

Myelodysplastic Neoplasms (MDS): Pathogenesis and Therapeutic Prospects


Abstract
1. Introduction
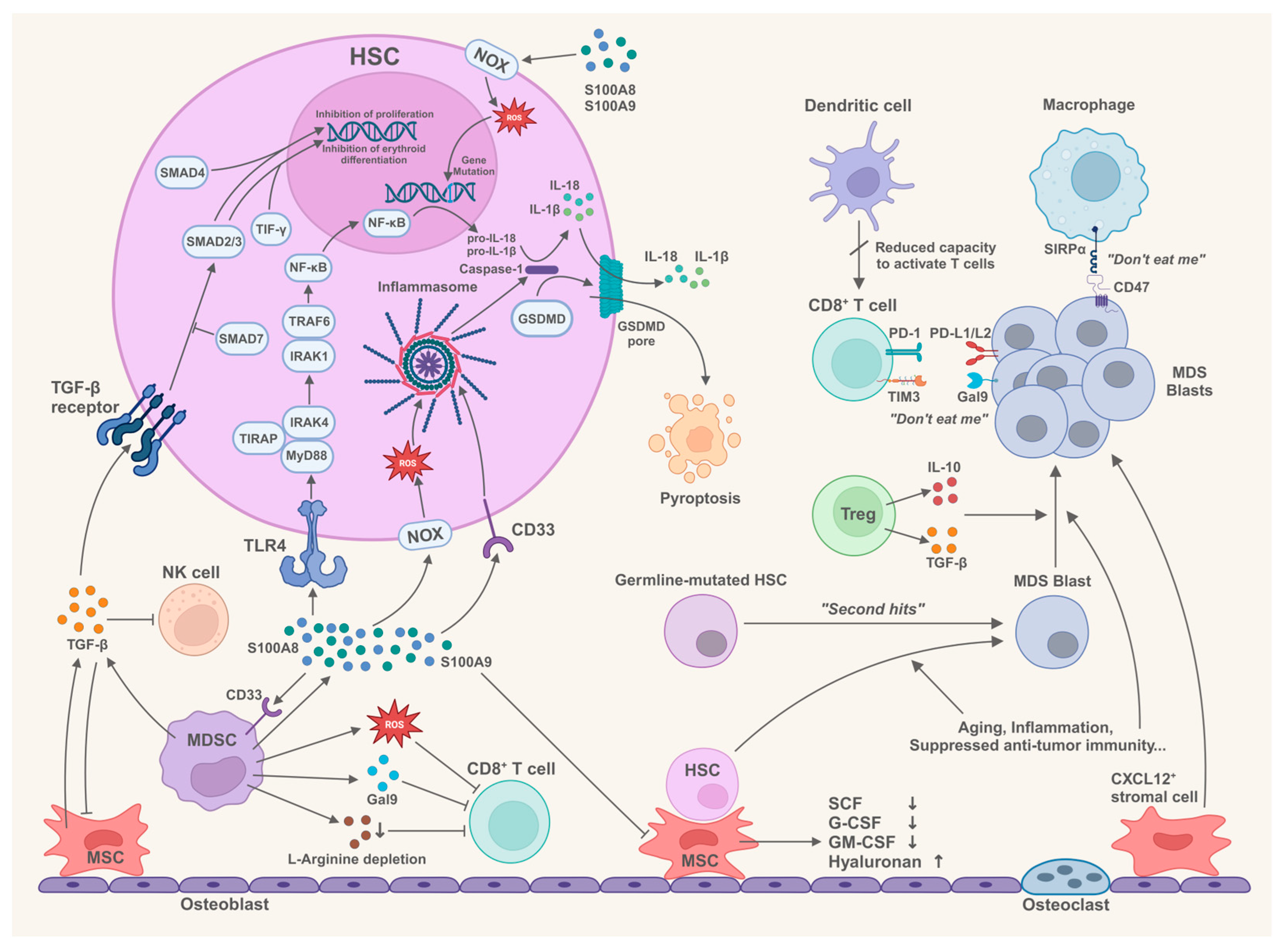
2. Bone Marrow Microenvironment and the Pathogenesis of MDS
2.1. Components in the Bone Marrow Microenvironment Trigger Innate Immune Responses
2.1.1. Innate Immune Signaling in HSCs of MDS
2.1.2. S100A8 and S100A9 Drive the Innate Immune Responses in MDS
2.1.3. Multifunctionality of MDSCs in the Pathogenesis of MDS
2.2. Immune Cells in the Bone Marrow Microenvironment Participate in the Pathogenesis of MDS
2.2.1. Status of CD4+ T Cell Subsets in MDS
2.2.2. Suppression of CD8+ T Cells in MDS
2.2.3. Dysfunction of NK Cells in MDS
2.2.4. Abnormal Macrophages Contribute to the Immune Invasion in MDS
2.2.5. Dysfunction of Dendritic Cells in MDS
2.3. The Role of Bone Marrow Mesenchymal Stem Cells/Stromal Cells in the Pathogenesis of MDS
2.3.1. Impaired Function of MDS-Derived BM-MSCs to Support Normal Hematopoiesis
2.3.2. BM-MSCs Correlate with the Prognosis and Progression of MDS
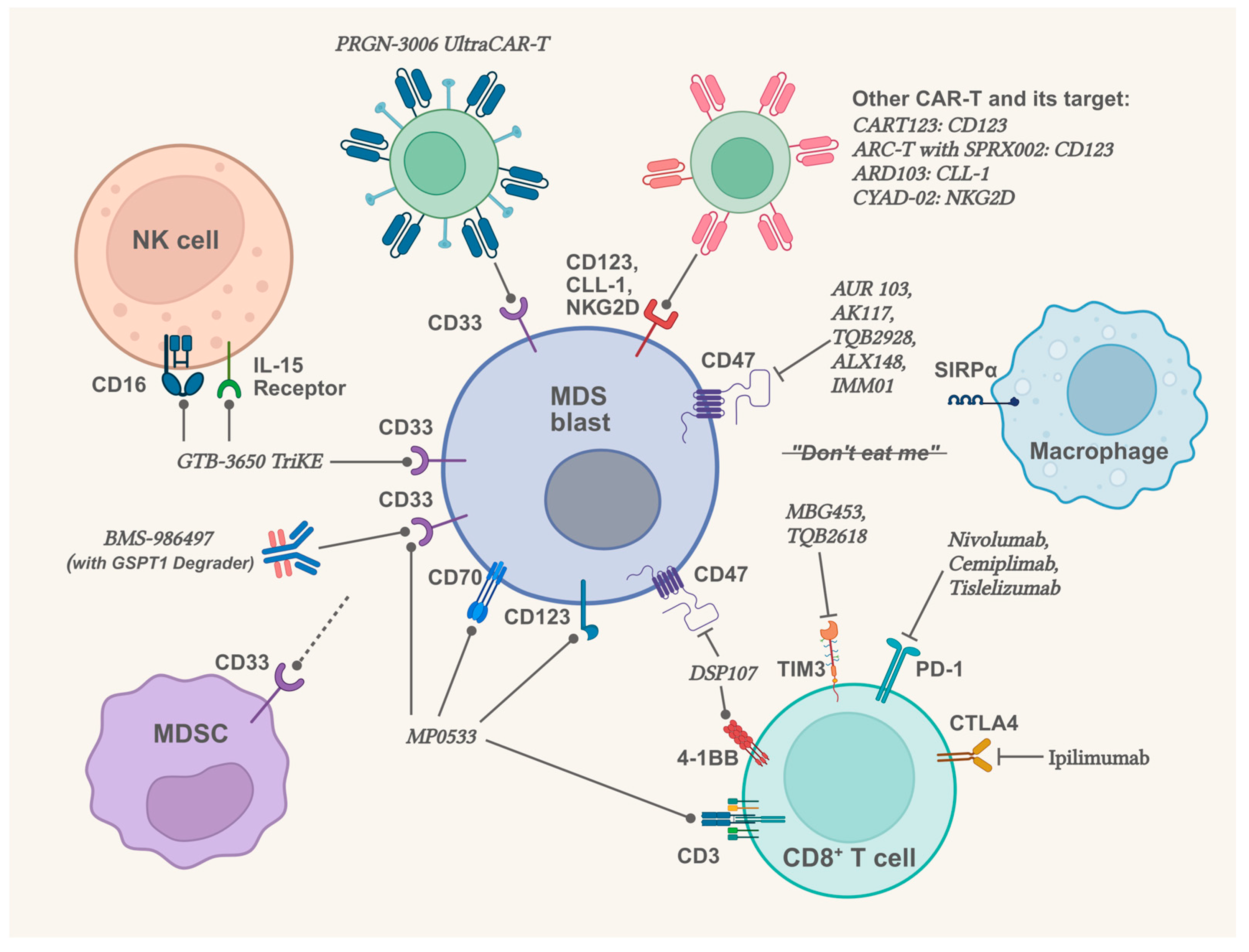
2.4. Therapeutic Prospects in Targeting the Bone Marrow Microenvironment
2.4.1. Therapeutic Prospects Related to Innate Immunity and CD33

{kind=link}
{kind=link}
| Drug | Mechanism | Phase | Results or Interim Reports | Register No. or Reference |
|---|---|---|---|---|
| CA-4948 | Inhibitor of IRAK4 | Phase 1 | All (3 of 3) patients (higher-risk MDS or AML) with spliceosome mutations achieved a marrow CR or better. | NCT04278768 [129] |
| BI 836858 | Inhibitor of CD33 | Phase 1/2 | Failed to meet expected outcomes in low- and intermediate-1-risk MDS patients. | NCT02240706 [133] |
| GTB-3550 TriKE | Tri-specific drug (CD33 x CD16 x IL-15) | Phase 1/2 | 3 of 11 patients (higher-risk MDS or AML) had blast cell decreases, with dose-dependent NK cell activity. | NCT03214666 [134] |
2.4.2. Therapeutic Prospects Related to Immune Abnormality in Bone Marrow Microenvironment
| Drug | Mechanism | Phase | Results or Interim Reports | Register No. or Reference |
|---|---|---|---|---|
| Sabatolimab | TIM3 monoclonal antibody | Phase 2 | Sabatolimab plus HMA failed to meet the primary efficacy objectives in higher-risk MDS patients compared to placebo plus HMA (CR: 21.5% vs. 17.7%; median PFS: 11.07 vs. 8.48 months; both p > 0.05). | NCT03946670 |
