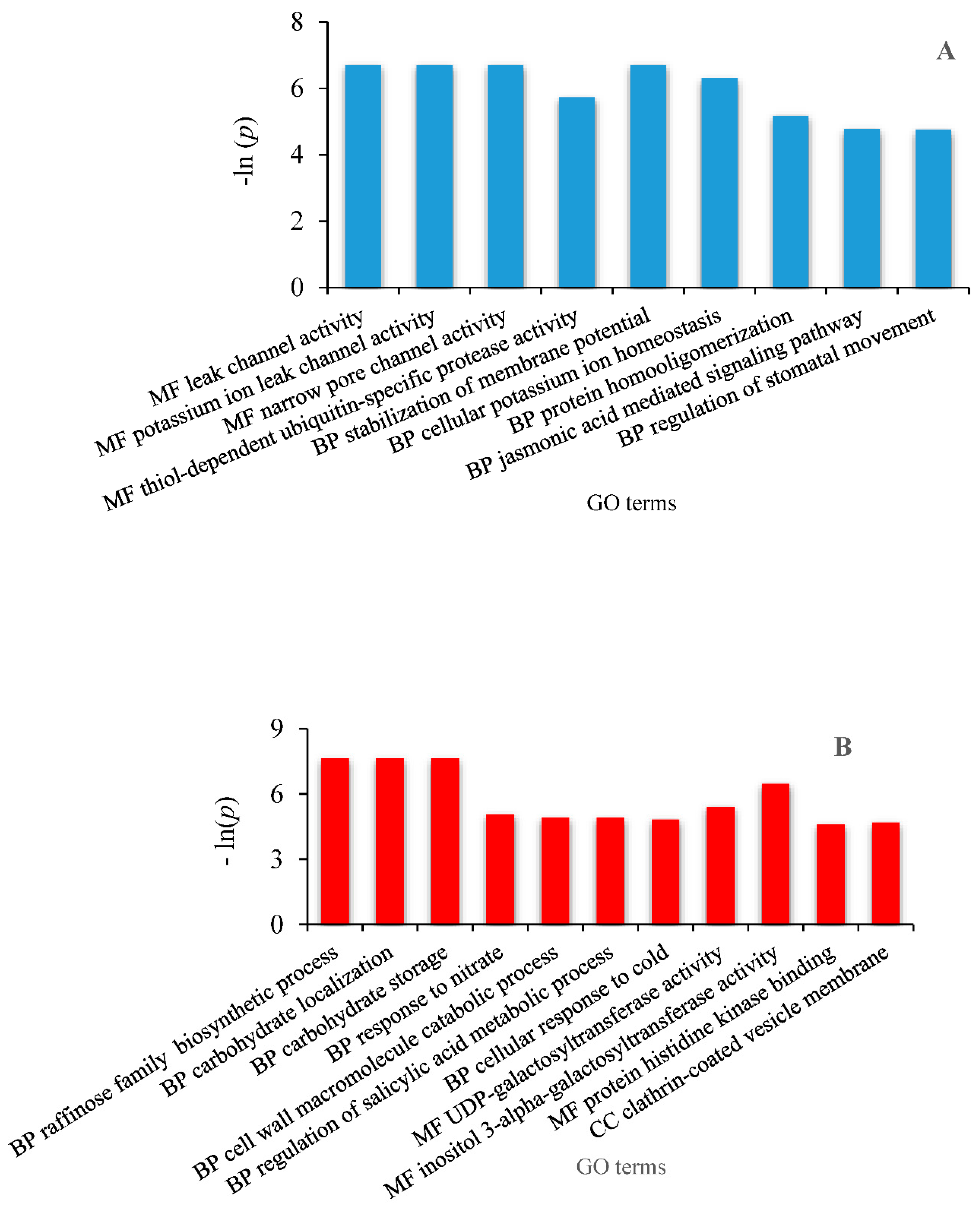
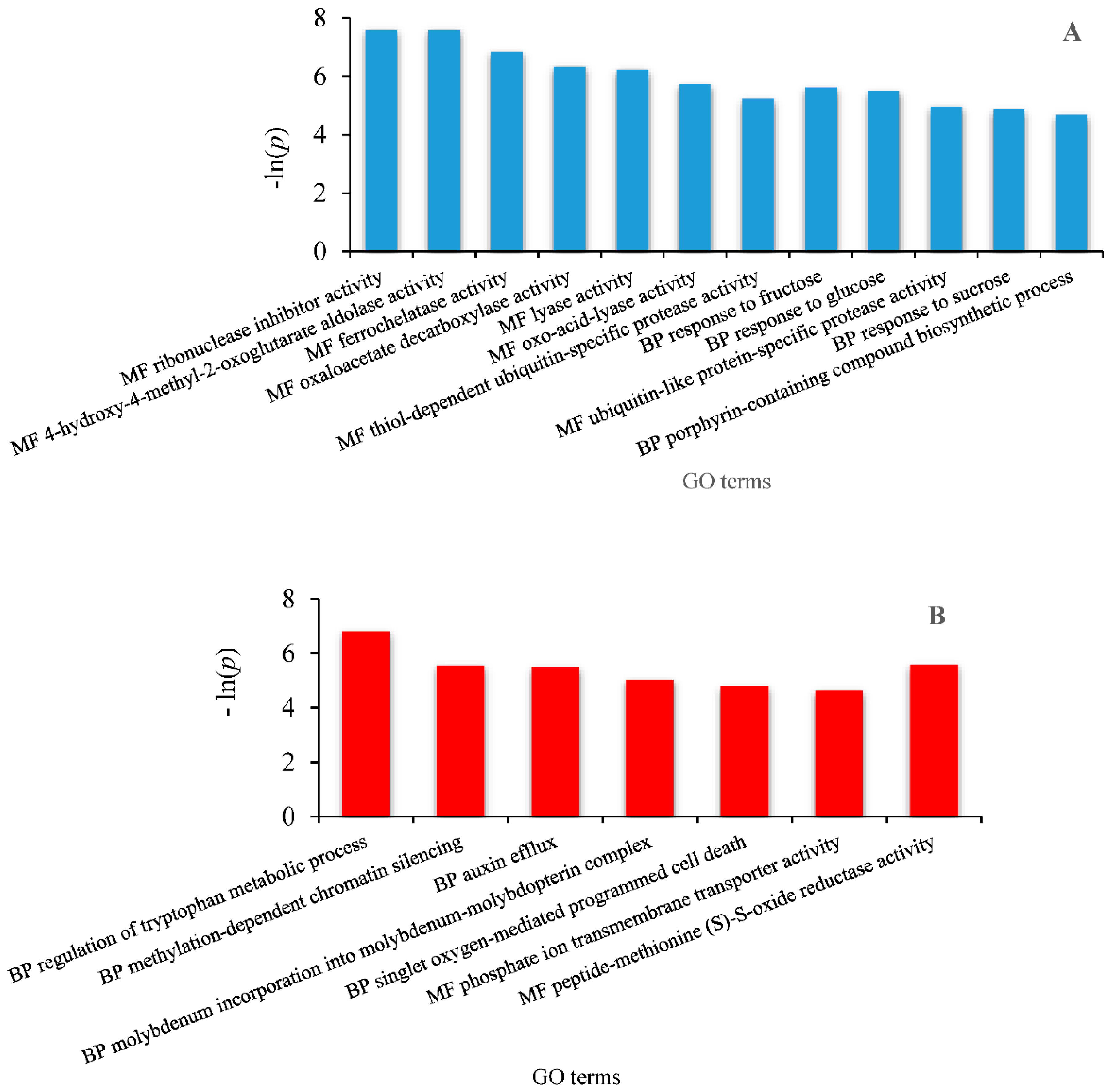
2.6. Identification of Differentially Expressed Genes Involved in Drought Tolerance and Gene Ontology Enrichment
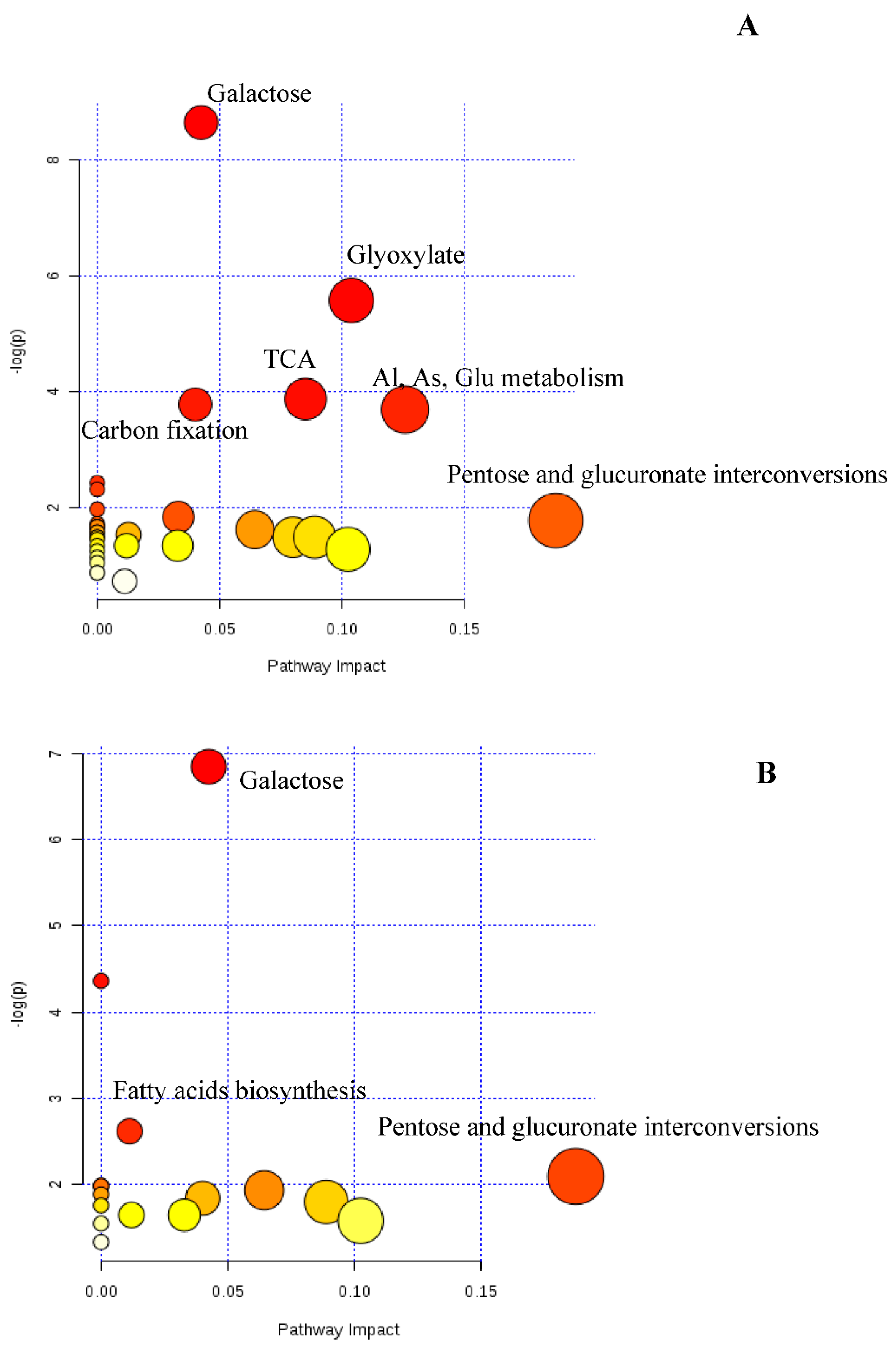
2.7. Targeted Metabolomics by Gas Chromatography/Mass Spectrometry (GC/MS) Analysis
2.8. Data Accessibility
3. Results
3.1. High Throughput Sequencing Output and De Novo Transcriptome Assembly
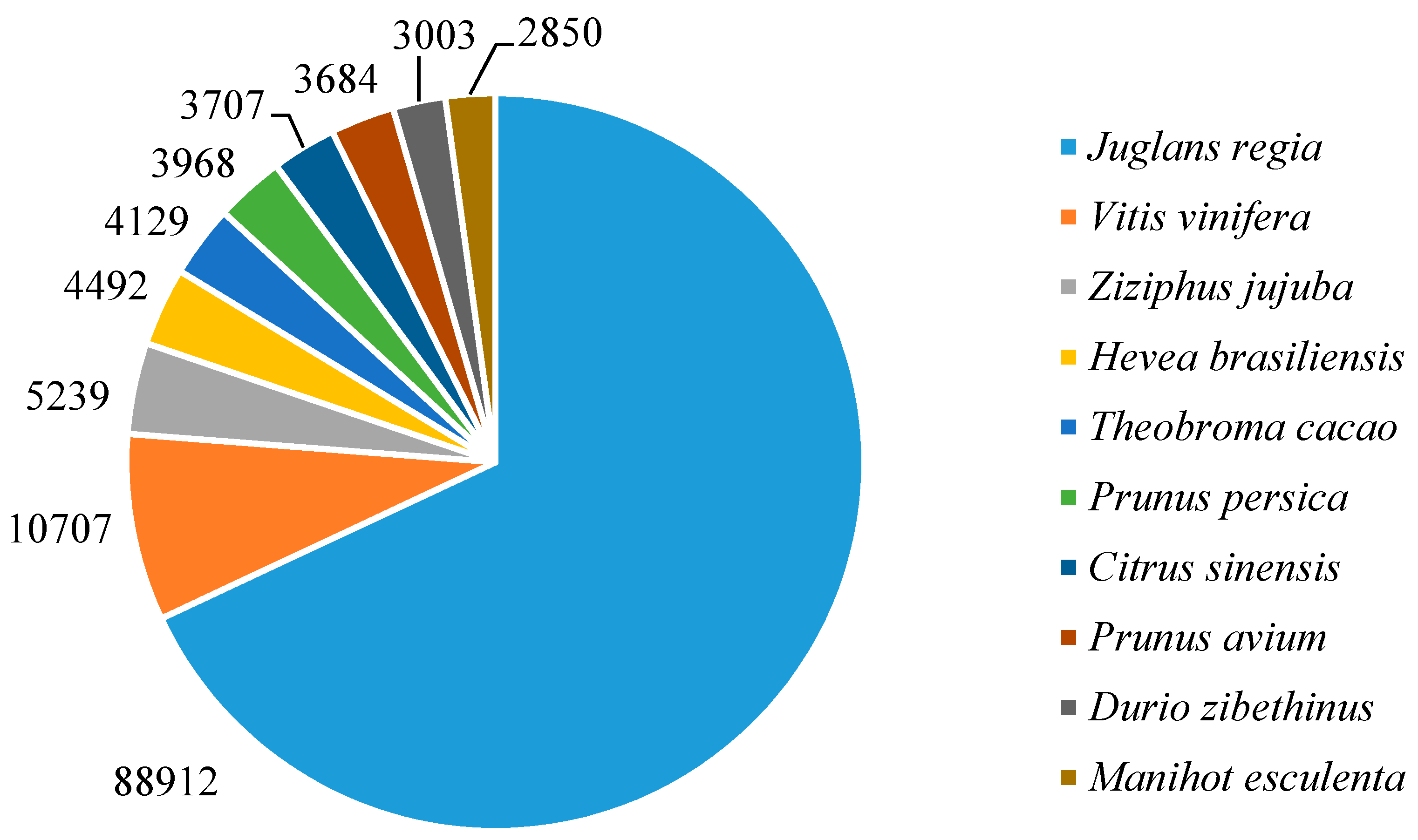
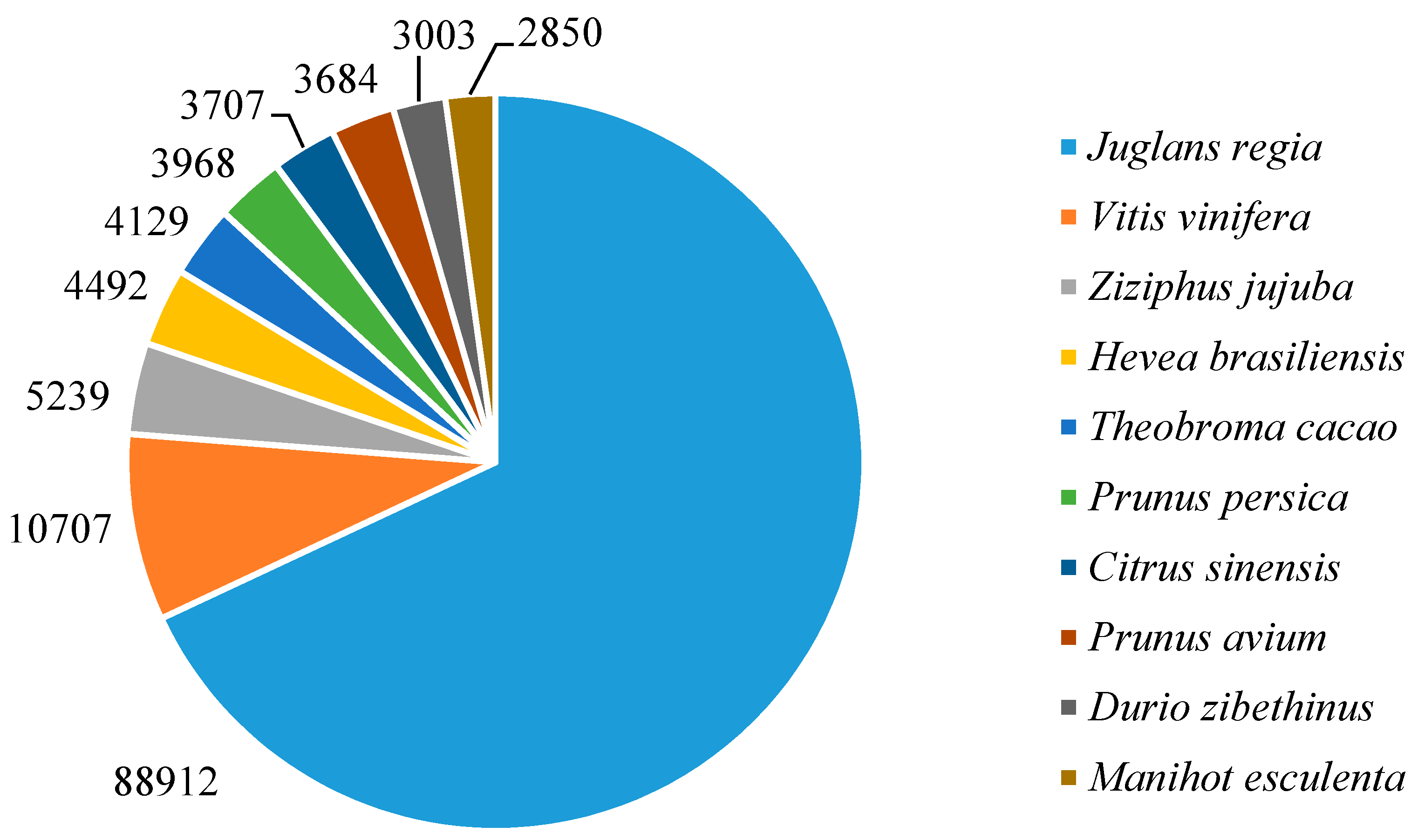
3.2. Functional Annotation of the Assembled Contigs
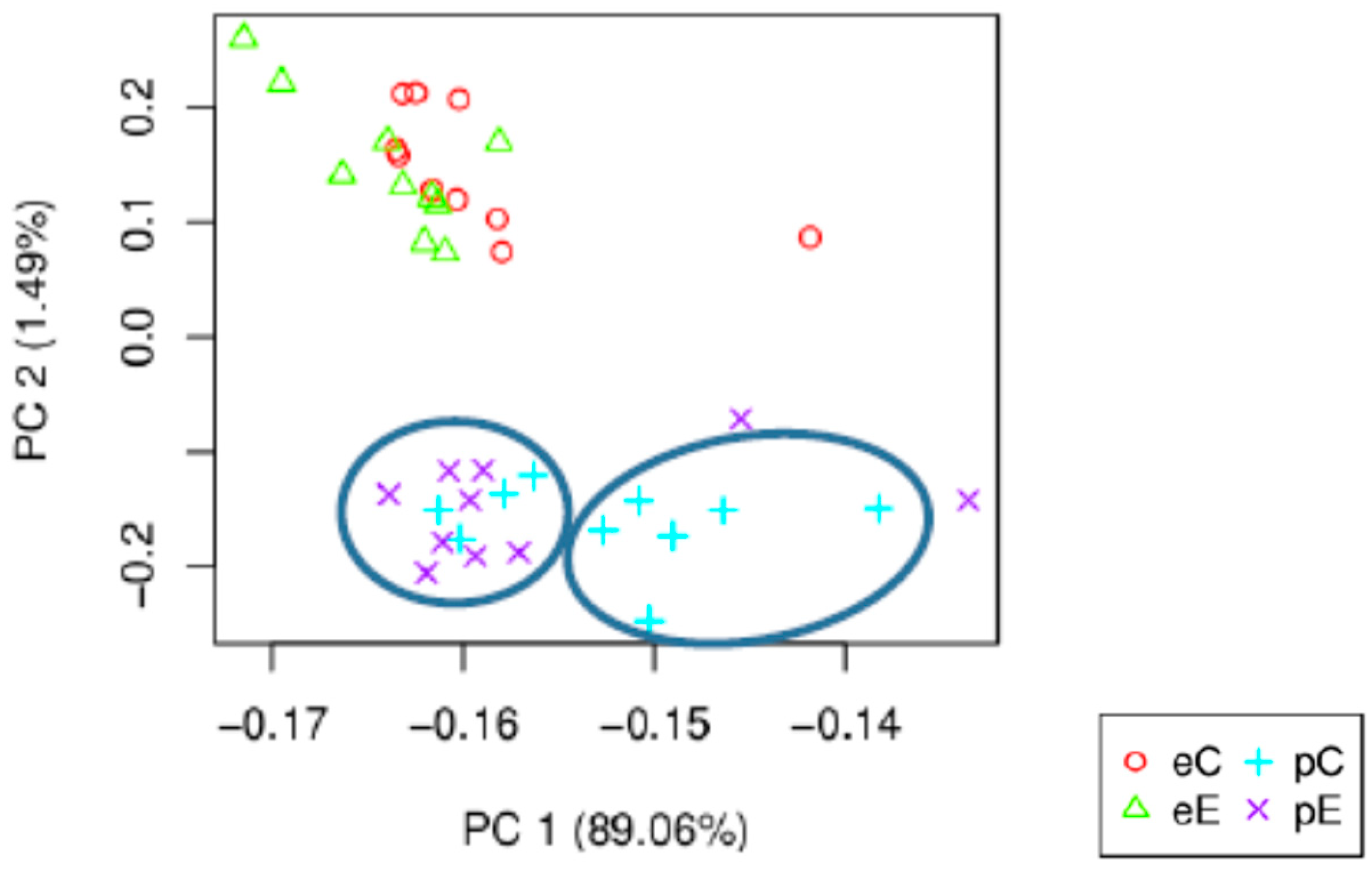
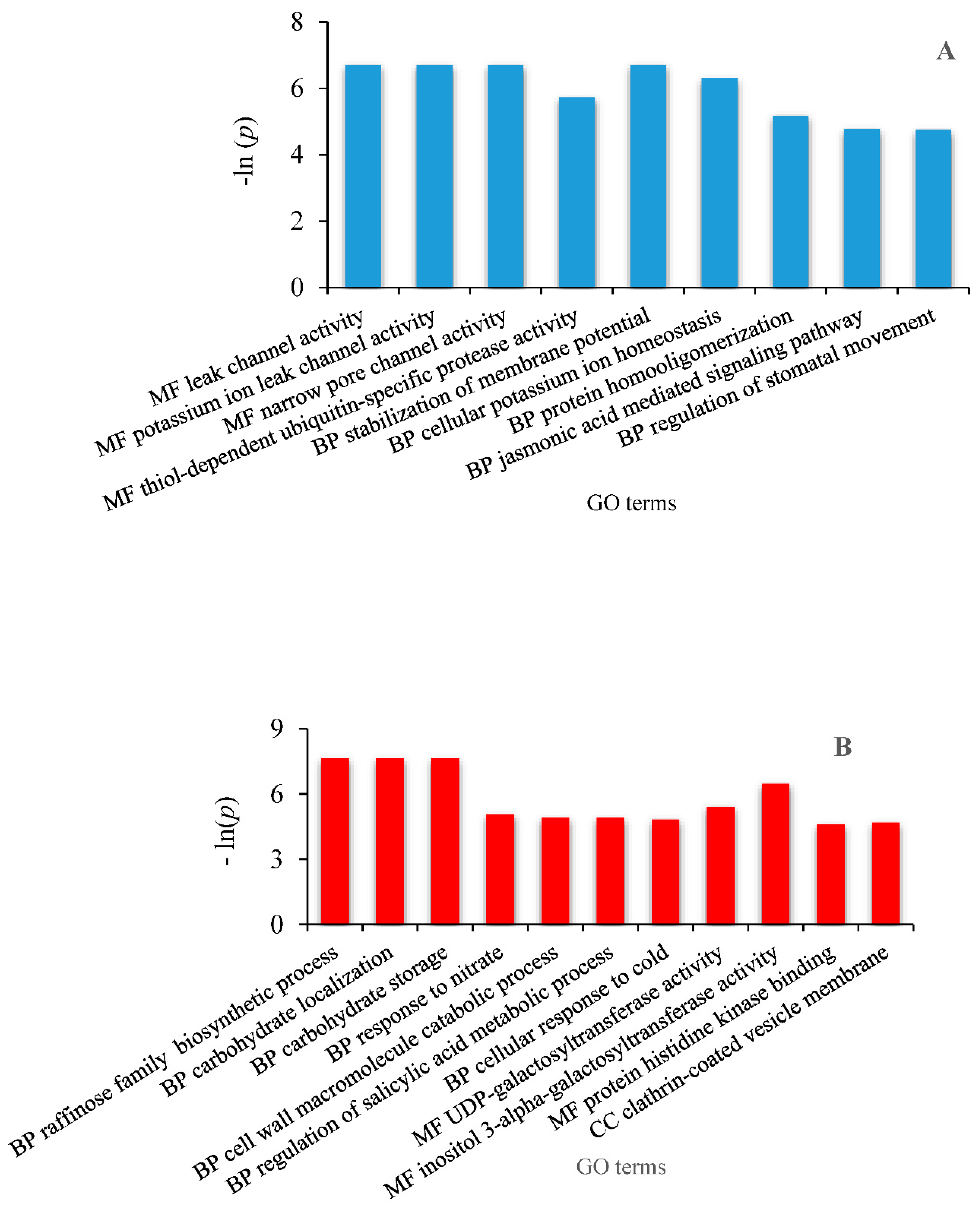
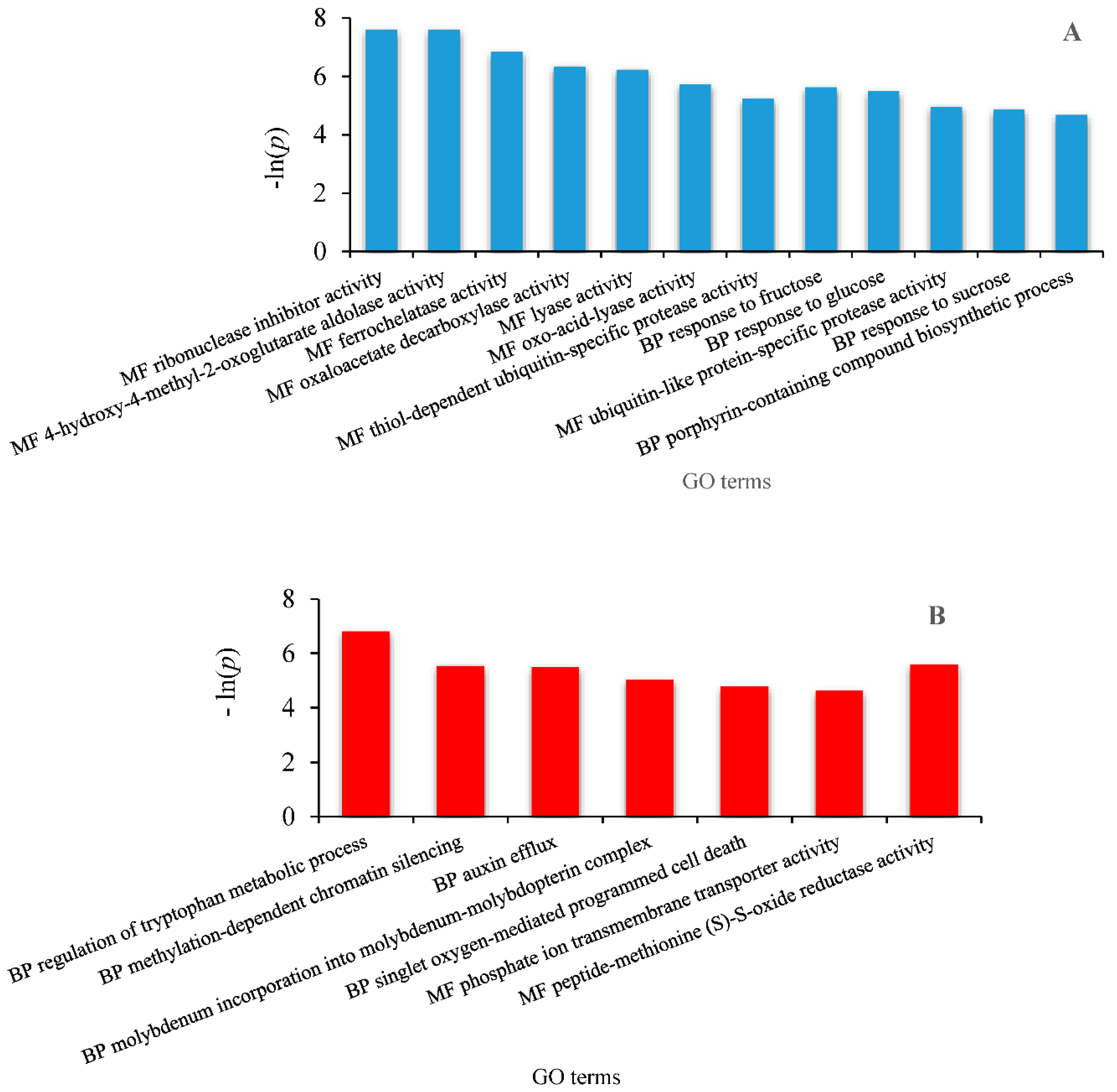
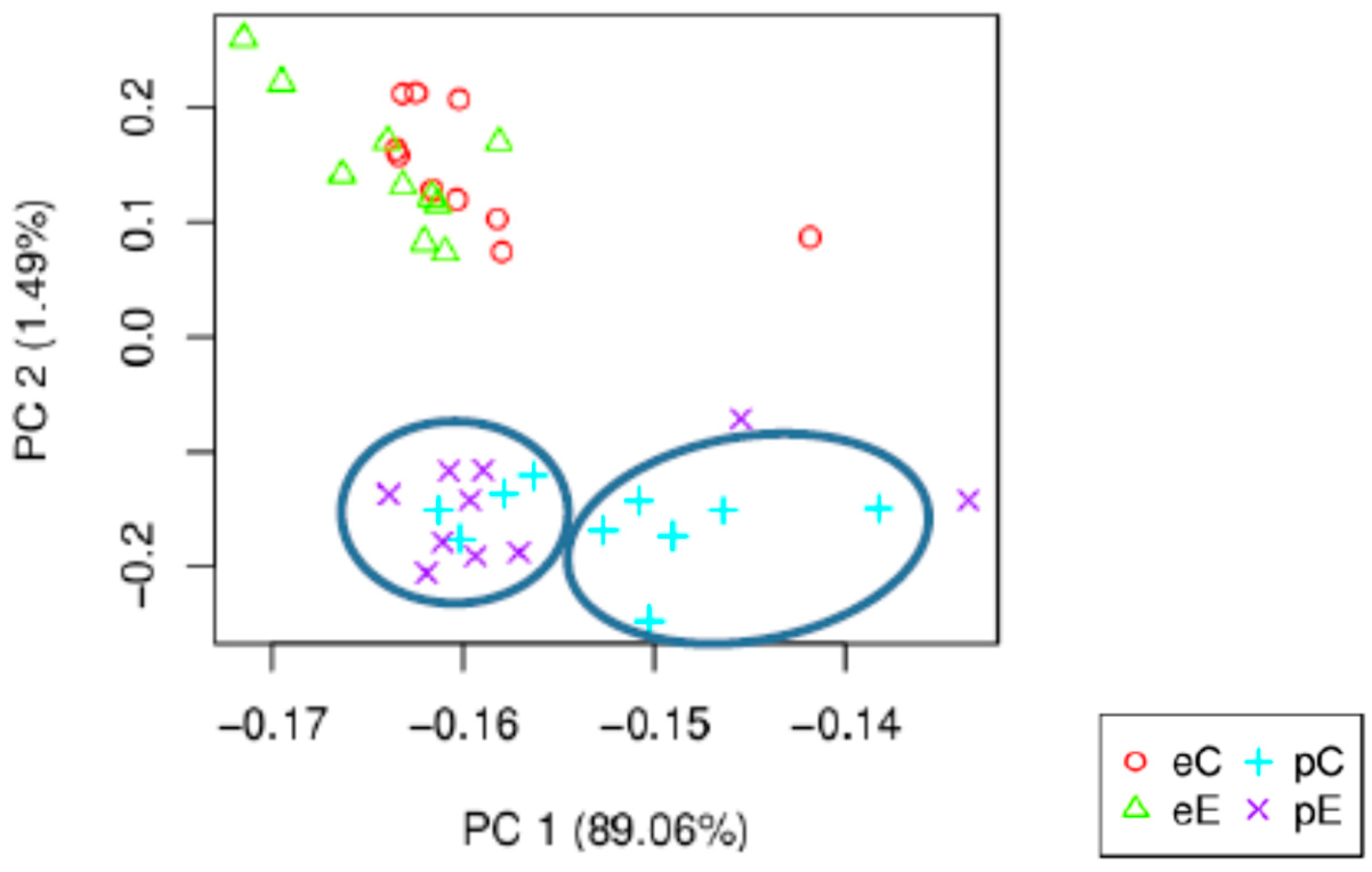
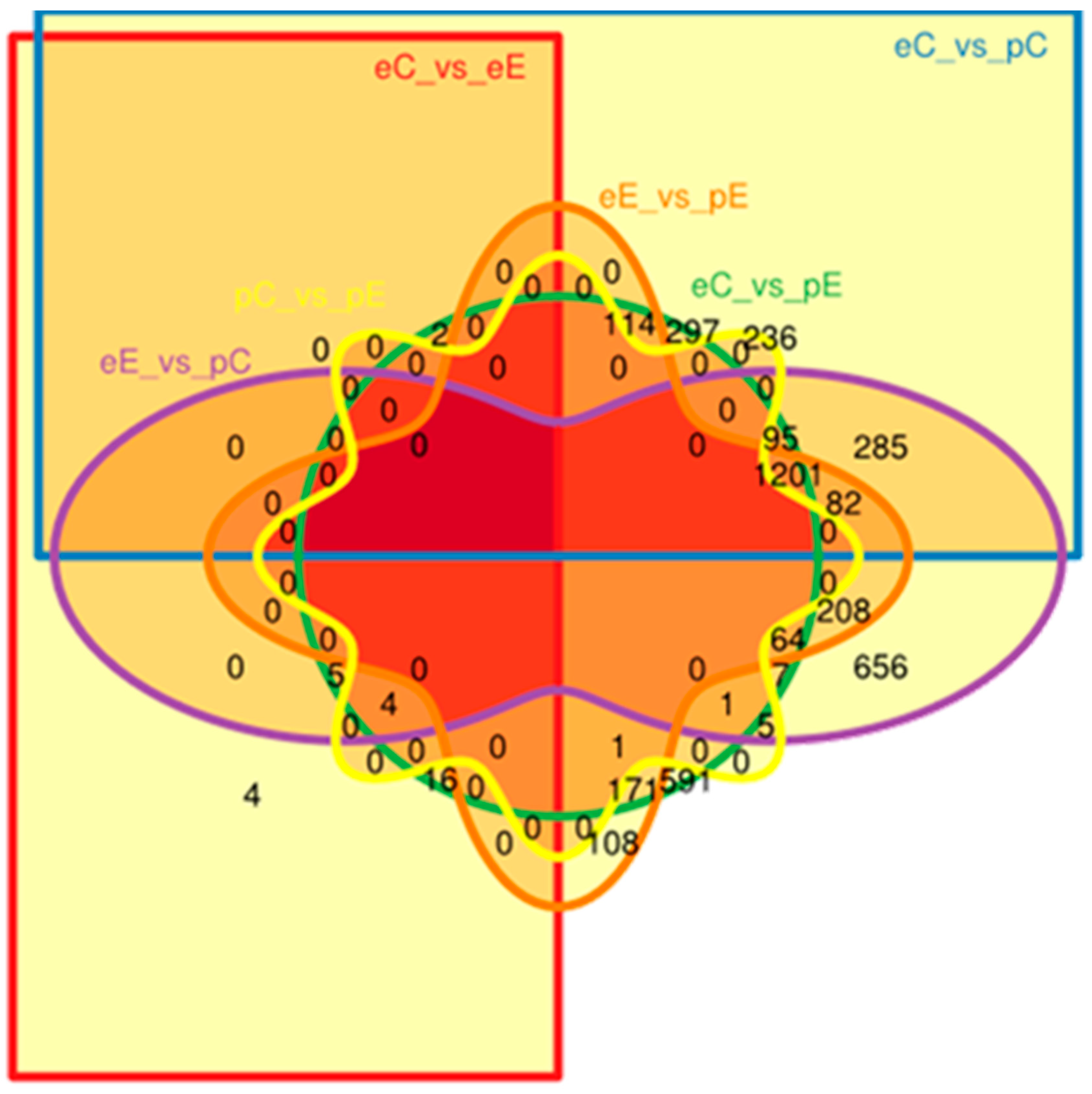
3.3. Differentially Expressed Genes (DEG) Analysis
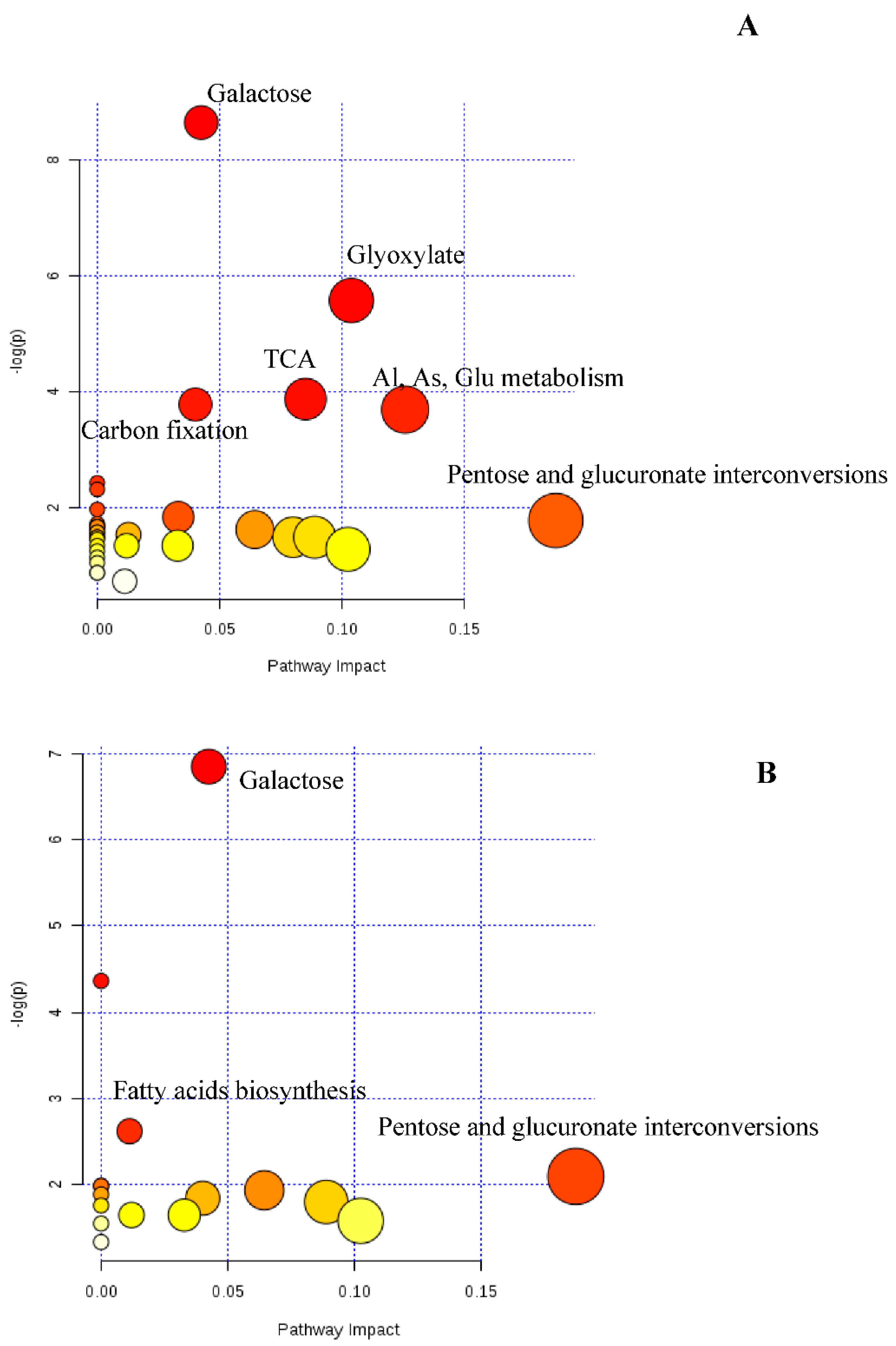
