

Exercise-Mediated Skeletal Muscle-Derived IL-6 Regulates Bone Metabolism: A New Perspective on Muscle–Bone Crosstalk

{kind=link}
{kind=link}
Abstract
1. Introduction
2. Effects of IL-6 on Bone Homeostasis
2.1. IL-6 Structure and Function
2.2. IL-6 Regulates Bone Homeostasis
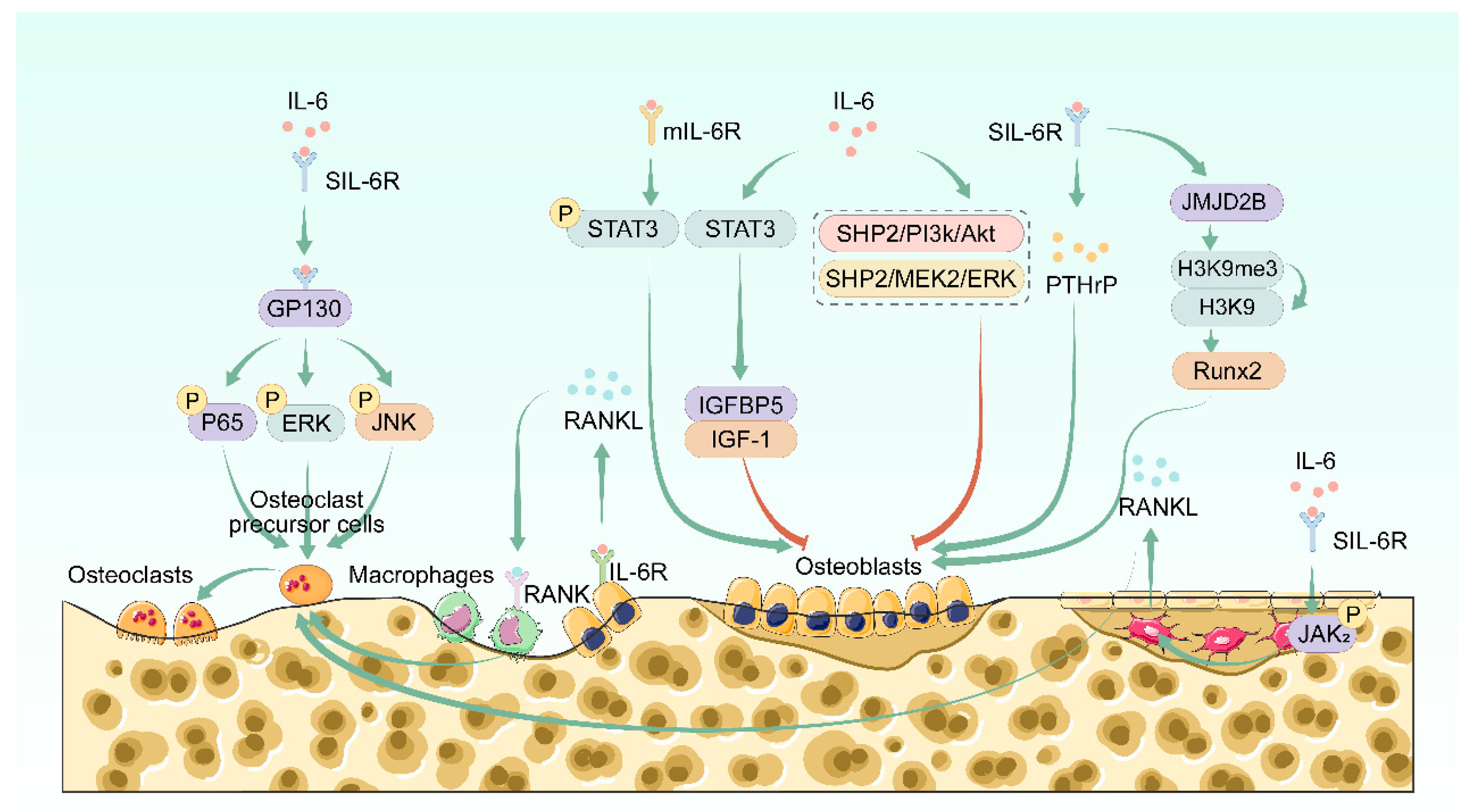
2.2.1. IL-6 Regulates Bone Resorption
2.2.2. IL-6 Regulates Bone Formation
3. Effects of Other Muscle-Derived Factors on Bone Homeostasis
3.1. Irisin
3.2. IGF-1
3.3. Myostatin
4. Exercise-Mediated IL-6 Pathway Regulates Bone Metabolism
4.1. Effects of Exercise on IL-6 in the Skeletal Muscle
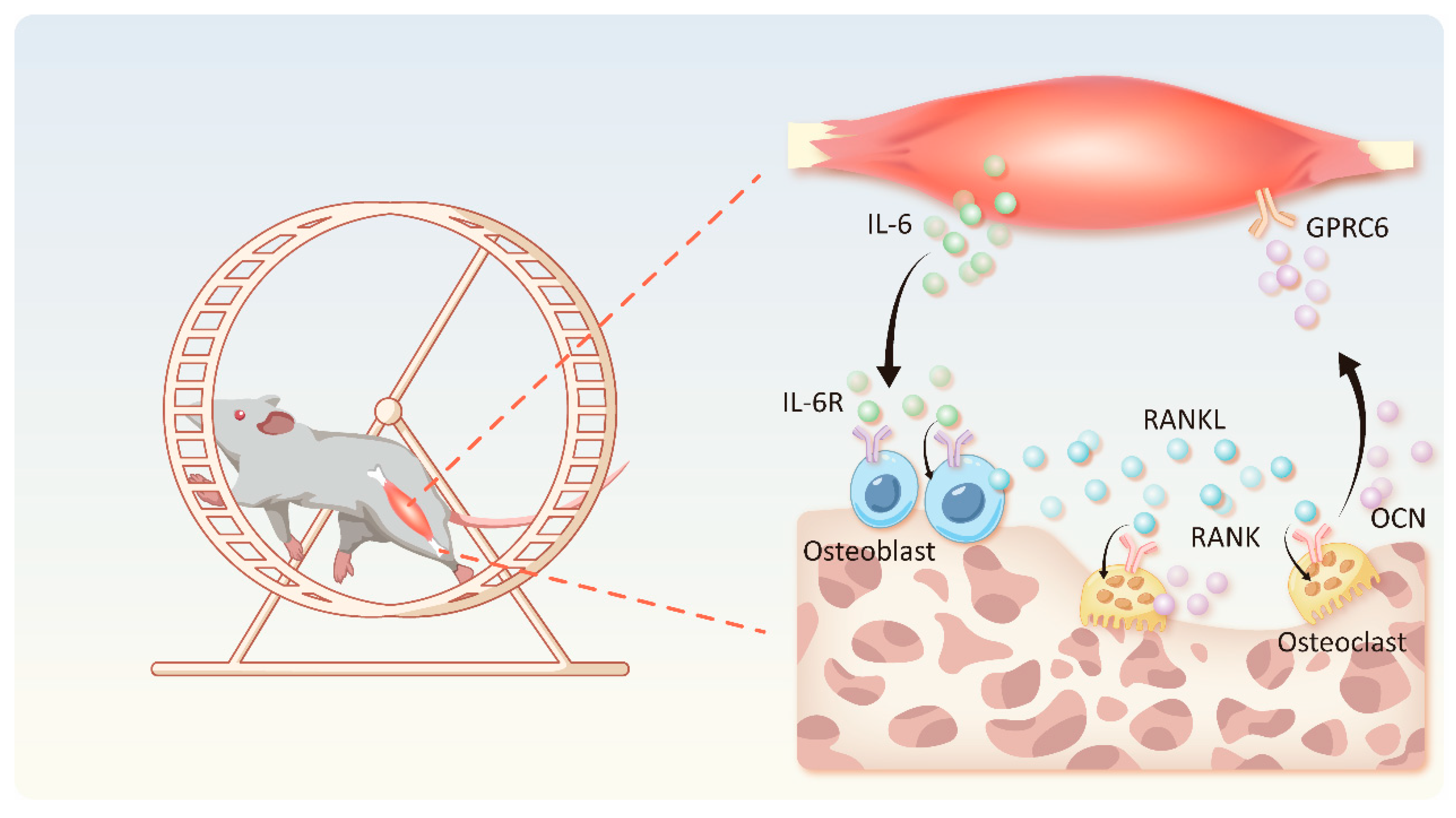
4.2. IL-6-OCN Cycle in the Musculoskeletal System Mediated by Exercise
4.3. Exercise-Mediated IL-6 Promotes Fatty Acid Oxidation
4.4. IL-6 Pro-Inflammatory Effect Mediated by Exercise
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bull, F.C.; Al-Ansari, S.S.; Biddle, S.; Borodulin, K.; Buman, M.P.; Cardon, G.; Carty, C.; Chaput, J.P.; Chastin, S.; Chou, R.; et al. World Health Organization 2020 guidelines on physical activity and sedentary behaviour. Br. J. Sports Med. 2020, 54, 1451–1462. [Google Scholar] [CrossRef] [PubMed]
- Lobelo, F.; Stoutenberg, M.; Hutber, A. The Exercise is Medicine Global Health Initiative: A 2014 update. Br. J. Sports Med. 2014, 48, 1627–1633. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Yan, K.; Guan, Q.; Guo, Q.; Zhao, C. Mechanism and physical activities in bone-skeletal muscle crosstalk. Front. Endocrinol. 2023, 14, 1287972. [Google Scholar] [CrossRef]
- Steensberg, A.; van Hall, G.; Osada, T.; Sacchetti, M.; Saltin, B.; Pedersen, B.K. Production of interleukin-6 in contracting human skeletal muscles can account for the exercise-induced increase in plasma interleukin-6. J. Physiol. 2000, 529 Pt 1, 237–242. [Google Scholar] [CrossRef]
- Gomarasca, M.; Banfi, G.; Lombardi, G. Myokines: The endocrine coupling of skeletal muscle and bone. Adv. Clin. Chem. 2020, 94, 155–218. [Google Scholar]
- Dong, Y.; Yuan, H.; Ma, G.; Cao, H. Bone-muscle crosstalk under physiological and pathological conditions. Cell. Mol. Life Sci. 2024, 81, 310. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Febbraio, M.A. Muscle as an endocrine organ: Focus on muscle-derived interleukin-6. Physiol. Rev. 2008, 88, 1379–1406. [Google Scholar] [CrossRef]
- Kishimoto, T. Interleukin-6: Discovery of a pleiotropic cytokine. Arthritis Res. Ther. 2006, 8 (Suppl. 2), S2. [Google Scholar] [CrossRef]
- Kang, S.; Narazaki, M.; Metwally, H.; Kishimoto, T. Historical overview of the interleukin-6 family cytokine. J. Exp. Med. 2020, 217, e20190347. [Google Scholar] [CrossRef]
- Jones, S.A.; Jenkins, B.J. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 2018, 18, 773–789. [Google Scholar] [CrossRef]
- Varghese, J.N.; Moritz, R.L.; Lou, M.-Z.; van Donkelaar, A.; Ji, H.; Ivancic, N.; Branson, K.M.; Hall, N.E.; Simpson, R.J. Structure of the extracellular domains of the human interleukin-6 receptor α-chain. Proc. Natl. Acad. Sci. USA 2002, 99, 15959–15964. [Google Scholar] [CrossRef] [PubMed]
