
Applied Barcoding: The Practicalities of DNA Testing for Herbals


Abstract
:
1. Introduction
2. Results
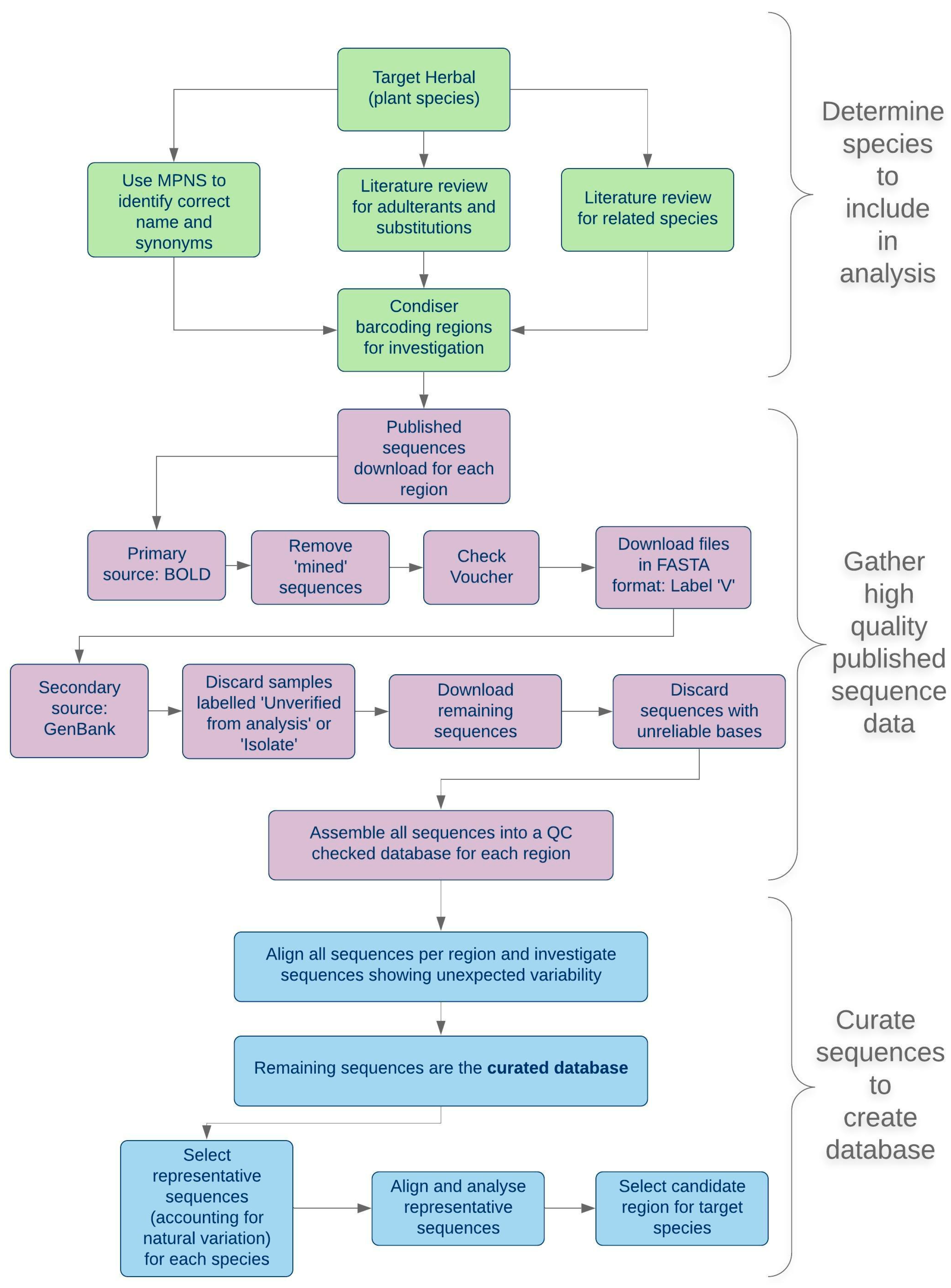
2.1. A Scheme for Applied Barcoding
2.1.1. Nomenclature, Taxonomy and Adulteration
2.1.2. Selection of a Panel of Potential Adulterant Hypericum Species
2.1.3. Public Databases of DNA Barcode Sequences
2.1.4. Reference Samples for Pilot Studies and Assay Standards
2.1.5. Test Samples for Barcode Assay Validation
2.2. The nrITS Barcode Region
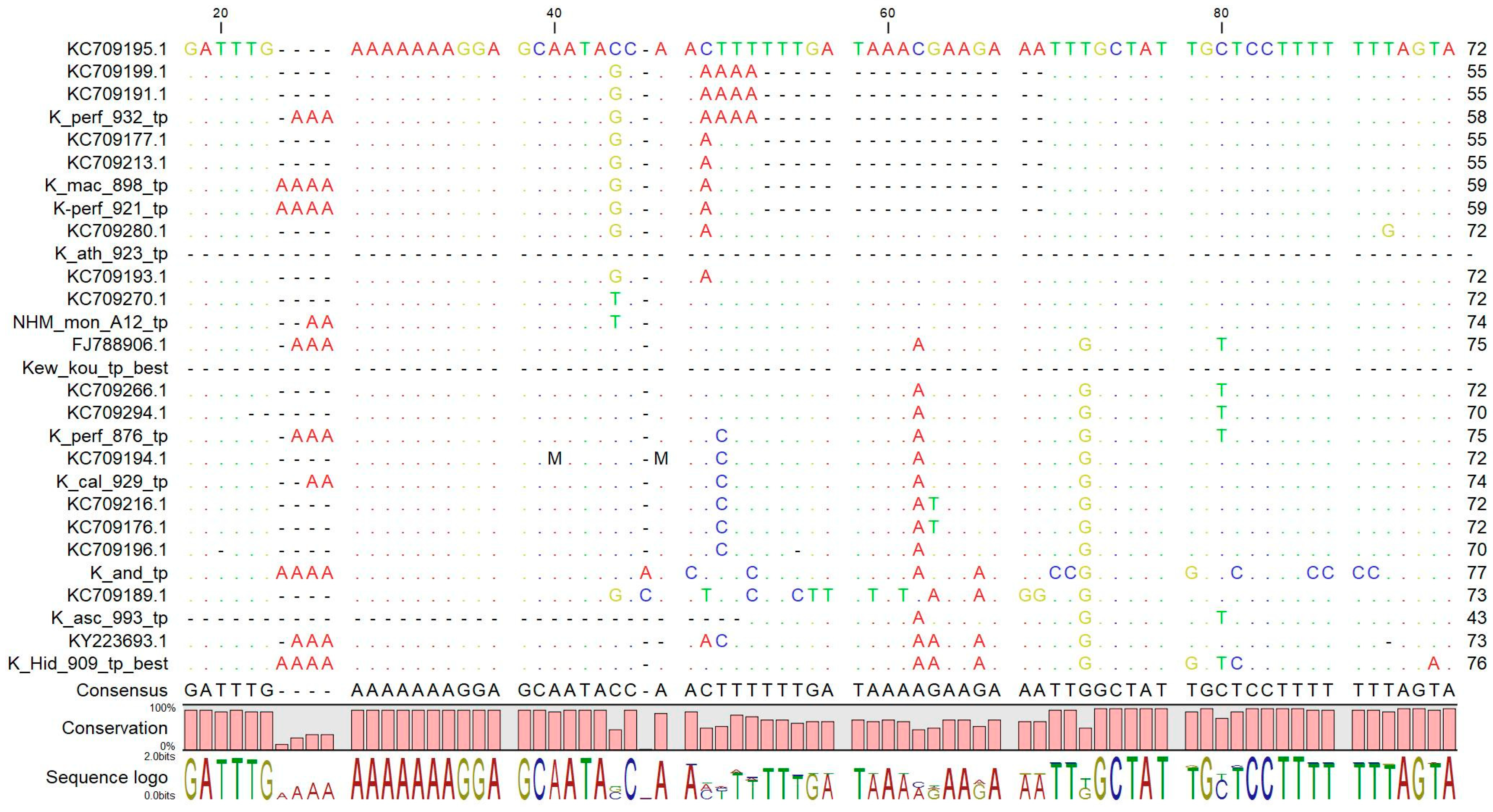
2.2.1. A Curated Database of ITS Sequences
2.2.2. ITS Barcoding of Reference Samples
2.3. The ITS2 Barcode Region
2.3.1. ITS2 Sequences
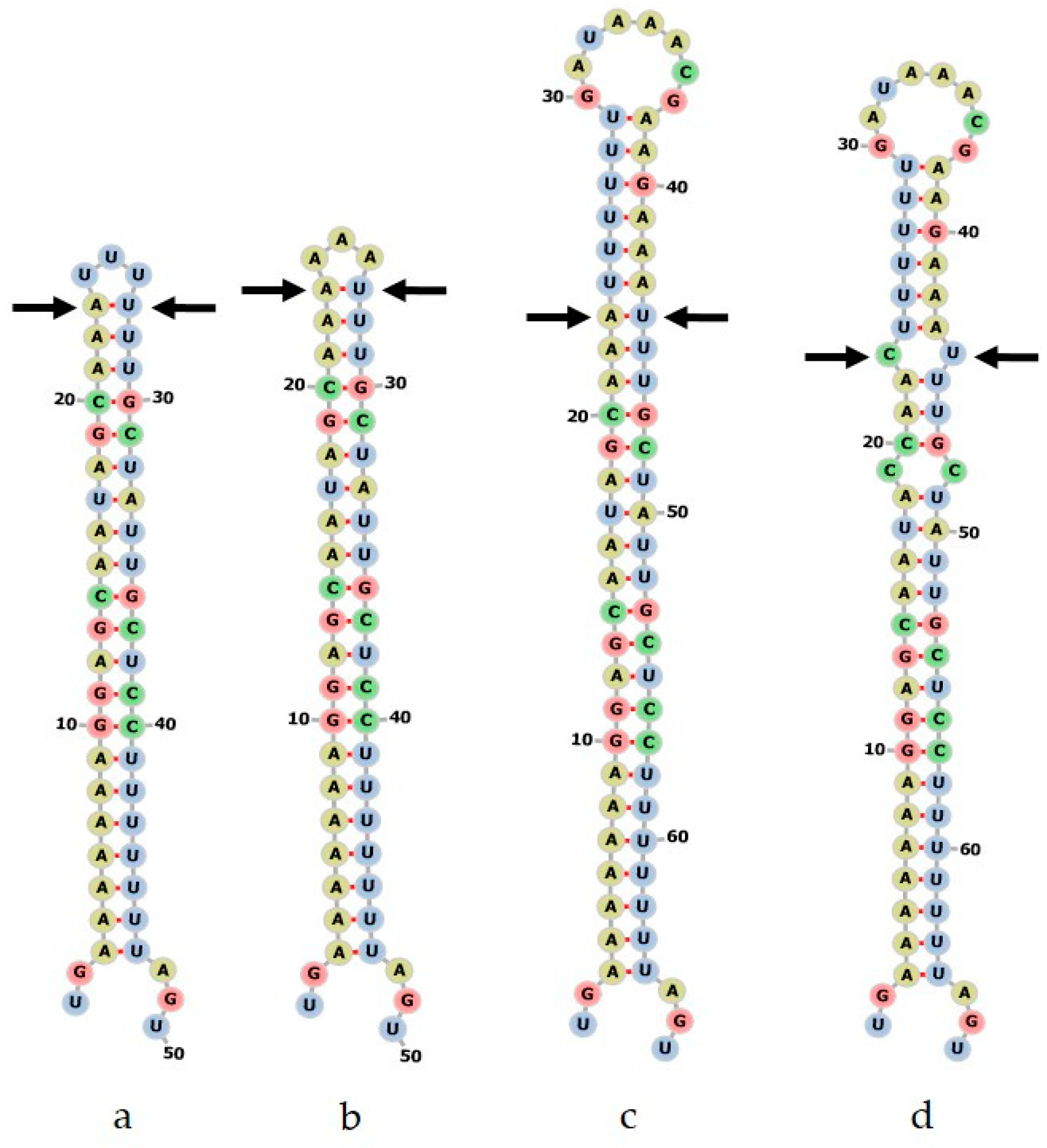
2.3.2. Secondary Structure of ITS2 Sequences
2.4. The rbcLa Barcode Region
2.4.1. A Curated Database of rbcLa Sequences
2.4.2. Haplotype Mapping of Reference Samples
2.5. The matK Barcode Region
2.6. The psbA-trnH Barcode Region
2.6.1. A Curated Database of psbA-trnH Sequences
2.6.2. Reference and Test psbA-trnH Sequences
2.7. Choosing the Optimal Barcode
2.7.1. ITS Barcoding of Test Samples
2.7.2. Supporting Evidence from rbcL Haplotype and psbA-trnH Stem-Loop
3. Discussion
3.1. Applied Barcoding for Herbals
3.2. Choosing a Barcode for Species Identification
3.2.1. Defining a Reference Barcode Sequence: Sensitivity and Specificity
3.2.2. Matching a Reference Barcode Sequence: Accuracy and Precision
3.3. Recommended Barcodes for H. perforatum Authentication
3.4. Current Trends in Applied Barcoding
4. Materials and Methods
4.1. A panel of Potential Adulterant Hypericum Species
- Close relatives of H. perforatum;
- Common in commercial trade as ornamental or medicinal plants;
- Reported as adulterants of H. perforatum.