3.4. Targeted Metabolomic
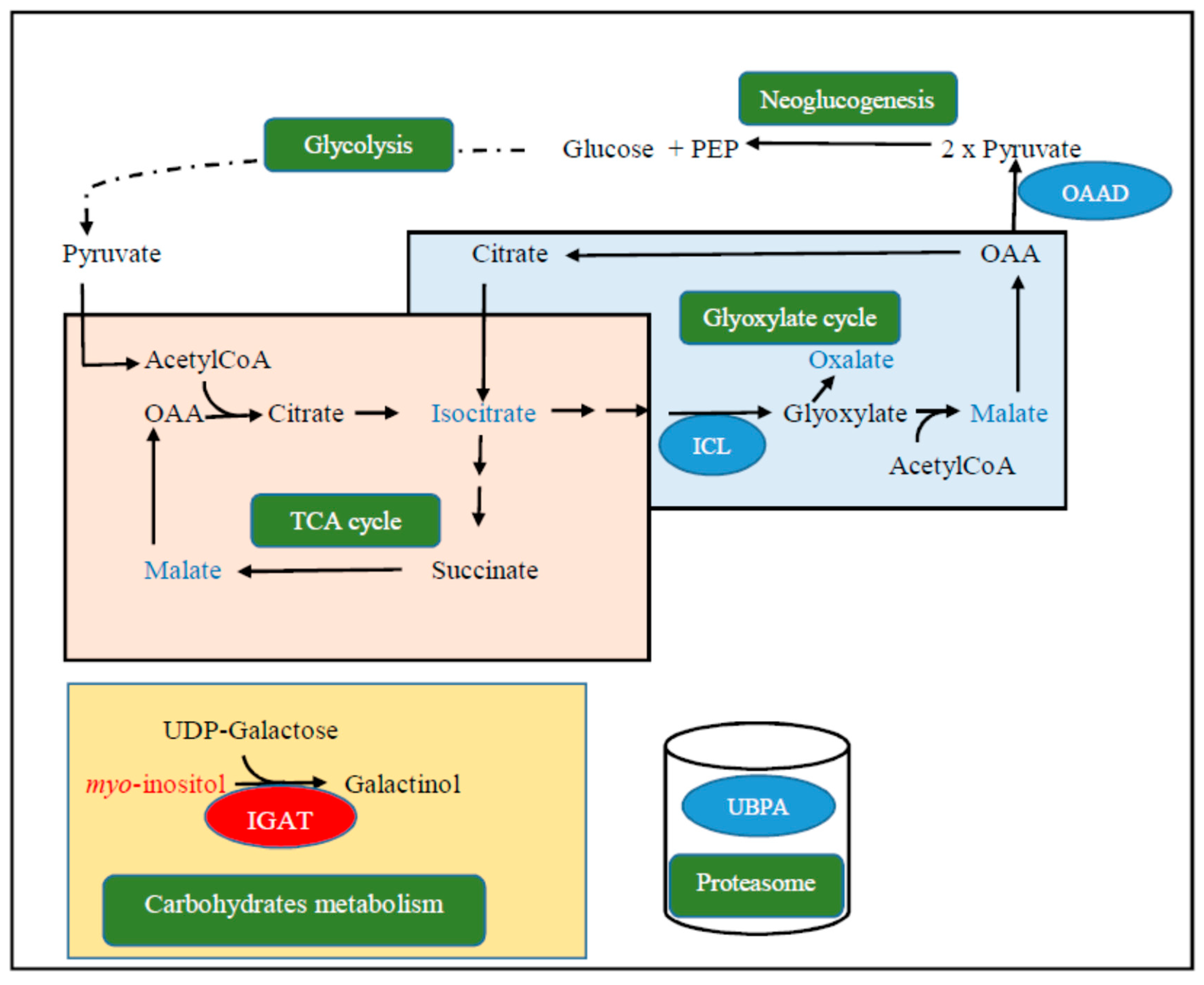
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- AR4 Climate Change 2007: The Physical Science Basis. Available online: https://www.ipcc.ch/report/ar4/wg1/ (accessed on 10 June 2020).
- Myers, N.; Mittermeier, R.A.; Mittermeier, C.G.; Fonseca, G.A.B.; Kent, J. Biodiversity hotspots for conservation priorities. Nature 2000, 403, 853–858. [Google Scholar] [CrossRef]
- Giorgi, F.; Lionello, P. Climate change projections for the Mediterranean region. Glob. Planet. Chang. 2008, 63, 90–104. [Google Scholar] [CrossRef]
- Polade, S.D.; Pierce, D.W.; Cayan, D.R.; Gershunov, A.; Dettinger, M.D. The key role of dry days in changing regional climate and precipitation regimes. Sci. Rep. 2014, 4, 4364. [Google Scholar] [CrossRef] [Green Version]