- Briso, E.M.; Dienz, O.; Rincon, M. Cutting edge: Soluble IL-6R is produced by IL-6R ectodomain shedding in activated CD4 T cells. J. Immunol. 2008, 180, 7102–7106. [Google Scholar] [CrossRef] [PubMed]
- Chalaris, A.; Garbers, C.; Rabe, B.; Rose-John, S.; Scheller, J. The soluble Interleukin 6 receptor: Generation and role in inflammation and cancer. Eur. J. Cell Biol. 2011, 90, 484–494. [Google Scholar] [CrossRef]
- Taga, T.; Hibi, M.; Hirata, Y.; Yamasaki, K.; Yasukawa, K.; Matsuda, T.; Hirano, T.; Kishimoto, T. Interleukin-6 triggers the association of its receptor with a possible signal transducer, gp130. Cell 1989, 58, 573–581. [Google Scholar] [CrossRef]
- Masjedi, A.; Hashemi, V.; Hojjat-Farsangi, M.; Ghalamfarsa, G.; Azizi, G.; Yousefi, M.; Jadidi-Niaragh, F. The significant role of interleukin-6 and its signaling pathway in the immunopathogenesis and treatment of breast cancer. Biomed. Pharmacother. 2018, 108, 1415–1424. [Google Scholar] [CrossRef]
- Rose-John, S.; Jenkins, B.J.; Garbers, C.; Moll, J.M.; Scheller, J. Targeting IL-6 trans-signalling: Past, present and future prospects. Nat. Rev. Immunol. 2023, 23, 666–681. [Google Scholar] [CrossRef]
- Garbers, C.; Hermanns, H.M.; Schaper, F.; Müller-Newen, G.; Grötzinger, J.; Rose-John, S.; Scheller, J. Plasticity and cross-talk of interleukin 6-type cytokines. Cytokine Growth Factor Rev. 2012, 23, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Lamertz, L.; Rummel, F.; Polz, R.; Baran, P.; Hansen, S.; Waetzig, G.H.; Moll, J.M.; Floss, D.M.; Scheller, J. Soluble gp130 prevents interleukin-6 and interleukin-11 cluster signaling but not intracellular autocrine responses. Sci. Signal. 2018, 11, eaar7388. [Google Scholar] [CrossRef]
- Heink, S.; Yogev, N.; Garbers, C.; Herwerth, M.; Aly, L.; Gasperi, C.; Husterer, V.; Croxford, A.L.; Möller-Hackbarth, K.; Bartsch, H.S.; et al. Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic TH17 cells. Nat. Immunol. 2017, 18, 74–85. [Google Scholar] [CrossRef]
- Kenkre, J.S.; Bassett, J.H.D. The bone remodelling cycle. Ann. Clin. Biochem. 2018, 55, 308–327. [Google Scholar] [CrossRef]
- Dirckx, N.; Moorer, M.C.; Clemens, T.L.; Riddle, R.C. The role of osteoblasts in energy homeostasis. Nat. Rev. Endocrinol. 2019, 15, 651–665. [Google Scholar] [CrossRef] [PubMed]
- Robling, A.G.; Bonewald, L.F. The Osteocyte: New Insights. Annu. Rev. Physiol. 2020, 82, 485–506. [Google Scholar] [CrossRef] [PubMed]
- Šućur, A.; Katavić, V.; Kelava, T.; Jajić, Z.; Kovačić, N.; Grčević, D. Induction of osteoclast progenitors in inflammatory conditions: Key to bone destruction in arthritis. Int. Orthop. 2014, 38, 1893–1903. [Google Scholar] [CrossRef]
- Peruzzi, B.; Cappariello, A.; Del Fattore, A.; Rucci, N.; De Benedetti, F.; Teti, A. c-Src and IL-6 inhibit osteoblast differentiation and integrate IGFBP5 signalling. Nat. Commun. 2012, 3, 630. [Google Scholar] [CrossRef]
- Liang, J.D.; Hock, J.M.; Sandusky, G.E.; Santerre, R.F.; Onyia, J.E. Immunohistochemical localization of selected early response genes expressed in trabecular bone of young rats given hPTH 1-34. Calcif. Tissue Int. 1999, 65, 369–373. [Google Scholar] [CrossRef]