4.2. Plant and DNA Materials
4.3. DNA Extraction and Amplification of DNA Barcode Regions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Primer Sequence | Program |
|---|---|---|
| ITS | ITS1- TCCGTAGGTGAACCTGCGG ITS4-TCCTCCGCTTATTGATATGC [101] | 7 min at 95 °C initial denaturation step, 30 cycles consisting of 1 min at 95 °C, 30 s at 60 °C and 1 min at 72 °C, final extension period of 7 min at 72 °C. |
| trnH-psbA | trnHf_05-CGCGCATGGTGGATTCACAATCC psbA3_f–GTTATGCATGAACGTAATGCTC [102] | 5 min at 95 °C initial denaturation step, 35 cycles consisting of 1 min at 95 °C, 30 s at touchdown temperature and 1 min at 72 °C, final extension period of 7 min at 72 °C. Touchdown temperature began at 58 °C, reduced by 1 °C per cycle until 48 °C, then continued at 48 °C for the remainder of the program. |
| rbcL | rbcLa_f -ATGTCACCACAAACAGAAAC rbcLa_rev-GTAAAATCAAGTCCACCRCG [103] | 5 min 95 °C initial denaturation step, 35 cycles consisting of 30 s at 95 °C, 20 s at 52 °C and 50 s at 72 °C, with a final extension period of 5 min at 72 °C. |
| matK | 390F-CGATCTATTCATTCAATATTTC 1326R–TCTAGCACACGAAAGTCGAAGT 2.1-CCTATCCATCTGGAAATCTTAG 2.1a–ATCCATCTGGAAATCTTAGTTC X F- TAATTTACGATCAATTCATTC 5-GTTCTAGCACAAGAAAGTCG 3.2–CTTCCTCTGTAAAGAATTC 3F_KIM f-CGTACAGTACTTTTGTGTTTACGAG 1R_KIM r -ACCCAGTCCATCTGGAAATCTTGGTTC [104,105,106] | Initial “touch-up” program, 5 min 94 °C initial denaturation step, 5 cycles consisting of 30 s at 94 °C, 40 s at 44 °C and 40 s at 72 °C, followed by 30 cycles consisting of 30 s at 94 °C, 40 s at 46 °C and 40 s at 72 °C, with a final extension period of 3 min at 72 °C. The second amplification contained 2 μL of the initial PCR product diluted 1:200 as the DNA template. Second matK program: 5 min 94 °C initial denaturation step, 35 cycles consisting of 30 s at 95 °C, 20 s at 46 °C and 40 s at 72 °C, with a final extension period of 3 min at 72 °C. |
4.4. DNA Sequence Analysis of Barcode Amplicons
4.5. Computer Analysis of Barcode Regions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Parveen, I.; Gafner, S.; Techen, N.; Murch, S.J.; Khan, I.A. DNA Barcoding for the identification of botanicals in herbal medicine and dietary supplements: Strengths and limitations. Planta Medica 2016, 82, 1225–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Techen, N.; Parveen, I.; Pan, Z.; Khan, I.A. DNA barcoding of medicinal plant material for identification. Curr. Opin. Biotechnol. 2014, 25, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Kumar, A.; Nagireddy, A.; Mani, D.N.; Shukla, A.K.; Tiwari, R.; Sundaresan, V. DNA barcoding: An efficient tool to overcome authentication challenges in the herbal market. Plant Biotechnol. J. 2016, 14, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Pang, X.; Liao, B.; Yao, H.; Song, J.; Chen, S. An authenticity survey of herbal medicines from markets in China using DNA barcoding. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pawar, R.S.; Handy, S.M.; Cheng, R.; Shyong, N.; Grundel, E. Assessment of the authenticity of herbal dietary supplements: Comparison of chemical and DNA barcoding methods. Planta Medica 2017, 83. [Google Scholar] [CrossRef] [Green Version]
- Raclariu, A.; Heinrich, M.; Ichim, M.; Boer, H. Benefits and Limitations of DNA Barcoding and Metabarcoding in Herbal Product Authentication. Phytochem. Anal. 2017. [Google Scholar] [CrossRef]
- Sgamma, T.; Lockie-Williams, C.; Kreuzer, M.; Williams, S.; Scheyhing, U.; Koch, E.; Slater, A.; Howard, C.; Mishra, P.; Kumar, A.; et al. DNA Barcoding for industrial quality assurance. Planta Medica 2017, 14, 8–21. [Google Scholar] [CrossRef] [Green Version]
- Ivanova, N.V. Authentication of herbal supplements using next-generation sequencing. PLoS ONE 2016, 11, e0156426. [Google Scholar] [CrossRef] [PubMed]
- Moraes, D.F.; Still, D.W.; Lum, M.R.; Hirsch, A.M. DNA-based authentication of botanicals and plant-derived dietary supplements: Where have we been and where are we going? Planta Medica 2015, 81, 687–695. [Google Scholar] [CrossRef] [Green Version]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Ostell, J.; Pruitt, K.D.; Sayers, E.W. GenBank. Nucleic Acids Res. 2018, 46, D41–D47. [Google Scholar] [CrossRef] [Green Version]
- Crockett, S.L.; Douglas, A.W.; Scheffler, B.E.; Khan, I.A. Genetic profiling of Hypericum (St. John’s wort) species by nuclear ribosomal ITS sequence analysis. Planta Medica 2004, 70, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Howard, C.; Hill, E.; Kreuzer, M.; Mali, P.; Masiero, E.; Slater, A.; Sgamma, T. DNA Authentication of St John’s wort (Hypericum perforatum L.) commercial products targeting the ITS region. Genes 2019, 10, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, C.; Bremner, P.D.; Fowler, M.R.; Isodo, B.; Scott, N.W.; Slater, A. Molecular identification of Hypericum perforatum by PCR amplification of the ITS and 5.8S rDNA region. Planta Medica 2009, 75, 864–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, C.; Socratous, E.; Williams, S.; Graham, E.; Fowler, M.R.; Scott, N.W.; Bremner, P.D.; Slater, A. PlantID - DNA-based identification of multiple medicinal plants in complex mixtures. Chin. Med. 2012, 7, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, J.; Campos, B.; Amaral, J.S.; Nunes, E.M.; Oliveira, B.M.; Mafra, I. HRM analysis targeting ITS1 and matK loci as potential DNA mini-barcodes for the authentication of Hypericum perforatum and Hypericum androsaemum in herbal infusions. Food Control 2016, 61, 105–114. [Google Scholar] [CrossRef]