| Pembrolizumab | PD-1 monoclonal antibody | Phase 2 | For untreated higher-risk MDS patients, Pembrolizumab plus azacitidine reached the OR rate of 76% and the CR rate of 18%; for patients failed prior HMA therapy, the OR rate was only 25%, and the CR rate was only 5%. | NCT03094637, [140] |
| Magrolimab | CD47 monoclonal antibody | Phase 3 | In untreated MDS patients, azacitidine plus magrolimab showed a lower CR rate and shorter OS compared to azacitidine plus placebo (CR: 21.3% vs. 23.6%; median OS: 15.9 vs. 18.6 months). | NCT03248479, NCT04313881, [147] |
| ALX148 (Evorpacept) | CD47-blocking fusion protein | Phase 1b | ALX148 plus azacitidine: in 5 newly diagnosed higher-risk MDS patients (all had TP53 mutation), 1 reached marrow CR, 2 reached cytogenetic response; in 5 relapsed/refractory MDS patients, 2 reached marrow CR. | NCT04417517, [148] |
| IMM01 | CD47-blocking fusion protein | Phase 2 | In 17 higher-risk MDS patients who received IMM01 plus azacitidine for ≥6 months, the OR rate was 88.2%, and the CR rate was 41.2%. | NCT05140811, [149] |
2.4.3. Therapeutic Prospects Related to BM-MSCs
3. Recurrent Gene Abnormalities in the Pathogenesis of MDS
3.1. Pathogenic Mechanisms of Recurrent Gene Abnormalities in MDS
3.1.1. Cohesin Complex Member STAG2 in MDS
3.1.2. RAS Signaling-Related Genes in MDS
3.1.3. TP53 Abnormalities in MDS
3.1.4. Germline Alterations in MDS
3.2. Therapeutic Prospects for Gene Abnormalities in MDS
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AML | Acute myeloid leukemia |
| BM-MSCs | Bone marrow mesenchymal stromal cells |
| DAMPs | Damage-associated molecular patterns |
| HSCs | Hematopoietic stem cells |
| HSPCs | Hematopoietic stem/progenitor cells |
| LFS | Leukemia-free survival |
| MDS | Myelodysplastic neoplasms |
| MDSCs | Myeloid-derived suppressor cells |
| OS | Overall survival |
| ROS | Reactive oxygen species |
| TLR | Toll-like receptor |
References
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Sauta, E.; Robin, M.; Bersanelli, M.; Travaglino, E.; Meggendorfer, M.; Zhao, L.P.; Caballero Berrocal, J.C.; Sala, C.; Maggioni, G.; Bernardi, M.; et al. Real-World Validation of Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. J. Clin. Oncol. 2023, 41, 2827–2842. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.M.; Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X. Epidemiology of myelodysplastic syndromes: Why characterizing the beast is a prerequisite to taming it. Blood Rev. 2019, 34, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Rollison, D.E.; Howlader, N.; Smith, M.T.; Strom, S.S.; Merritt, W.D.; Ries, L.A.; Edwards, B.K.; List, A.F. Epidemiology of myelodysplastic syndromes and chronic myeloproliferative disorders in the United States, 2001-2004, using data from the NAACCR and SEER programs. Blood 2008, 112, 45–52. [Google Scholar] [CrossRef]
- Barreyro, L.; Chlon, T.M.; Starczynowski, D.T. Chronic immune response dysregulation in MDS pathogenesis. Blood 2018, 132, 1553–1560. [Google Scholar] [CrossRef]
- Ogawa, S. Genetics of MDS. Blood 2019, 133, 1049–1059. [Google Scholar] [CrossRef]
- Sallman, D.A.; List, A. The central role of inflammatory signaling in the pathogenesis of myelodysplastic syndromes. Blood 2019, 133, 1039–1048. [Google Scholar] [CrossRef]
- Mundle, S. Interleukin-1β converting enzyme-like protease may be involved in the intramedullary apoptotic death in the marrows of patients with myelodysplasia. Proc. Am. Soc. Hematol. Blood 1995, 86, 334. [Google Scholar]
- Maratheftis, C.I.; AndreakosS, E.; Moutsopoulos, H.M.; Voulgarelis, M. Toll-like receptor-4 is up-regulated in hematopoietic progenitor cells and contributes to increased apoptosis in myelodysplastic syndromes. Clin. Cancer Res. 2007, 13, 1154–1160. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Sallman, D.A.; Cluzeau, T.; Basiorka, A.A.; List, A. Unraveling the Pathogenesis of MDS: The NLRP3 Inflammasome and Pyroptosis Drive the MDS Phenotype. Front. Oncol. 2016, 6, 151. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000, 103, 351–361. [Google Scholar] [CrossRef]
- Kawagoe, T.; Sato, S.; Matsushita, K.; Kato, H.; Matsui, K.; Kumagai, Y.; Saitoh, T.; Kawai, T.; Takeuchi, O.; Akira, S. Sequential control of Toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat. Immunol. 2008, 9, 684–691. [Google Scholar] [CrossRef]
- Gopal, A.; Ibrahim, R.; Fuller, M.; Umlandt, P.; Parker, J.; Tran, J.; Chang, L.; Wegrzyn-Woltosz, J.; Lam, J.; Li, J.; et al. TIRAP drives myelosuppression through an Ifnγ-Hmgb1 axis that disrupts the endothelial niche in mice. J. Exp. Med. 2022, 219, e20200731. [Google Scholar] [CrossRef]
- Gebhardt, C.; Nemeth, J.; Angel, P.; Hess, J. S100A8 and S100A9 in inflammation and cancer. Biochem. Pharmacol. 2006, 72, 1622–1631. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.W.; Song, R.; Wang, Z.Y.; Jing, Z.C.; Wang, S.X.; Ma, J. S100A8/A9 in Inflammation. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef]