- Poli, V.; Balena, R.; Fattori, E.; Markatos, A.; Yamamoto, M.; Tanaka, H.; Ciliberto, G.; Rodan, G.; Costantini, F. Interleukin-6 deficient mice are protected from bone loss caused by estrogen depletion. EMBO J. 1994, 13, 1189–1196. [Google Scholar] [CrossRef]
- Jilka, R.L.; Hangoc, G.; Girasole, G.; Passeri, G.; Williams, D.C.; Abrams, J.S.; Boyce, B.; Broxmeyer, H.; Manolagas, S.C. Increased osteoclast development after estrogen loss: Mediation by interleukin-6. Science 1992, 257, 88–91. [Google Scholar] [CrossRef]
- Kurihara, N.; Bertolini, D.; Suda, T.; Akiyama, Y.; Roodman, G.D. IL-6 stimulates osteoclast-like multinucleated cell formation in long term human marrow cultures by inducing IL-1 release. J. Immunol. 1990, 144, 4226–4230. [Google Scholar] [CrossRef]
- Udagawa, N.; Takahashi, N.; Katagiri, T.; Tamura, T.; Wada, S.; Findlay, D.M.; Martin, T.J.; Hirota, H.; Taga, T.; Kishimoto, T.; et al. Interleukin (IL)-6 induction of osteoclast differentiation depends on IL-6 receptors expressed on osteoblastic cells but not on osteoclast progenitors. J. Exp. Med. 1995, 182, 1461–1468. [Google Scholar] [CrossRef]
- Hattersley, G.; Dorey, E.; Horton, M.A.; Chambers, T.J. Human macrophage colony-stimulating factor inhibits bone resorption by osteoclasts disaggregated from rat bone. J. Cell. Physiol. 1988, 137, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Schulz, L.C.; Palmisano, B.; Singh, P.; Berger, J.M.; Yadav, V.K.; Mera, P.; Ellingsgaard, H.; Hidalgo, J.; Brüning, J.C.; et al. Muscle-derived interleukin 6 increases exercise capacity by signaling in osteoblasts. J. Clin. Investig. 2020, 130, 2888–2902. [Google Scholar] [CrossRef] [PubMed]
- Kitaura, H.; Marahleh, A.; Ohori, F.; Noguchi, T.; Shen, W.-R.; Qi, J.; Nara, Y.; Pramusita, A.; Kinjo, R.; Mizoguchi, I. Osteocyte-Related Cytokines Regulate Osteoclast Formation and Bone Resorption. Int. J. Mol. Sci. 2020, 21, 5169. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Zhou, X.; Huang, D.; Ji, Y.; Kang, F. IL-6 Enhances Osteocyte-Mediated Osteoclastogenesis by Promoting JAK2 and RANKL Activity In Vitro. Cell. Physiol. Biochem. 2017, 41, 1360–1369. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, H.J.; Choi, Y.; Bae, M.K.; Hwang, D.S.; Shin, S.H.; Lee, J.Y. Zoledronate Enhances Osteocyte-Mediated Osteoclast Differentiation by IL-6/RANKL Axis. Int. J. Mol. Sci. 2019, 20, 1467. [Google Scholar] [CrossRef]
- Devlin, R.D.; Reddy, S.V.; Savino, R.; Ciliberto, G.; Roodman, G.D. IL-6 mediates the effects of IL-1 or TNF, but not PTHrP or 1,25(OH)2D3, on osteoclast-like cell formation in normal human bone marrow cultures. J. Bone Miner. Res. 1998, 13, 393–399. [Google Scholar] [CrossRef]
- Palmqvist, P.; Persson, E.; Conaway, H.H.; Lerner, U.H. IL-6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulate the expression of receptor activator of NF-kappa B ligand, osteoprotegerin, and receptor activator of NF-kappa B in mouse calvariae. J. Immunol. 2002, 169, 3353–3362. [Google Scholar] [CrossRef]