- Raclariu, A.C.; Paltinean, R.; Vlase, L.; Labarre, A.; Manzanilla, V.; Ichim, M.C.; Crisan, G.; Brysting, A.K.; De Boer, H. Comparative authentication of Hypericum perforatum herbal products using DNA metabarcoding, TLC and HPLC-MS. Sci. Rep. 2017, 7, 1291. [Google Scholar] [CrossRef]
- Chase, M.; Cowan, R.; Hollingsworth, P. A proposal for a standardised protocol to barcode all land plants. Taxon 2007, 56, 295–299. [Google Scholar] [CrossRef]
- Hollingsworth, P.M.; Graham, S.W.; Little, D.P. Choosing and using a plant DNA barcode. PLoS ONE 2011. [Google Scholar] [CrossRef]
- Hollingsworth, M.L.; Clark, A.; Forrest, L.L.; Richardson, J.; Pennington, R.; Long, D.G.; Cowan, R.; Chase, M.W.; Gaudeul, M.; Hollingsworth, P.M. Selecting barcoding loci for plants: Evaluation of seven candidate loci with species level sampling in three divergent groups of land plants. Mol. Ecol. Resour. 2009, 9. [Google Scholar] [CrossRef]
- Banchi, E.; Ametrano, C.G.; Greco, S.; Stanković, D.; Muggia, L.; Pallavicini, A. PLANiTS: A curated sequence reference dataset for plant ITS DNA metabarcoding. Database 2020, 2020. [Google Scholar] [CrossRef] [Green Version]
- Pentinsaari, M.; Ratnasingham, S.; Miller, S.E.; Hebert, P.D.N. BOLD and GenBank revisited—Do identification errors arise in the lab or in the sequence libraries? PLoS ONE 2020, 15, e0231814. [Google Scholar] [CrossRef] [PubMed]
- Carine, M.A.; Christenhusz, M.J.M. About this volume: The monograph of Hypericum by Norman Robson. Phytotaxa 2013, 4, 1–4. [Google Scholar] [CrossRef]
- Nürk, N.M.; Crockett, S.L. Morphological and Phytochemical Diversity among Hypericum Species of the Mediterranean Basin. Med. Aromat. Plant Sci. Biotechnol. 2011, 5, 14–28. [Google Scholar] [PubMed]
- Crockett, S.L.; Robson, N.K. Taxonomy and chemotaxonomy of the Genus hypericum. Med. Aromat. Plant Sci. Biotechnol. 2011, 5, 1–13. [Google Scholar]
- Nürk, N.M.; Madriñán, S.; Carine, M.A.; Chase, M.W.; Blattner, F.R. Molecular phylogenetics and morphological evolution of St. John’s wort (Hypericum; Hypericaceae). Mol. Phylogen. Evol. 2013, 66, 1–16. [Google Scholar] [CrossRef]
- Meseguer, A.; Aldasoro, J.; Sanmartín, I. Bayesian inference of phylogeny, morphology and range evolution reveals a complex evolutionary history in St. John’s wort (Hypericum). Mol. Phylogen. Evol. 2013, 67, 379–403. [Google Scholar] [CrossRef]
- Pilepić, K.H.; Balić, M.; Blažina, N. Estimation of phylogenetic relationships among some Hypericum (Hypericaceae) species using internal transcribed spacer sequences. Plant Biosyst. Int. J. Deal. Asp. Plant Biol. 2011, 145, 81–87. [Google Scholar] [CrossRef]
- ROBSON, N.K.B. And then came molecular phylogenetics—Reactions to a monographic study of Hypericum (Hypericaceae). Phytotaxa 2016, 255, 181–198. [Google Scholar] [CrossRef]
- Robson, N. Studies in the genus Hypericum L. (Guttiferae) 4(2). Section 9. Hypericum sensu lato (part 2): Subsection 1. Hypericum series 1. Hypericum. Bull. Nat. Hist. Mus. Bot. Ser. 2002, 32. [Google Scholar] [CrossRef]
- Brutovská, R.; Čellárová, E.; Schubert, I. Cytogenetic characterization of three Hypericum species by in situ hybridization. Theor. Appl. Genet. 2000, 101, 46–50. [Google Scholar] [CrossRef]
- McCutcheon, A. Adulteration of Hypericum perforatum. Bot. Adulterants Bull. 2017, 1–9. [Google Scholar]
- Dauncey, E.; Edwards, S. Kew’s Medicinal Plant Names Services (MPNS) enable effective information retrieval and communication. Planta Medica 2015, 81. [Google Scholar] [CrossRef]
- Dauncey, E.; Irving, J.; Allkin, R. A review of issues of nomenclature and taxonomy of Hypericum perforatum L. and Kew’s Medicinal Plant Names Services. J. Pharm. Pharmacol. 2019, 71, 4–14. [Google Scholar] [CrossRef] [Green Version]
- Mitra, S.; Kannan, R. A note on unintentional adulterations in Ayurvedic herbs. Ethnobot. Leaflets 2007, 11, 11–15. [Google Scholar]
- Huck-Pezzei, V.; Bittner, L.; Pallua, J.; Sonderegger, H.; Abel, G.; Popp, M.; Bonn, G.; Huck, C. A chromatographic and spectroscopic analytical platform for the characterization of St John’s wort extract adulterations. Anal. Methods 2012, 5. [Google Scholar] [CrossRef]
- Frommenwiler, D.A.A.; Reich, E.; Sudberg, S.; Sharaf, M.H.; Bzhelyansky, A.; Lucas, B. St. John’s wort versus counterfeit St. John’s wort: An HPTLC study. J. AOAC Int. 2016, 99, 1204–1212. [Google Scholar] [CrossRef]
- Ratnasingham, S.; Hebert, P.D. Bold: The barcode of life data system (http://www.barcodinglife.org). Mol. Ecol. Notes 2007. [Google Scholar] [CrossRef] [Green Version]
- Hodkinson, T.R.; Waldren, S.; Parnell, J.A.N.; Kelleher, C.T.; Salamin, K.; Salamin, N. DNA banking for plant breeding, biotechnology and biodiversity evaluation. J. Plant Res. 2007, 120, 17–29. [Google Scholar] [CrossRef]
- Gemeinholzer, B.; Dröge, G.; Zetzsche, H.; Haszprunar, G.; Klenk, H.-P.; Güntsch, A.; Berendsohn, W.G.; Wägele, J.-W. The DNA bank network: The start from a german initiative. Biopreserv. Biobank 2011, 9, 51–55. [Google Scholar] [CrossRef]
- Droege, G.; Barker, K.; Astrin, J.J.; Bartels, P.; Butler, C.; Cantrill, D.; Coddington, J.; Forest, F.; Gemeinholzer, B.; Hobern, D.; et al. The Global Genome Biodiversity Network (GGBN) data portal. Nucleic Acids Res. 2014, 42, D607–D612. [Google Scholar] [CrossRef] [Green Version]