- Li, X.; Li, Q.; Xiang, X.; Zhang, X.; Wu, Y. The diagnostic value and clinical correlations of bone marrow supernatant S100A8 and S100A9 in myelodysplastic neoplasms. Cytokine 2025, 187, 156856. [Google Scholar] [CrossRef] [PubMed]
- Ehrchen, J.M.; Sunderkötter, C.; Foell, D.; Vogl, T.; Roth, J. The endogenous Toll-like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J. Leukoc. Biol. 2009, 86, 557–566. [Google Scholar] [CrossRef]
- Simard, J.-C.; Cesaro, A.; Chapeton-Montes, J.; Tardif, M.; Antoine, F.; Girard, D.; Tessier, P.A. S100A8 and S100A9 induce cytokine expression and regulate the NLRP3 inflammasome via ROS-dependent activation of NF-κB1. PLoS ONE 2013, 8, e72138. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef]
- Kajla, S.; Mondol, A.S.; Nagasawa, A.; Zhang, Y.; Kato, M.; Matsuno, K.; Yabe-Nishimura, C.; Kamata, T. A crucial role for Nox 1 in redox-dependent regulation of Wnt-beta-catenin signaling. FASEB J. 2012, 26, 2049–2059. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Eksioglu, E.A.; Zhou, J.; Zhang, L.; Djeu, J.; Fortenbery, N.; Epling-Burnette, P.; Van Bijnen, S.; Dolstra, H.; Cannon, J.; et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J. Clin. Investig. 2013, 123, 4595–4611. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Okoro, C.; Foell, D.; Freeze, H.H.; Ostrand-Rosenberg, S.; Srikrishna, G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J. Immunol. 2008, 181, 4666–4675. [Google Scholar] [CrossRef]
- Mies, A.; Platzbecker, U. Increasing the effectiveness of hematopoiesis in myelodysplastic syndromes: Erythropoiesis-stimulating agents and transforming growth factor-β superfamily inhibitors. Semin. Hematol. 2017, 54, 141–146. [Google Scholar] [CrossRef]
- Zermati, Y.; Fichelson, S.; Valensi, F.; Freyssinier, J.M.; Rouyer-Fessard, P.; Cramer, E.; Guichard, J.; Varet, B.; Hermine, O. Transforming growth factor inhibits erythropoiesis by blocking proliferation and accelerating differentiation of erythroid progenitors. Exp. Hematol. 2000, 28, 885–894. [Google Scholar] [CrossRef]
- Bewersdorf, J.P.; Zeidan, A.M. Transforming growth factor (TGF)-β pathway as a therapeutic target in lower risk myelodysplastic syndromes. Leukemia 2019, 33, 1303–1312. [Google Scholar] [CrossRef]