- Ohsaki, Y.; Takahashi, S.; Scarcez, T.; Demulder, A.; Nishihara, T.; Williams, R.; Roodman, G.D. Evidence for an autocrine/paracrine role for interleukin-6 in bone resorption by giant cells from giant cell tumors of bone. Endocrinology 1992, 131, 2229–2234. [Google Scholar] [CrossRef]
- Kudo, O.; Sabokbar, A.; Pocock, A.; Itonaga, I.; Fujikawa, Y.; Athanasou, N.A. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone 2003, 32, 1–7. [Google Scholar] [CrossRef]
- Feng, W.; Liu, H.; Luo, T.; Liu, D.; Du, J.; Sun, J.; Wang, W.; Han, X.; Yang, K.; Guo, J.; et al. Combination of IL-6 and sIL-6R differentially regulate varying levels of RANKL-induced osteoclastogenesis through NF-κB, ERK and JNK signaling pathways. Sci. Rep. 2017, 7, 41411. [Google Scholar] [CrossRef]
- Rose-John, S.; Neurath, M.F. IL-6 trans-signaling: The heat is on. Immunity 2004, 20, 2–4. [Google Scholar] [CrossRef] [PubMed]
- De Benedetti, F.; Rucci, N.; Del Fattore, A.; Peruzzi, B.; Paro, R.; Longo, M.; Vivarelli, M.; Muratori, F.; Berni, S.; Ballanti, P.; et al. Impaired skeletal development in interleukin-6-transgenic mice: A model for the impact of chronic inflammation on the growing skeletal system. Arthritis Rheum. 2006, 54, 3551–3563. [Google Scholar] [CrossRef] [PubMed]
- Kaneshiro, S.; Ebina, K.; Shi, K.; Higuchi, C.; Hirao, M.; Okamoto, M.; Koizumi, K.; Morimoto, T.; Yoshikawa, H.; Hashimoto, J. IL-6 negatively regulates osteoblast differentiation through the SHP2/MEK2 and SHP2/Akt2 pathways in vitro. J. Bone Miner. Metab. 2014, 32, 378–392. [Google Scholar] [CrossRef]
- Yoshida, H.; Suzuki, M.; Tanaka, K.; Takeda, S.; Yogo, K.; Matsumoto, Y. Anti-interleukin-6 receptor antibody prevents loss of bone structure and bone strength in collagen-induced arthritis mice. Scand. J. Rheumatol. 2018, 47, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bäckesjö, C.-M.; Haldosén, L.-A.; Lindgren, U. IL-6 receptor expression and IL-6 effects change during osteoblast differentiation. Cytokine 2008, 43, 165–173. [Google Scholar] [CrossRef]
- Xie, Z.; Tang, S.; Ye, G.; Wang, P.; Li, J.; Liu, W.; Li, M.; Wang, S.; Wu, X.; Cen, S.; et al. Interleukin-6/interleukin-6 receptor complex promotes osteogenic differentiation of bone marrow-derived mesenchymal stem cells. Stem Cell Res. Ther. 2018, 9, 13. [Google Scholar] [CrossRef]
- Ye, L.; Fan, Z.; Yu, B.; Chang, J.; Al Hezaimi, K.; Zhou, X.; Park, N.-H.; Wang, C.-Y. Histone demethylases KDM4B and KDM6B promotes osteogenic differentiation of human MSCs. Cell Stem Cell 2012, 11, 50–61. [Google Scholar] [CrossRef]
- Sheng, R.; Cao, M.; Song, M.; Wang, M.; Zhang, Y.; Shi, L.; Xie, T.; Li, Y.; Wang, J.; Rui, Y. Muscle-bone crosstalk via endocrine signals and potential targets for osteosarcopenia-related fracture. J. Orthop. Transl. 2023, 43, 36–46. [Google Scholar] [CrossRef]
- Colaianni, G.; Storlino, G.; Sanesi, L.; Colucci, S.; Grano, M. Myokines and Osteokines in the Pathogenesis of Muscle and Bone Diseases. Curr. Osteoporos. Rep. 2020, 18, 401–407. [Google Scholar] [CrossRef]