- Kazi, T.; Hussain, N.; Bremner, P.; Slater, A.; Howard, C. The application of a DNA-based identification technique to over-the-counter herbal medicines. Fitoterapia 2013, 87, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Pang, X.; Song, J.; Shi, L.; Yao, H.; Han, J.; Leon, C. A renaissance in herbal medicine identification: From morphology to DNA. Biotechnol. Adv. 2014, 32, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yao, H.; Han, J.; Liu, C.; Song, J.; Shi, L.; Zhu, Y.; Ma, X.; Gao, T.; Pang, X.; et al. Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species. PLoS ONE 2010, 5, e8613. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Song, J.; Liu, C.; Luo, K.; Han, J.; Li, Y.; Pang, X.; Xu, H.; Zhu, Y.; Xiao, P.; et al. Use of ITS2 region as the universal DNA barcode for plants and animals. PLoS ONE 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Yuan, Y.; Yang, S.; Huang, J.; Huang, L. ITS2 secondary structure improves discrimination between medicinal “Mu Tong” species when using DNA barcoding. PLoS ONE 2015, 10, e0131185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koetschan, C.; Hackl, T.; Müller, T.; Wolf, M.; Förster, F.; Schultz, J. ITS2 Database IV: Interactive taxon sampling for internal transcribed spacer 2 based phylogenies. Mol. Phylogenet. Evol. 2012. [Google Scholar] [CrossRef]
- Wolf, M.; Koetschan, C.; Müller, T. ITS2, 18S, 16S or any other RNA—Simply aligning sequences and their individual secondary structures simultaneously by an automatic approach. Gene 2014, 546, 145–149. [Google Scholar] [CrossRef]
- Schultz, J.; Wolf, M. ITS2 sequence–structure analysis in phylogenetics: A how-to manual for molecular systematics. Mol. Phylogenet. Evol. 2009. [Google Scholar] [CrossRef]
- Müller, T.; Philippi, N.; Dandekar, T.; Schultz, J.; Wolf, M. Distinguishing species. RNA 2007, 13, 1469–1472. [Google Scholar] [CrossRef] [Green Version]
- Wolf, M.; Chen, S.; Song, J.; Ankenbrand, M.; Müller, T. Compensatory base changes in ITS2 secondary structures correlate with the biological species concept despite intragenomic variability in ITS2 sequences—A proof of concept. PLoS ONE 2013, 8, e66726. [Google Scholar] [CrossRef]
- Seibel, P.N.; Müller, T.; Dandekar, T.; Wolf, M. Synchronous visual analysis and editing of RNA sequence and secondary structure alignments using 4SALE. BMC Res. Notes 2008, 1, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Štorchová, H.; Olson, M. The architecture of the chloroplast psbA-trnH non-coding region in angiosperms. Plant Syst. Evol. 2007, 268, 235–256. [Google Scholar] [CrossRef]
- Degtjareva, G.; Logacheva, M.; Samigullin, T.; Terentieva, E.; Valiejo-Roman, C. Organization of chloroplast psbA-trnH intergenic spacer in dicotyledonous angiosperms of the family umbelliferae. Biochemistry (Moscow) 2012, 77, 1056–1064. [Google Scholar] [CrossRef]
- Deoxyribonucleic Acid (DNA) Based identification techniques for herbal drugs. In British Pharmacopoeia; British Pharmacopoeia Commission: London, UK, 2017; Appendix XI V.
- DNA barcoding as a tool for botanical identification. In British Pharmacopoeia; British Pharmacopoeia Supplementary Chapter SC VII D; British Pharmacopoeia Commission: London, UK, 2017.
- Radusiene, J.; Judzentiene, A.; Bernotiene, G. Essential oil composition and variability of Hypericum perforatum L. growing in Lithuania. Biochem. Syst. Ecol. 2005, 33, 113–124. [Google Scholar] [CrossRef]
- Sgamma, T.; Masiero, E.; Mali, P.; Mahat, M.; Slater, A. Sequence-specific detection of aristolochia DNA—A simple test for contamination of herbal products. Front. Plant Sci. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Little, D. Authentication of Ginkgo biloba herbal dietary supplements using DNA barcoding. Genome 2014. [Google Scholar] [CrossRef] [Green Version]
- Hajibabaei, M.; McKenna, C. DNA mini-barcodes. In Methods in Molecular Biology; Humana Press: Clifton, NJ, USA, 2012; pp. 339–353. [Google Scholar] [CrossRef]
- Summerbell, R.; Lévesque, C.; Seifert, K.; Bovers, M.; Fell, J.; Diaz, M.; Boekhout, T.; de Hoog, G.; Stalpers, J.; Crous, P. Microcoding: The second step in DNA barcoding. Phil. Transact. R. Soc. London Ser. B Biol. Sci. 2005, 360, 1897–1903. [Google Scholar] [CrossRef] [Green Version]
- Newmaster, S.G.; Shanmughanandhan, D.; Kesanakurti, P.; Shehata, H.; Faller, A.; Noce, I.; Lee, J.; Rudzinski, P.; Lu, Z.; Zhang, Y.; et al. Recommendations for validation of real-time PCR methods for molecular diagnostic identification of botanicals. J. AOAC Int. 2019. [Google Scholar] [CrossRef]
- Masiero, E.; Banik, D.; Abson, J.; Greene, P.; Slater, A.; Sgamma, T. Genus-specific real-time PCR and HRM assays to distinguish liriope from ophiopogon samples. Plants 2017, 6, 53. [Google Scholar] [CrossRef] [Green Version]
- Koch, M.A.; Scheriau, C.; Betzin, A.; Hohmann, N.; Sharbel, T.F. Evolution of cryptic gene pools in Hypericum perforatum: The influence of reproductive system and gene flow. Ann. Bot. 2013, 111, 1083–1094. [Google Scholar] [CrossRef] [Green Version]
- Little, D.P.; Stevenson, D. A comparison of algorithms for the identification of specimens using DNA barcodes: Examples from gymnosperms. Cladistics 2007, 23. [Google Scholar] [CrossRef]
- Little, D.P. DNA Barcode sequence identification incorporating taxonomic hierarchy and within taxon variability. PLoS ONE 2011, 6, e20552. [Google Scholar] [CrossRef]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Individual sequences in large sets of gene sequences may be distinguished efficiently by combinations of shared sub-sequences. BMC Bioinform. 2005, 6, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DasGupta, B.; Konwar, K.; Mandoiu, I.; Shvartsman, A. DNA-BAR: Distinguisher selection for DNA barcoding. Bioinformatics 2005, 21, 3424–3426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rash, S.; Gusfield, D. String barcoding: Uncovering optimal virus signatures. In Proceedings of the 6th Annual International Conference on Computational Biology, Washington, DC, USA, 18–21 April 2002; pp. 254–261. [Google Scholar] [CrossRef]