- He, W.; Dorn, D.C.; Erdjument-Bromage, H.; Tempst, P.; Moore, M.A.; Massague, J. Hematopoiesis controlled by distinct TIF1gamma and Smad4 branches of the TGFbeta pathway. Cell 2006, 125, 929–941. [Google Scholar] [CrossRef]
- Blank, U.; Karlsson, S. TGF-beta signaling in the control of hematopoietic stem cells. Blood 2015, 125, 3542–3550. [Google Scholar] [CrossRef]
- Zhou, L.; McMahon, C.; Bhagat, T.; Alencar, C.; Yu, Y.T.; Fazzari, M.; Sohal, D.; Heuck, C.; Gundabolu, K.; Ng, C.; et al. Reduced SMAD7 Leads to Overactivation of TGF-β Signaling in MDS that Can Be Reversed by a Specific Inhibitor of TGF-β Receptor I Kinase. Cancer Res. 2011, 71, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Kordasti, S.Y.; Afzali, B.; Lim, Z.; Ingram, W.; Hayden, J.; Barber, L.; Matthews, K.; Chelliah, R.; Guinn, B.; Lombardi, G.; et al. IL-17-producing CD4(+) T cells, pro-inflammatory cytokines and apoptosis are increased in low risk myelodysplastic syndrome. Br. J. Haematol. 2009, 145, 64–72. [Google Scholar] [CrossRef]
- Kasamatsu, T.; Saitoh, T.; Minato, Y.; Shimizu, H.; Yokohama, A.; Tsukamoto, N.; Handa, H.; Sakura, T.; Murakami, H. Polymorphisms of IL-10 affect the severity and prognosis of myelodysplastic syndrome. Eur. J. Haematol. 2016, 96, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Sevilla, J.J.; Colla, S. T-cell dysfunctions in myelodysplastic syndromes. Blood 2024, 143, 1329–1343. [Google Scholar] [CrossRef] [PubMed]
- Barakos, G.P.; Georgoulis, V.; Koumpis, E.; Hatzimichael, E. Elucidating the Role of the T Cell Receptor Repertoire in Myelodysplastic Neoplasms and Acute Myeloid Leukemia. Diseases 2025, 13, 19. [Google Scholar] [CrossRef]
- Ruterbusch, M.; Pruner, K.B.; Shehata, L.; Pepper, M. In Vivo CD4(+) T Cell Differentiation and Function: Revisiting the Th1/Th2 Paradigm. Annu. Rev. Immunol. 2020, 38, 705–725. [Google Scholar] [CrossRef]
- Sun, L.; Su, Y.; Jiao, A.; Wang, X.; Zhang, B. T cells in health and disease. Signal Transduct. Target. Ther. 2023, 8, 235. [Google Scholar] [CrossRef]
- Wu, L.; Li, X.; Chang, C.; Ying, S.; He, Q.; Pu, Q. Deviation of type I and type II T cells and its negative effect on hematopoiesis in myelodysplastic syndrome. Int. J. Lab. Hematol. 2008, 30, 390–399. [Google Scholar] [CrossRef]