- Colaianni, G.; Cuscito, C.; Mongelli, T.; Pignataro, P.; Buccoliero, C.; Liu, P.; Lu, P.; Sartini, L.; Di Comite, M.; Mori, G.; et al. The myokine irisin increases cortical bone mass. Proc. Natl. Acad. Sci. USA 2015, 112, 12157–12162. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Hu, S.; Chen, C.; He, J.; Sun, J.; Jin, Y.; Zhang, Y.; Zhu, G.; Shi, Q.; Rui, Y. Myokine Irisin promotes osteogenesis by activating BMP/SMAD signaling via αV integrin and regulates bone mass in mice. Int. J. Biol. Sci. 2022, 18, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Wu, H.; Lu, J.; Zhang, R.; Shen, X.; Gu, Y.; Shi, C.; Zhang, Y.; Yuan, W. Irisin reduces bone fracture by facilitating osteogenesis and antagonizing TGF-β/Smad signaling in a growing mouse model of osteogenesis imperfecta. J. Orthop. Transl. 2023, 38, 175–189. [Google Scholar] [CrossRef]
- Ma, Y.; Qiao, X.; Zeng, R.; Cheng, R.; Zhang, J.; Luo, Y.; Nie, Y.; Hu, Y.; Yang, Z.; Zhang, J.; et al. Irisin promotes proliferation but inhibits differentiation in osteoclast precursor cells. FASEB J. 2018, 32, 5813–5823. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Xue, Y.; He, J.; Chen, C.; Sun, J.; Jin, Y.; Zhang, Y.; Shi, Q.; Rui, Y. Irisin recouples osteogenesis and osteoclastogenesis to protect wear-particle-induced osteolysis by suppressing oxidative stress and RANKL production. Biomater. Sci. 2021, 9, 5791–5801. [Google Scholar] [CrossRef]
- Zhang, Y.; He, X.; Wang, K.; Xue, Y.; Hu, S.; Jin, Y.; Zhu, G.; Shi, Q.; Rui, Y. Irisin alleviates obesity-induced bone loss by inhibiting interleukin 6 expression via TLR4/MyD88/NF-κB axis in adipocytes. J. Adv. Res. 2025, 69, 343–359. [Google Scholar] [CrossRef]
- Schoenle, E.; Zapf, J.; Humbel, R.E.; Froesch, E.R. Insulin-like growth factor I stimulates growth in hypophysectomized rats. Nature 1982, 296, 252–253. [Google Scholar] [CrossRef]
- Goldspink, G. Mechanical signals, IGF-I gene splicing, and muscle adaptation. Physiology 2005, 20, 232–238. [Google Scholar] [CrossRef]
- Zhao, G.; Monier-Faugere, M.C.; Langub, M.C.; Geng, Z.; Nakayama, T.; Pike, J.W.; Chernausek, S.D.; Rosen, C.J.; Donahue, L.R.; Malluche, H.H.; et al. Targeted overexpression of insulin-like growth factor I to osteoblasts of transgenic mice: Increased trabecular bone volume without increased osteoblast proliferation. Endocrinology 2000, 141, 2674–2682. [Google Scholar] [CrossRef]
- Hill, P.A.; Tumber, A.; Meikle, M.C. Multiple extracellular signals promote osteoblast survival and apoptosis. Endocrinology 1997, 138, 3849–3858. [Google Scholar] [CrossRef]
- Wang, Y.; Nishida, S.; Sakata, T.; Elalieh, H.Z.; Chang, W.; Halloran, B.P.; Doty, S.B.; Bikle, D.D. Insulin-like growth factor-I is essential for embryonic bone development. Endocrinology 2006, 147, 4753–4761. [Google Scholar] [CrossRef] [PubMed]
- Kirk, B.; Feehan, J.; Lombardi, G.; Duque, G. Muscle, Bone, and Fat Crosstalk: The Biological Role of Myokines, Osteokines, and Adipokines. Curr. Osteoporos. Rep. 2020, 18, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Cariati, I.; Scimeca, M.; Bonanni, R.; Triolo, R.; Naldi, V.; Toro, G.; Marini, M.; Tancredi, V.; Iundusi, R.; Gasbarra, E.; et al. Role of Myostatin in Muscle Degeneration by Random Positioning Machine Exposure: An in vitro Study for the Treatment of Sarcopenia. Front. Physiol. 2022, 13, 782000. [Google Scholar] [CrossRef]