- Holland, V.; Koller, S.; Lukas, S.; Brüggemann, W. Drought- and frost-induced accumulation of soluble carbohydrates during accelerated senescence in Quercus pubescens. Trees 2016, 30, 215–226. [Google Scholar] [CrossRef]
- Sperlich, D.; Chang, C.T.; Penuelas, J.; Gracia, C.; Sabate, S. Seasonal variability of foliar photosynthetic and morphological traits and drought impacts in a Mediterranean mixed forest. Tree Physiol. 2015, 35, 501–520. [Google Scholar] [CrossRef] [PubMed]
- Contran, N.; Günthardt-Goerg, M.S.; Kuster, T.M.; Cerana, R.; Crosti, P.; Paoletti, E. Physiological and biochemical responses of Quercus pubescens to air warming and drought on acidic and calcareous soils. Plant Biol. 2013, 15, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Peñuelas, J.; Llusià, J. Linking photorespiration, monoterpenes and thermotolerance in Quercus. New Phytol. 2002, 155, 227–237. [Google Scholar] [CrossRef] [Green Version]
- Prieto, P.; Peñuelas, J.; Llusià, J.; Asensio, D.; Estiarte, M. Effects of long-term experimental night-time warming and drought on photosynthesis, Fv/Fm and stomatal conductance in the dominant species of a Mediterranean shrubland. Acta Physiol. Plant 2009, 31, 729–739. [Google Scholar] [CrossRef]
- Gugger, P.F.; Penaloza-Ramirez, J.M.; Wright, J.W.; Sork, V.L. Whole-transcriptome response to water stress in a California endemic oak, Quercus lobata. Tree Physiol. 2017, 37, 632–644. [Google Scholar] [CrossRef]
- Torre, S.; Tattini, M.; Brunetti, C.; Fineschi, S.; Fini, A.; Ferrini, F.; Sebastiani, F. RNA-Seq Analysis of Quercus pubescens Leaves: De Novo Transcriptome Assembly, Annotation and Functional Markers Development. PLoS ONE 2014, 9, e112487. [Google Scholar] [CrossRef] [Green Version]
- Yates, S.A.; Chernukhin, I.; Alvarez-Fernandez, R.; Bechtold, U.; Baeshen, M.; Baeshen, N.; Mutwakil, M.Z.; Sabir, J.; Lawson, T.; Mullineaux, P.M. The temporal foliar transcriptome of the perennial C3 desert plant Rhazya stricta in its natural environment. BMC Plant Biol. 2014, 14, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungerer, M.C.; Johnson, L.C.; Herman, M.A. Ecological genomics: Understanding gene and genome function in the natural environment. Heredity 2008, 100, 178–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinform. Oxf. Engl. 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Li, G.; Chang, Z.; Yu, T.; Liu, B.; McMullen, R.; Chen, P.; Huang, X. BinPacker: Packing-Based De Novo Transcriptome Assembly from RNA-seq Data. PLoS Comput. Biol. 2016, 12, e1004772. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 10 July 2020).