- Liu, C.; Liang, D.; Gao, T.; Pang, X.; Song, J.; Yao, H.; Han, J.; Liu, Z.; Guan, X.; Jiang, K.; et al. PTIGS-IdIt, a system for species identification by DNA sequences of the psbA-trnH intergenic spacer region. BMC Bioinform. 2011, 12 (Suppl. 13), S4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitlock, A.B.; Hale, A.M.; Groff, P.A. Intraspecific inversions pose a challenge for the trnH-psbA plant DNA barcode. PLoS ONE 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Ledesma, P.; Bárcenas, R.T. Phylogenetic utility of the trnH–psbA IGR and stem-loop diversity of the 3′ UTR in Cactaceae (Caryophyllales). Plant Syst. Evol. 2017, 303, 299–315. [Google Scholar] [CrossRef]
- Bain, J.F.; Jansen, R.K. A chloroplast DNA hairpin structure provides useful phylogenetic data within tribe Senecioneae (Asteraceae). Can. J. Bot. 2006, 84, 862–868. [Google Scholar] [CrossRef]
- Molins, M.; Corral, J.M.; Aliyu, O.; Koch, M.A.; Betzin, A.; Maron, J.L.; Sharbel, T.F. Biogeographic variation in genetic variability, apomixis expression and ploidy of St. John’s wort (Hypericum perforatum) across its native and introduced range. Ann. Bot. 2014, 113, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Barcaccia, G.; Arzenton, F.; Sharbel, T.; Varotto, S.; Parrini, P.; Lucchin, M. Genetic diversity and reproductive biology in ecotypes of the facultative apomict Hypericum perforatum L. Heredity 2006, 96, 322–334. [Google Scholar] [CrossRef]
- Murch, S.J.; Saxena, P.K. St. John’s wort (Hypericum perforatum L.): Challenges and strategies for production of chemically-consistent plants. Can J. Plant Sci. 2006, 86, 765–771. [Google Scholar] [CrossRef]
- Pank, F.; Matzk, F.; Kästner, U.; Blüthner, W.; de Garcia, F.E.; Meister, A.; Ryschka, U.; Schumann, G. Reproductive diversity and strategies for breeding in St. John’s wort (Hypericum perforatum L.). Euphytica 2003, 134, 77–84. [Google Scholar] [CrossRef]
- Matzk, F.; Hammer, K.; Schubert, I. Coevolution of apomixis and genome size within the genus Hypericum. Sex. Plant Reprod. 2003, 16, 51–58. [Google Scholar] [CrossRef]
- Galla, G.; Barcaccia, G.; Schallau, A.; Molins, P.M.; Bäumlein, H.; Sharbel, T. The cytohistological basis of apospory in Hypericum perforatum L. Sex. Plant Reprod. 2011, 24, 47–61. [Google Scholar] [CrossRef]
- Tsitrone, A.; Kirkpatrick, M.; Levin, D.A. A model for chloroplast capture. Evolution 2003, 57, 1776–1782. [Google Scholar] [CrossRef] [PubMed]
- Mariac, C.; Scarcelli, N.; Pouzadou, J.; Barnaud, A.; Billot, C.; Faye, A.; Kougbeadjo, A.; Maillol, V.; Martin, G.; Sabot, F.; et al. Cost-effective enrichment hybridization capture of chloroplast genomes at deep multiplexing levels for population genetics and phylogeography studies. Mol. Ecol. Resour. 2014. [Google Scholar] [CrossRef]
- Stegemann, S.; Keuthe, M.; Greiner, S.; Bock, R. Horizontal transfer of chloroplast genomes between plant species. Proc Natil. Acad. Sci USA 2012, 109, 2434–2438. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.-B.; Campbell, C.S.; Hong, D.-Y.; Wen, J. Phylogenetic relationships and chloroplast capture in the Amelanchier-Malacomeles-Peraphyllum clade (Maleae, Rosaceae): Evidence from chloroplast genome and nuclear ribosomal DNA data using genome skimming. Mol. Phylogenet. Evol. 2020, 147, 106784. [Google Scholar] [CrossRef]
- Milne, R.I.; Davies, C.; Prickett, R.; Inns, L.H.; Chamberlain, D.F. Phylogeny of Rhododendron subgenus Hymenanthes based on chloroplast DNA markers: Between-lineage hybridisation during adaptive radiation? Plant Syst. Evol. 2010, 285. [Google Scholar] [CrossRef]
- Kreuzer, M.; Howard, C.; Adhikari, B.; Pendry, C.A.; Hawkins, J.A. Phylogenomic approaches to DNA barcoding of herbal medicines: Developing clade-specific diagnostic characters for berberis. Front. Plant Sci. 2019, 10. [Google Scholar] [CrossRef]
- Vaughn, J.N.; Chaluvadi, S.R.; Tushar; Rangan, L.; Bennetzen, J.L. Whole plastome sequences from five ginger species facilitate marker development and define limits to barcode methodology. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, Y.; Henry, R.J.; Rossetto, M.; Wang, Y.; Chen, S. Plant DNA barcoding: From gene to genome. Biol. Rev. 2014. [Google Scholar] [CrossRef] [PubMed]
- Zhokhova, E.; Rodionov, A.; Povydysh, M.; Goncharov, Y.M.; Protasova, Y.A.; Yakovlev, G. Current state and prospects of DNA barcoding and DNA fingerprinting in the analysis of the quality of plant raw materials and plant-derived drugs. Biol. Bull. Rev. 2019, 9, 301–314. [Google Scholar] [CrossRef]
- Seethapathy, G.; Raclariu-Manolica, A.-C.; Anmarkrud, J.; Wangensteen, H.; de Boer, H.J. DNA metabarcoding authentication of ayurvedic herbal products on the European market Raises concerns of quality and fidelity. Front. Plant Sci. 2019, 10, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raclariu, A.; Ţebrencu, C.; Ichim, M.; Ciupercǎ, O.; Brysting, A.; de Boer, H. What’s in the box? Authentication of Echinacea herbal products using DNA metabarcoding and HPTLC. Phytomedicine 2018. [Google Scholar] [CrossRef]