- Hamrick, M.; Shi, X.; Zhang, W.; Pennington, C.; Thakore, H.; Haque, M.; Kang, B.; Isales, C.; Fulzele, S.; Wenger, K. Loss of myostatin (GDF8) function increases osteogenic differentiation of bone marrow-derived mesenchymal stem cells but the osteogenic effect is ablated with unloading. Bone 2007, 40, 1544–1553. [Google Scholar] [CrossRef]
- Dankbar, B.; Fennen, M.; Brunert, D.; Hayer, S.; Frank, S.; Wehmeyer, C.; Beckmann, D.; Paruzel, P.; Bertrand, J.; Redlich, K.; et al. Myostatin is a direct regulator of osteoclast differentiation and its inhibition reduces inflammatory joint destruction in mice. Nat. Med. 2015, 21, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Mera, P.; Laue, K.; Ferron, M.; Confavreux, C.; Wei, J.; Galán-Díez, M.; Lacampagne, A.; Mitchell, S.J.; Mattison, J.A.; Chen, Y.; et al. Osteocalcin Signaling in Myofibers Is Necessary and Sufficient for Optimum Adaptation to Exercise. Cell Metab. 2017, 25, 218. [Google Scholar] [CrossRef]
- Fischer, C.P.; Hiscock, N.J.; Penkowa, M.; Basu, S.; Vessby, B.; Kallner, A.; Sjöberg, L.; Pedersen, B.K. Supplementation with vitamins C and E inhibits the release of interleukin-6 from contracting human skeletal muscle. J. Physiol. 2004, 558 Pt 2, 633–645. [Google Scholar] [CrossRef]
- Fischer, C.P. Interleukin-6 in acute exercise and training: What is the biological relevance? Exerc. Immunol. Rev. 2006, 12, 6–33. [Google Scholar]
- Du, J.; Yang, J.; He, Z.; Cui, J.; Yang, Y.; Xu, M.; Qu, X.; Zhao, N.; Yan, M.; Li, H.; et al. Osteoblast and Osteoclast Activity Affect Bone Remodeling Upon Regulation by Mechanical Loading-Induced Leukemia Inhibitory Factor Expression in Osteocytes. Front. Mol. Biosci. 2020, 7, 585056. [Google Scholar] [CrossRef]
- Lazzaro, L.; Tonkin, B.A.; Poulton, I.J.; McGregor, N.E.; Ferlin, W.; Sims, N.A. IL-6 trans-signalling mediates trabecular, but not cortical, bone loss after ovariectomy. Bone 2018, 112, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kirschenbaum, A.; Yao, S.; Levine, A.C. Interactive effect of interleukin-6 and prostaglandin E2 on osteoclastogenesis via the OPG/RANKL/RANK system. Ann. N. Y. Acad. Sci. 2006, 1068, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Fasshauer, M.; Blüher, M. Adipokines in health and disease. Trends Pharmacol. Sci. 2015, 36, 461–470. [Google Scholar] [CrossRef]
- Karaytug, K.; Arzu, U.; Ergin, O.N.; Bilgili, F.; Unverengil, G.; Bayram, S.; Sen, C. Effects of Collagen- and Arginine-Fortified Osteokine Supplementation on Fracture Healing. Cureus 2021, 13, e19072. [Google Scholar] [CrossRef]
- Yang, X.; Yang, J.; Lei, P.; Wen, T. LncRNA MALAT1 shuttled by bone marrow-derived mesenchymal stem cells-secreted exosomes alleviates osteoporosis through mediating microRNA-34c/SATB2 axis. Aging 2019, 11, 8777–8791. [Google Scholar] [CrossRef]
- Hojo, H. Emerging RUNX2-Mediated Gene Regulatory Mechanisms Consisting of Multi-Layered Regulatory Networks in Skeletal Development. Int. J. Mol. Sci. 2023, 24, 2979. [Google Scholar] [CrossRef]