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plomion, C.; Aury, J.-M.; Amselem, J.; Alaeitabar, T.; Barbe, V.; Belser, C.; Bergès, H.; Bodénès, C.; Boudet, N.; Boury, C.; et al. Decoding the oak genome: Public release of sequence data, assembly, annotation and publication strategies. Mol. Ecol. Resour. 2016, 16, 254–265. [Google Scholar] [CrossRef]
- OAK GENOME SEQUENCING: Sequencing of the Oak Genome and Identification of Genes that Matter for Forest Tree Adaptation. Available online: http://www.oakgenome.fr/ (accessed on 10 July 2020).
- McGettigan, P.A. Transcriptomics in the RNA-seq era. Curr. Opin. Chem. Biol. 2013, 17, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Saunier, A.; Ormeño, E.; Havaux, M.; Wortham, H.; Ksas, B.; Temime-Roussel, B.; Blande, J.D.; Lecareux, C.; Mévy, J.-P.; Bousquet-Mélou, A.; et al. Resistance of native oak to recurrent drought conditions simulating predicted climatic changes in the Mediterranean region. Plant Cell Environ. 2018, 41, 2299–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosy, E.; Vavasseur, A.; Mouline, K.; Dreyer, I.; Gaymard, F.; Porée, F.; Boucherez, J.; Lebaudy, A.; Bouchez, D.; Véry, A.-A.; et al. The Arabidopsis outward K+ channel GORK is involved in regulation of stomatal movements and plant transpiration. Proc. Natl. Acad. Sci. USA 2003, 100, 5549–5554. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Ren, H.-M.; Tan, Y.-Q.; Qi, G.-N.; Yao, F.-Y.; Wu, G.-L.; Yang, L.-W.; Hussain, J.; Sun, S.-J.; Wang, Y.-F. S-Type Anion Channels SLAC1 and SLAH3 Function as Essential Negative Regulators of Inward K+ Channels and Stomatal Opening in Arabidopsis. Plant Cell 2016, 28, 949–965. [Google Scholar] [CrossRef]
- Jin, Z.; Wang, Z.; Ma, Q.; Sun, L.; Zhang, L.; Liu, Z.; Liu, D.; Hao, X.; Pei, Y. Hydrogen sulfide mediates ion fluxes inducing stomatal closure in response to drought stress in Arabidopsis thaliana. Plant Soil 2017, 419, 141–152. [Google Scholar] [CrossRef]
- Luan, S. Signalling drought in guard cells. Plant Cell Environ. 2002, 25, 229–237. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Song, C.-P. Guard-cell signalling for hydrogen peroxide and abscisic acid. New Phytol. 2008, 178, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Adachi, Y.; Nakamura, Y.; Munemasa, S.; Mori, I.C.; Murata, Y. Involvement of OST1 Protein Kinase and PYR/PYL/RCAR Receptors in Methyl Jasmonate-Induced Stomatal Closure in Arabidopsis Guard Cells. Plant Cell Physiol. 2016, 57, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Mukherjee, S.; Basak, P.; Majumder, A.L. Significance of galactinol and raffinose family oligosaccharide synthesis in plants. Front. Plant Sci. 2015, 6, 656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharfenberg, M.; Mittermayr, L.; Roepenack-Lahaye, E.V.; Schlicke, H.; Grimm, B.; Leister, D.; Kleine, T. Functional characterization of the two ferrochelatases in Arabidopsis thaliana. Plant Cell Environ. 2015, 38, 280–298. [Google Scholar] [CrossRef]
- Wu, H.; Shabala, L.; Zhou, M.; Shabala, S. Chloroplast-generated ROS dominate NaCl- induced K+ efflux in wheat leaf mesophyll. Plant Signal. Behav. 2015, 10. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Jin, X.; Liao, W.; Hu, L.; Dawuda, M.M.; Zhao, X.; Tang, Z.; Gong, T.; Yu, J. 5-Aminolevulinic Acid (ALA) Alleviated Salinity Stress in Cucumber Seedlings by Enhancing Chlorophyll Synthesis Pathway. Front. Plant Sci. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Kong, D.D.; Fu, X.K.; Ali, B.; Xu, L.; Zhou, W.J. Influence of exogenous 5-aminolevulinic acid on chlorophyll synthesis and related gene expression in oilseed rape de-etiolated cotyledons under water-deficit stress. Photosynthetica 2016, 54, 468–474. [Google Scholar] [CrossRef]
- Yan, N.; Doelling, J.H.; Falbel, T.G.; Durski, A.M.; Vierstra, R.D. The Ubiquitin-Specific Protease Family from Arabidopsis.AtUBP1 and 2 Are Required for the Resistance to the Amino Acid Analog Canavanine. Plant Physiol. 2000, 124, 1828–1843. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Wang, M.H. Expression and Biological Properties of a Novel Methionine Sulfoxide Reductase A in Tobacco (Nicotiana tabacum). Protein J. 2013, 32, 266–274. [Google Scholar] [CrossRef]