- Omelchenko, D.O.; Speranskaya, A.S.; Ayginin, A.A.; Khafizov, K.; Krinitsina, A.A.; Fedotova, A.V.; Pozdyshev, D.V.; Shtratnikova, V.Y.; Kupriyanova, E.V.; Shipulin, G.A.; et al. Improved protocols of ITS1-based metabarcoding and their application in the analysis of plant-containing products. Genes 2019, 10, 122. [Google Scholar] [CrossRef] [Green Version]
- Hoban, C.L.; Musgrave, I.F.; Coghlan, M.L.; Power, M.W.P.; Byard, R.W.; Nash, C.; Farrington, R.; Maker, G.; Crighton, E.; Trengove, R.; et al. Adulterants and contaminants in psychotropic herbal medicines detected with mass spectrometry and next-generation DNA sequencing. Pharm. Med. 2018, 32, 429–444. [Google Scholar] [CrossRef]
- Coghlan, M.L.; Maker, G.; Crighton, E.; Haile, J.; Murray, D.C.; White, N.E.; Byard, R.W.; Bellgard, M.I.; Mullaney, I.; Trengove, R.; et al. Combined DNA, toxicological and heavy metal analyses provides an auditing toolkit to improve pharmacovigilance of traditional Chinese medicine (TCM). Sci. Rep. 2015, 5, 17475. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Li, X.; Zhou, H.; Hu, H.; Zhang, H.; Sun, W.; Wang, Y.; Yao, H. Molecular identification of chinese materia medica and its adulterants using its2 and psba-trnh barcodes: A case study on rhizoma menispermi. Chin. Med. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Au, K.-Y.; Lam, H.; Cheng, L.; But, P.; Shaw, P.-C. Molecular identification and cytotoxicity study of herbal medicinal materials that are confused by Aristolochia herbs. Food Chem. 2014, 147, 332–339. [Google Scholar] [CrossRef]
- Wu, L.; Wang, B.; Zhao, M.; Liu, W.; Zhang, P.; Shi, Y.; Xiong, C.; Wang, P.; Sun, W.; Chen, S. Rapid Identification of officinal akebiae caulis and its toxic adulterant Aristolochiae manshuriensis caulis (Aristolochia manshuriensis) By loop-mediated isothermal amplification. Front. Plant Sci. 2016, 7, 887. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Sun, W.; Wang, B.; Zhao, H.; Li, Y.; Cai, S.; Xiang, L.; Zhu, Y.; Yao, H.; Song, J.; et al. An integrated system for identifying the hidden assassins in traditional medicines containing aristolochic acids. Sci. Rep. 2015, 5, 11318. [Google Scholar] [CrossRef] [PubMed]
- Juul, S.; Izquierdo, F.; Hurst, A.; Dai, X.; Wright, A.; Kulesha, E.; Pettett, R.; Turner, D.J. What’s in my pot? Real-time species identification on the MinIONTM. BioRxiv 2015. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Accuracy of taxonomy prediction for 16S rRNA and fungal ITS sequences. PeerJ 2018, 6, e4652. [Google Scholar] [CrossRef] [PubMed]
- Staats, M.; Arulandhu, A.J.; Gravendeel, B.; Holst-Jensen, A.; Scholtens, I.; Peelen, T.; Prins, T.W.; Kok, E. Advances in DNA metabarcoding for food and wildlife forensic species identification. Anal. Bioanal. Chem. 2016, 408, 4615–4630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, C. The Development of Deoxyribonucleic Acid (DNA) Based Methods for the Identification and Authentication of Medicinal Plant Material. Ph.D. Thesis, De Montfort University, Leicester, UK, 2010. [Google Scholar]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J.W.; Innis, M.A.; Gelfand, D.H.; Sninsky, J.J.; White, T.J. Amplification and Direct Sequencing of Fungal Ribosomal Rna Genes for Phylogenetics; Academic Press Inc.: Cambridge, MA, USA, 1990. [Google Scholar]
- Kress, J.W.; Wurdack, K.J.; Zimmer, E.A.; Weigt, L.A.; Janzen, D.H. Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci. USA 2005, 102, 8369–8374. [Google Scholar] [CrossRef] [Green Version]
- Lahaye, R.; van der Bank, M.; Bogarin, D.; Warner, J.; Pupulin, F.; Gigot, G.; Maurin, O.; Duthoit, S.; Barraclough, T.G.; Savolainen, V. DNA barcoding the floras of biodiversity hotspots. Proc. Natl. Acad. Sci. USA 2008, 105, 2923–2928. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Ji, Y.; Choi, G.; Kang, Y.; Yang, S.; Moon, B. Molecular identification and phylogenetic analysis of important medicinal plant species in genus Paeonia based on rDNA-ITS, matK, and rbcL DNA barcode sequences. Genet. Mol. Res. GMR 2016, 15. [Google Scholar] [CrossRef]
- Fazekas, A.J.; Burgess, K.S.; Kesanakurti, P.R.; Graham, S.W.; Newmaster, S.G.; Husband, B.C.; Percy, D.M.; Hajibabaei, M.; Barrett, S.C. Multiple multilocus DNA barcodes from the plastid genome discriminate plant species equally well. PLoS ONE 2008, 3, e2802. [Google Scholar] [CrossRef] [Green Version]
- Chatrou, L.; Grayer, R.; Chase, M. Molecular phylogenetics of Caryophyllales based on nuclear 18S rDNA and plastid rbcL, atpB, and matK DNA sequences. Am. J. Bot. 2002, 89, 132–144. [Google Scholar] [CrossRef]



| BOLD | GenBank | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Species | rbcL | matK | trnH-psbA | ITS | ITS2 | rbcL | matK | trnH-psbA | ITS | ITS2 |
| H. acmosepalum | 1 | 1 | 5 | |||||||
| H. androsaemum | 5 (6) | 1 | 1 | 7 | 1 | 1 | 7 | 4 | ||
| H. ascyron | 1 (2) | 1 | 1 | 2 (4) | 5 | 3 | 1 | 16 | 3 | |
| H. athoum | 1 | 1 | 4 | |||||||
| H. attenuatum | 2 | 7 | ||||||||
| H. barbatum | 1 | 2 | ||||||||
| H. calycinum | 1 | 1 | 2 | 1 | 1 | 1 | 7 | |||
| H. crux-andreae | 2 | 1 | 2 | |||||||
| H. delphicum | 2 | 6 | ||||||||
| H. elegans | 1 | |||||||||
| H. hirsutum | 4 (5) | 1 | (1) | 1 (3) | 6 | 1 | 5 | 2 | ||
| H. japonicum | 2 | 3 | 9 | 5 | 10 | 7 | ||||
| H. kouytchense | 2 | 1 | 4 | |||||||
| H. maculatum | 7 (8) | (1) | 1 | 1 (9) | 11 | 3 | 14 | 3 | ||
| H. montanum | 2 (3) | 1 | 1 | 2 | 3 | 1 | 6 | 1 | ||
| H. olympicum | 1 | 2 | 9 | |||||||
| H. patulum | 2 | 1 | 8 | 2 | ||||||
| H. perforatum | 22 (40) | 3 (6) | (3) | (1) | 9 (27) | 38 | 11 | 7 | 40 | 17 |
| H. tetrapterum | 2 (4) | 1 (2) | 4 | 3 | 2 | 2 | 5 | 2 | ||
| H. undulatum | 3 (4) | 1 | 1 | 4 | 1 | 1 | 9 | 4 | ||