- Han, Y.; Gao, H.; Gao, J.; Yang, Y.; He, C. Low-intensity pulsed ultrasound regulates bone marrow mesenchymal stromal cells differentiation and inhibits bone loss by activating the IL-11-Wnt/β-catenin signaling pathway. Int. Immunopharmacol. 2024, 143 Pt 1, 113380. [Google Scholar] [CrossRef]
- Lee, W.-C.; Guntur, A.R.; Long, F.; Rosen, C.J. Energy Metabolism of the Osteoblast: Implications for Osteoporosis. Endocr. Rev. 2017, 38, 255–266. [Google Scholar] [CrossRef]
- Adamek, G.; Felix, R.; Guenther, H.L.; Fleisch, H. Fatty acid oxidation in bone tissue and bone cells in culture. Characterization and hormonal influences. Biochem. J. 1987, 248, 129–137. [Google Scholar] [CrossRef]
- Frey, J.L.; Li, Z.; Ellis, J.M.; Zhang, Q.; Farber, C.R.; Aja, S.; Wolfgang, M.J.; Clemens, T.L.; Riddle, R.C. Wnt-Lrp5 signaling regulates fatty acid metabolism in the osteoblast. Mol. Cell. Biol. 2015, 35, 1979–1991. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, P.; Wolfgang, M.J.; Riddle, R.C. Fatty acid metabolism by the osteoblast. Bone 2018, 115, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Long, F. Energy Metabolism and Bone. Bone 2018, 115, 1. [Google Scholar] [CrossRef]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005, 1, 15–25. [Google Scholar] [CrossRef]
- Carey, A.L.; Steinberg, G.R.; Macaulay, S.L.; Thomas, W.G.; Holmes, A.G.; Ramm, G.; Prelovsek, O.; Hohnen-Behrens, C.; Watt, M.J.; James, D.E.; et al. Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty Acid oxidation in vitro via AMP-activated protein kinase. Diabetes 2006, 55, 2688–2697. [Google Scholar] [CrossRef]
- Petersen, E.W.; Carey, A.L.; Sacchetti, M.; Steinberg, G.R.; Macaulay, S.L.; Febbraio, M.A.; Pedersen, B.K. Acute IL-6 treatment increases fatty acid turnover in elderly humans in vivo and in tissue culture in vitro. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E155–E162. [Google Scholar] [CrossRef]
- Li, H.; Xiao, Y.; Tang, L.; Zhong, F.; Huang, G.; Xu, J.-M.; Xu, A.-M.; Dai, R.-P.; Zhou, Z.-G. Adipocyte Fatty Acid-Binding Protein Promotes Palmitate-Induced Mitochondrial Dysfunction and Apoptosis in Macrophages. Front. Immunol. 2018, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Roodman, G.D.; Kurihara, N.; Ohsaki, Y.; Kukita, A.; Hosking, D.; Demulder, A.; Smith, J.F.; Singer, F.R. Interleukin 6. A potential autocrine/paracrine factor in Paget’s disease of bone. J. Clin. Investig. 1992, 89, 46–52. [Google Scholar] [CrossRef]
- Sims, N.A. Cell-specific paracrine actions of IL-6 family cytokines from bone, marrow and muscle that control bone formation and resorption. Int. J. Biochem. Cell Biol. 2016, 79, 14–23. [Google Scholar] [CrossRef]
- Wong, P.K.; Quinn, J.M.; Sims, N.A.; van Nieuwenhuijze, A.; Campbell, I.K.; Wicks, I.P. Interleukin-6 modulates production of T lymphocyte-derived cytokines in antigen-induced arthritis and drives inflammation-induced osteoclastogenesis. Arthritis Rheum. 2006, 54, 158–168. [Google Scholar] [CrossRef]