- Romero, H.M.; Berlett, B.S.; Jensen, P.J.; Pell, E.J.; Tien, M. Investigations into the Role of the Plastidial Peptide Methionine Sulfoxide Reductase in Response to Oxidative Stress in Arabidopsis. Plant Physiol. 2004, 136, 3784–3794. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.-K.; Baek, K.-H.; Seong, E.S.; Joung, Y.H.; Choi, G.-J.; Park, J.M.; Cho, H.S.; Kim, E.A.; Lee, S.; Choi, D. CaMsrB2, Pepper Methionine Sulfoxide Reductase B2, Is a Novel Defense Regulator against Oxidative Stress and Pathogen Attack. Plant Physiol. 2010, 154, 245–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Zhu, J.; Ding, P.; Chen, F.; Xia, G. The Brachypodium distachyon methionine sulfoxide reductase gene family. Genes Genom. 2017, 39, 975–985. [Google Scholar] [CrossRef]
- Forestan, C.; Varotto, S. The Role of PIN Auxin Efflux Carriers in Polar Auxin Transport and Accumulation and Their Effect on Shaping Maize Development. Mol. Plant 2012, 5, 787–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.I.; Baek, D.; Park, H.C.; Chun, H.J.; Oh, D.-H.; Lee, M.K.; Cha, J.-Y.; Kim, W.-Y.; Kim, M.C.; Chung, W.S.; et al. Overexpression of Arabidopsis YUCCA6 in Potato Results in High-Auxin Developmental Phenotypes and Enhanced Resistance to Water Deficit. Mol. Plant 2013, 6, 337–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecube, M.L.; Noriega, G.O.; Cruz, D.M.S.; Tomaro, M.L.; Batlle, A.; Balestrasse, K.B. Indole acetic acid is responsible for protection against oxidative stress caused by drought in soybean plants: The role of heme oxygenase induction. Redox Rep. 2014, 19, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Danon, A.; Coll, N.S.; Apel, K. Cryptochrome-1-dependent execution of programmed cell death induced by singlet oxygen in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2006, 103, 17036–17041. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Kou, S.; Zhang, C.; Zou, Q.; Li, P.; Yuan, P. DNA Methylation and Plant Stress Responses. J. Plant Physiol. Pathol. 2018, 2018. [Google Scholar] [CrossRef]
- Maruyama, K.; Urano, K.; Yoshiwara, K.; Morishita, Y.; Sakurai, N.; Suzuki, H.; Kojima, M.; Sakakibara, H.; Shibata, D.; Saito, K.; et al. Integrated Analysis of the Effects of Cold and Dehydration on Rice Metabolites, Phytohormones, and Gene Transcripts. Plant Physiol. 2014, 164, 1759–1771. [Google Scholar] [CrossRef] [Green Version]
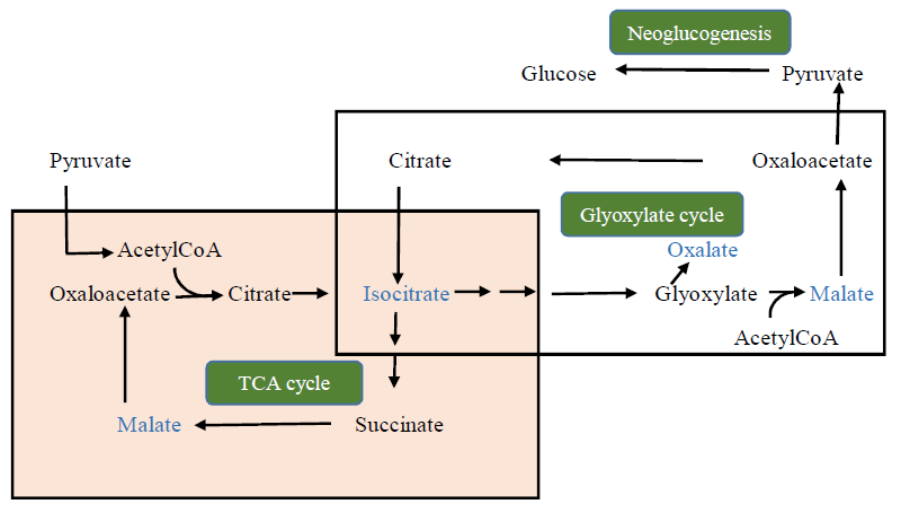
- Yu, L.; Jiang, J.; Zhang, C.; Jiang, L.; Ye, N.; Lu, Y.; Yang, G.; Liu, E.; Peng, C.; He, Z.; et al. Glyoxylate rather than ascorbate is an efficient precursor for oxalate biosynthesis in rice. J. Exp. Bot. 2010, 61, 1625–1634. [Google Scholar] [CrossRef] [Green Version]
- Jiménez-Arias, D.; García-Machado, F.J.; Morales-Sierra, S.; Luis, J.C.; Suarez, E.; Hernández, M.; Valdés, F.; Borges, A.A. Lettuce plants treated with L-pyroglutamic acid increase yield under water deficit stress. Environ. Exp. Bot. 2019, 158, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Purwaha, P.; Silva, L.P.; Hawke, D.H.; Weinstein, J.N.; Lorenzi, P.L. An artifact in LC-MS/MS measurement of glutamine and glutamic acid: In-source cyclization to pyroglutamic acid. Anal. Chem. 2014, 86, 5633–5637. [Google Scholar] [CrossRef] [PubMed]

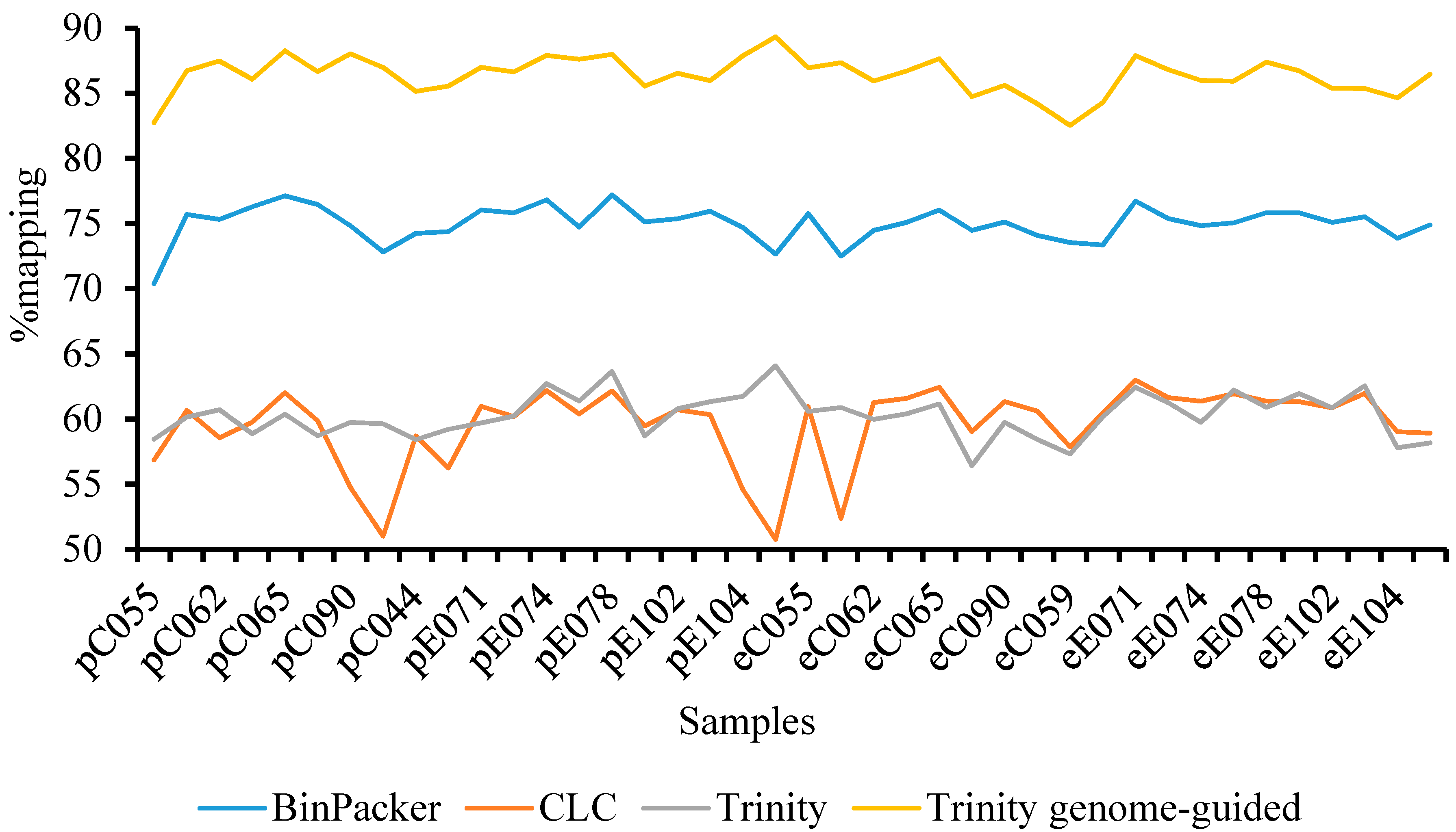


{kind=link}
{kind=link}
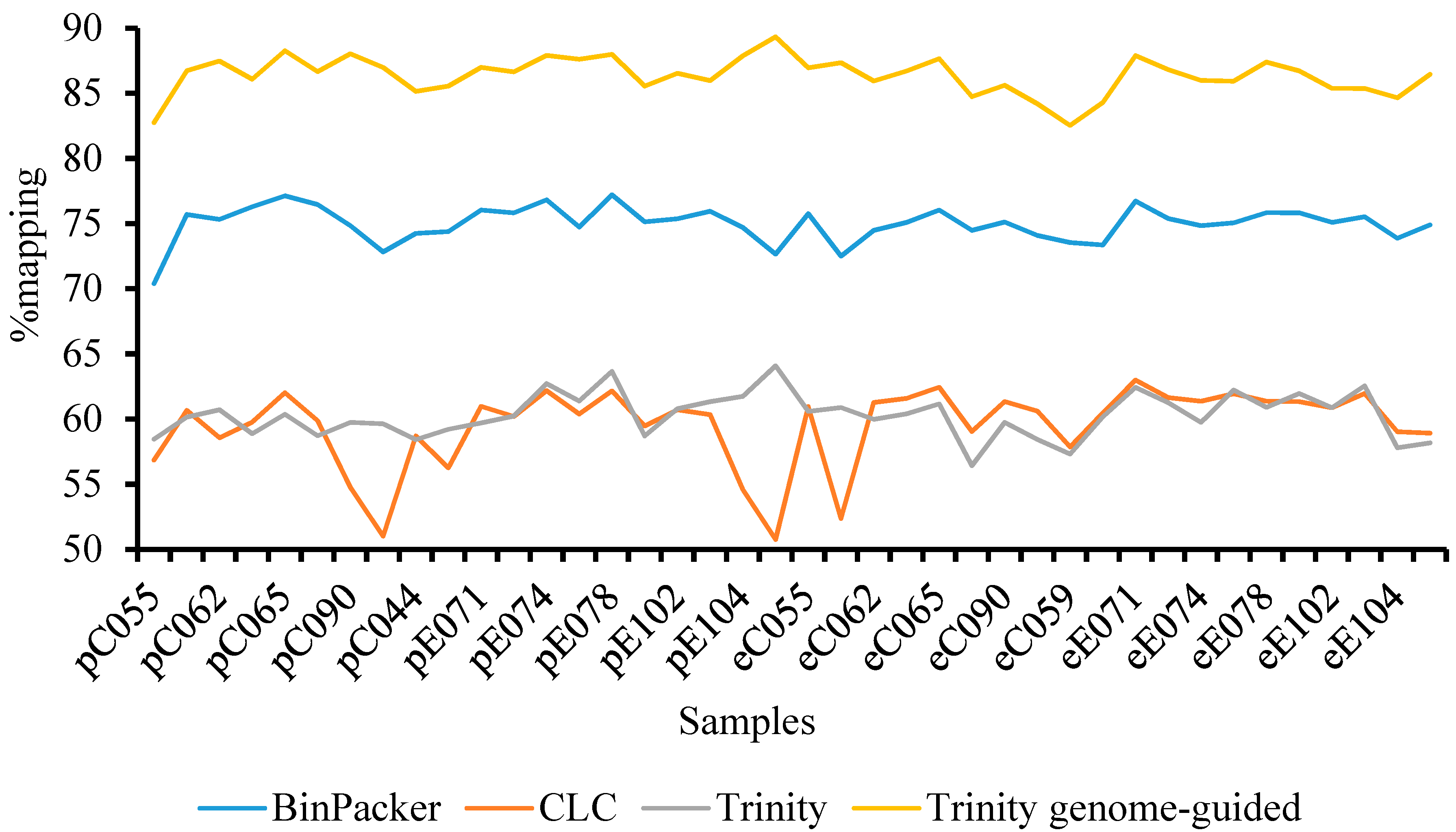{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
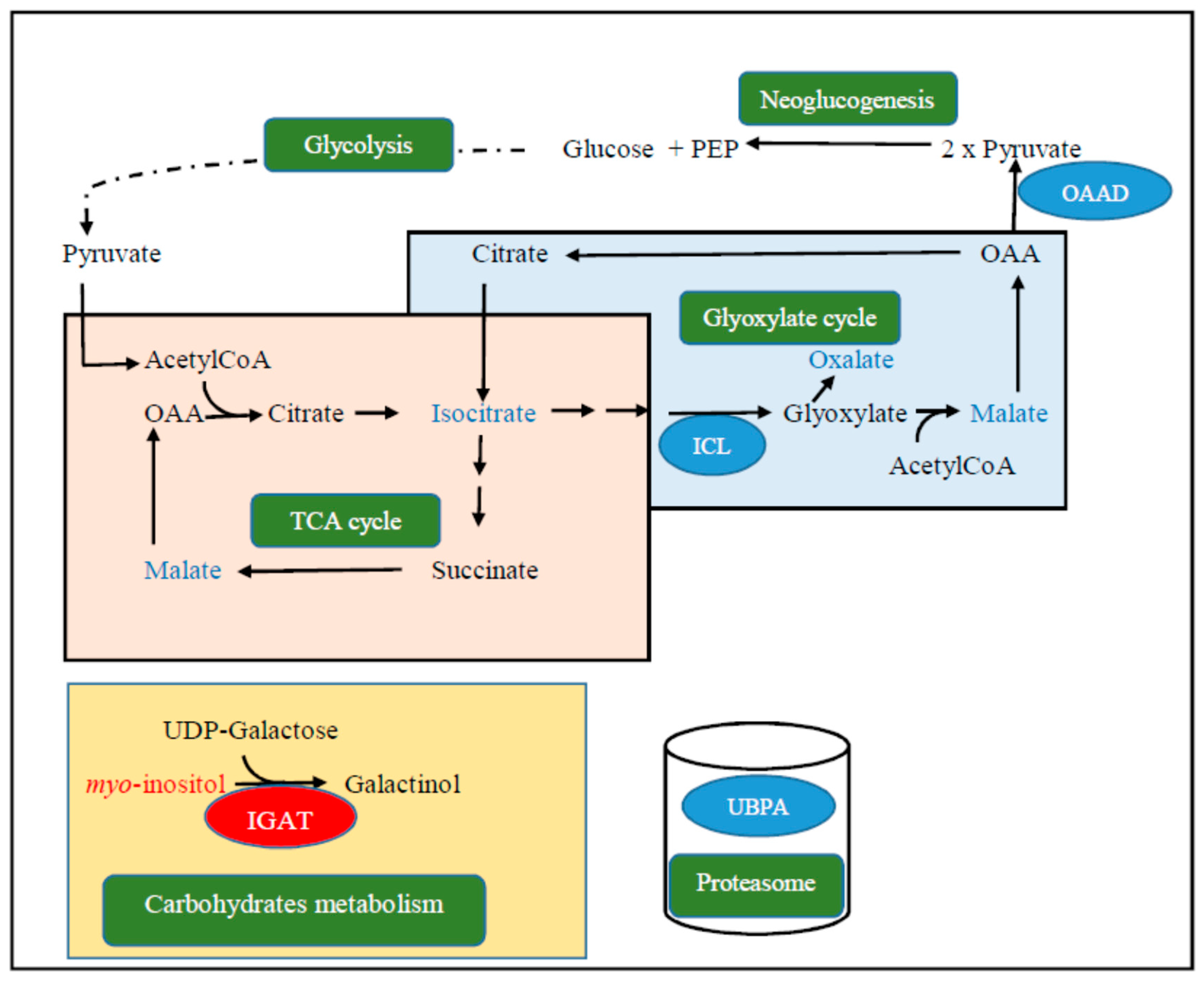{kind=link}
| Total raw paired-end reads | 1,971,431,570 × 2 |
| Total clean paired-end reads | 1,794,615,817 × 2 |
| Number of transcripts/trinity “genes”—raw assembly | 530,080/395,969 |
| Mean contig length (bp)/N50—raw assembly | 595/780 |
| E90N50/E90 number of transcripts | 1544/34,572 |
| Number of transcripts/trinity “genes”—filtered transcriptome (TPM < 1) | 156,986/126,106 |
| Mean contig length (bp)/N50—filtered transcriptome (TPM < 1) | 720/1133 |
| Transcriptomes Assemblies | Q. pubescens Filtered | Q. pubescens Raw | Q. robur | Q. pubescens Filtered | Q. pubescens Raw | Q. robur |