| Species | Section | Robson Clade [24] | Meseguer Clade [26] | CBC/Perf |
|---|---|---|---|---|
| H. delphicum | Adenosepalum | 27 | E | 0 |
| H. montanum | Adenosepalum | 27 | E | 0 |
| H. barbatum | Drosocarpium | 13 | E | 0 |
| H. attenuatum | Hypericum | 9 | E | 0 |
| H. maculatum | Hypericum | 9 | E | 0 |
| H. perforatum | Hypericum | 9 | E | 0 |
| H. tetrapterum | Hypericum | 9 | E | 0 |
| H. undulatum | Hypericum | 9 | E | 0 |
| H. hirsutum | Taenocarpium | 18 | E | 0 |
| H. olympicum | Oligostema | 14 | E | 0 |
| H. acmosepalum | Ascyreia | 3 | D | 1 |
| H. calycinum | Ascyreia | 3 | D | 1 |
| H. kouytchense | Ascyreia | 3 | D | 1 |
| H. patulum | Ascyreia | 3 | D | 1 |
| H. androsaemum | Androsaemum | 5 | C | 2 |
| H. crux-andreae | Myriandra | 20 | B | 4 |
| H. ascyron | Roscyna | 7 | D | 5 |
| H. japonicum | Trignobrathys | 30 | B | 5 |
| Species | Type | 66 | 180 | 263 | 300 | 372 | 378 | 492 |
|---|---|---|---|---|---|---|---|---|
| Consensus | C | C | A | A | A | A | A | |
| H. perforatum | 1p | T | T | T | G | C | . | . |
| H. maculatum | 1 | . | T | . | G | C | . | . |
| H. tetrapterum | 1 | . | T | . | G | C | . | . |
| H. undulatum | 1 | . | T | . | G | C | . | . |
| H. montanum | 1 | . | T | . | G | C | . | . |
| H. hirsutum | 1 | . | . | . | G | C | . | . |
| H. androsaemum | 2 | . | . | . | . | . | G | T |
| H. ascyron | 2 | . | . | . | . | . | G | T |
| H. calycinum | 2 | . | . | . | . | . | G | T |
| H. crux-andreae | 2 | . | . | . | . | . | G | T |
| H. japonicum | 2 | . | . | . | . | . | G | T |
| Reference | Type | 66 | 180 | 263 | 300 | 372 | 378 | 492 |
|---|---|---|---|---|---|---|---|---|
| Consensus | C | C | A | A | A | A | A | |
| H. perforatum 13876 | 1p | T | T | T | G | C | . | . |
| H. delphicum 13938 | 1 | . | T | . | G | C | . | . |
| H. montanum A12F | 1 | . | T | . | G | C | G | . |
| H. kouytchense 13866 | 1–2 | . | . | . | . | . | . | . |
| H. patulum 13908 | 1–2 | . | . | . | . | . | . | . |
| H. perforatum 13921 | 1–2 | . | . | . | . | . | . | . |
| H. perforatum 13932 | 1–2 | . | . | . | . | . | . | . |
| H. androsaemum 13854 | 1–2 | . | . | . | . | . | . | . |
| H. ascyron 13993 | 2 | . | . | . | . | . | G | T |
| H. athoum 13923 | 2 | . | . | . | . | . | G | T |
| H. calycinum 13929 | 2 | . | . | . | . | . | G | T |
| H. maculatum 13896 | 2 | . | . | . | . | . | G | T |
| Sample No. | DNA No. | ITS Match to H. perforatum | trnH-psbA Stem | rbcL Type | ID |
|---|---|---|---|---|---|
| mac 01 | 007 | No | L | 1 | M |
| mac 02 | 011 | No | L | 1(p) | M |
| mac 03 | 012 | No | L | 1p | M |
| mac 04 | 018 | No | L | 1p | M |
| mac 05 | 030 | L | 1p | m | |
| mac 06 | 045 | No | L | 1-2 | m |
| mac 07 | 001 | L | 1 | m | |
| mac 08 | 031 | No | L | 1p | M |
| mac 09 | 027 | No | L | 1 | M |
| mac 10 | 022 | L | 1(p) | m | |
| mac 11 | 014 | S | 1p | p | |
| mac 12 | 034 | No | L | 1p | M |
| mac 13 | 025 | No | L | 1p | M |
| mac 14 | 019 | No | L | 1 | M |
| mac 15 | 036 | No | s | 1p | m |
| mac 16 | 023 | No | L | 1 | M |
| perf 01 | 005 | S | 1p | p | |
| perf 02 | 043 | Yes | S | 1-2 | P |
| perf 03 | 017 | No | Sa | 1 | U |
| perf 04 | 026 | Yes | S | 1p | P |
| perf 05 | 016 | S | 1p | p | |
| perf 06 | 038 | L | 1p | U | |
| perf 07 | 044 | Yes | S | 1p | P |
| perf 08 | 035 | Yes | S | 1 | P |
| perf 09 | 029 | No | S | 1p | U |
| perf 10 | 015 | S | 1p | p | |
| perf 11 | 032 | Yes | S | 1 | P |
| perf 12 | 041 | S | 1p | p | |
| perf 13 | 028 | No | Sa | 1p | U |
| perf 14 | 024 | S | 1p | p | |
| perf 15 | 013 | L | 1p | U | |
| perf 16 | 037 | Yes | S | 1p | P |
| perf 17 | 039 | Yes | S | 1p | P |
| perf 18 | 020 | Yes | Sa | 1p | P |
| perf 19 | 042 | Yes | Sa | 1p | P |
| perf 20 | 040 | Yes | Sa | 1p | P |
| perf 21 | 033 | Yes | S | 1p | P |
| perf 22 | 021 | L | 1(p) | U |
| Species | Attribution | Section | Rel | MPNS | Cult | Adult |
|---|---|---|---|---|---|---|
| H. acmosepalum | N.Robson | Ascyreia | - | Y | ||
| H. androsaemum | L. | Androsaemum | Y | Y | Y | |
| H. ascyron | L. | Roscyna | Y | Y | ||
| H. athoum | Boiss. & Orph | Adenosepalum | - | Y | ||
| H. attenuatum | Fisch. ex Choisy | Hypericum | Y | - | ||
| H. barbatum | Jacq. | Drosocarpium | - | Y | ||
| H. calycinum | L. | Ascyreia | Y | Y | ||
| H. crux-andreae | (L.) Crantz | Myriandra | Y | Y | ||
| H. delphicum | Boiss. & Heldr. | Adenosepalum | - | |||
| H. elegans | Stephan ex Willd. | Hypericum | Y | Y | ||
| H. hirsutum | L. | Taeniocarpium | - | Y | ||
| H. japonicum | Thunb. | Trigynobrathys | Y | Y | ||
| H. kouytchense | H.Lev | Ascyreia | - | Y | ||
| H. maculatum | Crantz | Hypericum | Y | Y | Y | |
| H. montanum | L. | Adenosepalum | Y | |||
| H. olympicum | L. | Oligostema | - | Y | ||
| H. patulum | Thunb. | Ascyreia | Y | Y | Y | |
| H. perforatum | L. | Hypericum | Y | Y | ||
| H. tetrapterum | Fr. | Hypericum | Y | Y | Y | Y |
| H. undulatum | Schousb. ex Willd. | Hypericum | Y | Y |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Howard, C.; Lockie-Williams, C.; Slater, A. Applied Barcoding: The Practicalities of DNA Testing for Herbals. Plants 2020, 9, 1150. https://doi.org/10.3390/plants9091150
Howard C, Lockie-Williams C, Slater A. Applied Barcoding: The Practicalities of DNA Testing for Herbals. Plants. 2020; 9(9):1150. https://doi.org/10.3390/plants9091150
Chicago/Turabian StyleHoward, Caroline, Claire Lockie-Williams, and Adrian Slater. 2020. "Applied Barcoding: The Practicalities of DNA Testing for Herbals" Plants 9, no. 9: 1150. https://doi.org/10.3390/plants9091150