- Takegahara, N.; Kim, H.; Choi, Y. Unraveling the intricacies of osteoclast differentiation and maturation: Insight into novel therapeutic strategies for bone-destructive diseases. Exp. Mol. Med. 2024, 56, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Mossetti, G.; Rendina, D.; De Filippo, G.; Viceconti, R.; Di Domenico, G.; Cioffi, M.; Postiglione, L.; Nunziata, V. Interleukin-6 and osteoprotegerin systems in Paget’s disease of bone: Relationship to risedronate treatment. Bone 2005, 36, 549–554. [Google Scholar] [CrossRef]
- Shim, J.-H.; Stavre, Z.; Gravallese, E.M. Bone Loss in Rheumatoid Arthritis: Basic Mechanisms and Clinical Implications. Calcif. Tissue Int. 2018, 102, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Shetty, A.; Hanson, R.; Korsten, P.; Shawagfeh, M.; Arami, S.; Volkov, S.; Vila, O.; Swedler, W.; Naser Shunaigat, A.; Smadi, S.; et al. Tocilizumab in the treatment of rheumatoid arthritis and beyond. Drug Des. Dev. Ther. 2014, 8, 349–364. [Google Scholar] [CrossRef]
- Kume, K.; Amano, K.; Yamada, S.; Kanazawa, T.; Ohta, H.; Hatta, K.; Kuwaba, N. The effect of tocilizumab on bone mineral density in patients with methotrexate-resistant active rheumatoid arthritis. Rheumatology 2014, 53, 900–903. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Schett, G.; Emery, P.; Harari, O.; Byrjalsen, I.; Kenwright, A.; Bay-Jensen, A.C.; Platt, A. IL-6 receptor inhibition positively modulates bone balance in rheumatoid arthritis patients with an inadequate response to anti-tumor necrosis factor therapy: Biochemical marker analysis of bone metabolism in the tocilizumab RADIATE study (NCT00106522). Semin. Arthritis Rheum. 2012, 42, 131–139. [Google Scholar] [CrossRef]
- Rebouissou, S.; Amessou, M.; Couchy, G.; Poussin, K.; Imbeaud, S.; Pilati, C.; Izard, T.; Balabaud, C.; Bioulac-Sage, P.; Zucman-Rossi, J. Frequent in-frame somatic deletions activate gp130 in inflammatory hepatocellular tumours. Nature 2009, 457, 200–204. [Google Scholar] [CrossRef]
- Li, N.; Grivennikov, S.I.; Karin, M. The unholy trinity: Inflammation, cytokines, and STAT3 shape the cancer microenvironment. Cancer Cell 2011, 19, 429–431. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, C.; Ding, X.; Chen, M.; Feng, J.; Zou, J.; Zhang, L. Exercise-Mediated Skeletal Muscle-Derived IL-6 Regulates Bone Metabolism: A New Perspective on Muscle–Bone Crosstalk. Biomolecules 2025, 15, 893. https://doi.org/10.3390/biom15060893
Zhu C, Ding X, Chen M, Feng J, Zou J, Zhang L. Exercise-Mediated Skeletal Muscle-Derived IL-6 Regulates Bone Metabolism: A New Perspective on Muscle–Bone Crosstalk. Biomolecules. 2025; 15(6):893. https://doi.org/10.3390/biom15060893
Chicago/Turabian StyleZhu, Chenyu, Xiaoqing Ding, Min Chen, Jie Feng, Jun Zou, and Lingli Zhang. 2025. "Exercise-Mediated Skeletal Muscle-Derived IL-6 Regulates Bone Metabolism: A New Perspective on Muscle–Bone Crosstalk" Biomolecules 15, no. 6: 893. https://doi.org/10.3390/biom15060893
APA StyleZhu, C., Ding, X., Chen, M., Feng, J., Zou, J., & Zhang, L. (2025). Exercise-Mediated Skeletal Muscle-Derived IL-6 Regulates Bone Metabolism: A New Perspective on Muscle–Bone Crosstalk. Biomolecules, 15(6), 893. https://doi.org/10.3390/biom15060893