|---|---|---|---|---|---|---|
| BUSCO sets | Eukaryota | Embryophyta | ||||
| Complete BUSCOs (C) | 280 | 288 | 140 | 1206 | 1266 | 554 |
| Complete BUSCOs% | 92.40 | 95.10 | 46.30 | 83.80 | 87.90 | 38.40 |
| Complete and single-copy BUSCOs (S) | 112 | 103 | 115 | 645 | 601 | 447 |
| Complete and duplicated BUSCOs (D) | 168 | 185 | 25 | 561 | 665 | 107 |
| Fragmented BUSCOs (F) | 14 | 12 | 20 | 105 | 113 | 104 |
| Missing BUSCOs (M) | 9 | 3 | 143 | 129 | 61 | 782 |
| Total BUSCO groups searched | 303 | 303 | 303 | 1440 | 1440 | 1440 |
| Database | Q. pubescens Raw | Q. pubescens Filtered | ||
|---|---|---|---|---|
| Number of transcripts | 530,080 | 156,986 | ||
| Number of transcripts/peptides annotated | Percentage of transcripts/peptides annotated | Number of transcripts/peptides annotated | Percentage of transcripts/peptides annotated | |
| Uniref90 DiamondX | 214,856 | 40.54 | 102,445 | 65.26 |
| SwissProt DiamondX | 155,684 | 29.37 | 79,367 | 50.56 |
| GO Extract from Sequence Similarity Search | 146,689 | 75,852 | ||
| Number of Protein Predicted | 135,957 | 71,166 | ||
| KEGG | 124,285 | 91.14 | 67,348 | 94.63 |
| Uniref90 DiamondP | 120,789 | 88.85 | 66,374 | 93.27 |
| EggNOG Function | 118,026 | 86.81 | 63,724 | 89.54 |
| SwissProt DiamondP | 95,098 | 69.94 | 54,967 | 77.24 |
| PFAM | 86,021 | 63.27 | 49,462 | 69.5 |
| GO extract from PFAM Analysis | 52,107 | 31,994 | ||
| TmHMM | 22,131 | 11,986 | ||
| RNAMMER | 110 | 68 | ||
| Samples | eC | eE | pC | pE |
|---|---|---|---|---|
| eC | / | 15 | 3122 | 3529 |
| eE | 31 | / | 3460 | 3709 |
| pC | 2312 | 2613 | / | 2 |
| pE | 2569 | 1949 | 11 | / |
| Name | Fold Change | |||
|---|---|---|---|---|
| Summer | Spring | Spring/Summer | ||
| E/C | E/C | C | E | |
| Lactic acid | 1.0 | 1.6 | 0.7 | 0.4 |
| Oxalic acid | 1.8 | 1.2 | 0.4 | 0.6 |
| Phosphoric acid | 1.2 | n.d. | n.d. | 0.8 |
| Glycerol | 1.0 | 1.3 | 0.9 | 0.7 |
| Succinic acid | 1.0 | 1.2 | 0.4 | 0.4 |
| Aspartic acid | n.d. | 1.1 | n.d. | 0.6 |
| Pyroglutamic acid | 1.8 | 2.1 | 1.3 | 1.1 |
| Xylulose | 0.9 | 1.8 | 9.8 | 4.9 |
| Isocitric acid | 1.0 | 1.6 | 0.9 | 0.6 |
| Sorbitol | 1.4 | 1.1 | 2.1 | 2.5 |
| Viburnitol * | 0.9 | 1.2 | 0.9 | 0.7 |
| Inositol, myo- | 1.2 | 0.6 | 0.7 | 1.2 |
| Sucrose | 1.1 | 1.1 | 0.6 | 0.6 |
| Quinic acid | 0.8 | 0.6 | 0.6 | 0.8 |
| D-Ketohexose sugars | 1.2 | 0.9 | 0.8 | 1.2 |
| D-Aldohexoses | 1.0 | 0.9 | 1.0 | 1.1 |
| Erythronic acid | 0.8 | 0.7 | 0.9 | 1.0 |
| Hexadecanoic acid | 0.8 | 1.1 | 1.5 | 1.2 |
| Octadecanoic acid | 0.8 | 1.0 | 1.0 | 0.8 |
| Malic acid | 1.6 | 1.3 | 1.2 | 1.5 |
| Shikimic acid | n.d. | 0.5 | n.d. | n.d. |
| Gallic acid | 1.0 | 1.6 | 0.6 | 0.4 |
| Catechin | 0.5 | 0.5 | 0.3 | 0.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mevy, J.-P.; Loriod, B.; Liu, X.; Corre, E.; Torres, M.; Büttner, M.; Haguenauer, A.; Reiter, I.M.; Fernandez, C.; Gauquelin, T. Response of Downy Oak (Quercus pubescens Willd.) to Climate Change: Transcriptome Assembly, Differential Gene Analysis and Targeted Metabolomics. Plants 2020, 9, 1149. https://doi.org/10.3390/plants9091149
Mevy J-P, Loriod B, Liu X, Corre E, Torres M, Büttner M, Haguenauer A, Reiter IM, Fernandez C, Gauquelin T. Response of Downy Oak (Quercus pubescens Willd.) to Climate Change: Transcriptome Assembly, Differential Gene Analysis and Targeted Metabolomics. Plants. 2020; 9(9):1149. https://doi.org/10.3390/plants9091149
Chicago/Turabian StyleMevy, Jean-Philippe, Beatrice Loriod, Xi Liu, Erwan Corre, Magali Torres, Michael Büttner, Anne Haguenauer, Ilja Marco Reiter, Catherine Fernandez, and Thierry Gauquelin. 2020. "Response of Downy Oak (Quercus pubescens Willd.) to Climate Change: Transcriptome Assembly, Differential Gene Analysis and Targeted Metabolomics" Plants 9, no. 9: 1149. https://doi.org/10.3390/plants9091149
APA StyleMevy, J.-P., Loriod, B., Liu, X., Corre, E., Torres, M., Büttner, M., Haguenauer, A., Reiter, I. M., Fernandez, C., & Gauquelin, T. (2020). Response of Downy Oak (Quercus pubescens Willd.) to Climate Change: Transcriptome Assembly, Differential Gene Analysis and Targeted Metabolomics. Plants, 9(9), 1149. https://doi.org/10.3390/plants9091149