- Hamdi, W.; Ogawara, H.; Handa, H.; Tsukamoto, N.; Murakami, H. Clinical significance of Th1/Th2 ratio in patients with myelodysplastic syndrome. Int. J. Lab. Hematol. 2009, 31, 630–638. [Google Scholar] [CrossRef]
- Wang, X.; Wu, D.P.; He, G.; Miao, M.; Sun, A. Research of Subset and Function of Th Cells in Bone Marrow of Myelodysplastic Syndrome Patients. Blood 2005, 106, 4913. [Google Scholar] [CrossRef]
- Plitas, G.; Rudensky, A.Y. Regulatory T Cells: Differentiation and Function. Cancer Immunol. Res. 2016, 4, 721–725. [Google Scholar] [CrossRef]
- Lopes, M.R.; Traina, F.; Campos Pde, M.; Pereira, J.K.; Machado-Neto, J.A.; Machado Hda, C.; Gilli, S.C.; Saad, S.T.; Favaro, P. IL10 inversely correlates with the percentage of CD8⁺ cells in MDS patients. Leuk. Res. 2013, 37, 541–546. [Google Scholar] [CrossRef]
- Kordasti, S.Y.; Ingram, W.; Hayden, J.; Darling, D.; Barber, L.; Afzali, B.; Lombardi, G.; Wlodarski, M.W.; Maciejewski, J.P.; Farzaneh, F.; et al. CD4+CD25high Foxp3+ regulatory T cells in myelodysplastic syndrome (MDS). Blood 2007, 110, 847–850. [Google Scholar] [CrossRef]
- Mailloux, A.W.; Sugimori, C.; Komrokji, R.S.; Yang, L.; Maciejewski, J.P.; Sekeres, M.A.; Paquette, R.; Loughran, T.P., Jr.; List, A.F.; Epling-Burnette, P.K. Expansion of effector memory regulatory T cells represents a novel prognostic factor in lower risk myelodysplastic syndrome. J. Immunol. 2012, 189, 3198–3208. [Google Scholar] [CrossRef] [PubMed]
- Giovazzino, A.; Leone, S.; Rubino, V.; Palatucci, A.T.; Cerciello, G.; Alfinito, F.; Pane, F.; Ruggiero, G.; Terrazzano, G. Reduced regulatory T cells (Treg) in bone marrow preferentially associate with the expansion of cytotoxic T lymphocytes in low risk MDS patients. Br. J. Haematol. 2019, 185, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Bouchliou, I.; Miltiades, P.; Nakou, E.; Spanoudakis, E.; Goutzouvelidis, A.; Vakalopoulou, S.; Garypidou, V.; Kotoula, V.; Bourikas, G.; Tsatalas, C.; et al. Th17 and Foxp3(+) T regulatory cell dynamics and distribution in myelodysplastic syndromes. Clin. Immunol. 2011, 139, 350–359. [Google Scholar] [CrossRef]
- Li, J.; Yue, L.; Wang, H.; Liu, C.; Liu, H.; Tao, J.; Qi, W.; Wang, Y.; Zhang, W.; Fu, R.; et al. Th17 Cells Exhibit Antitumor Effects in MDS Possibly through Augmenting Functions of CD8+ T Cells. J. Immunol. Res. 2016, 2016, 9404705. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, X.; Guo, J.; Xu, F.; He, Q.; Zhao, Y.; Yang, Y.; Gu, S.; Zhang, Y.; Wu, L.; et al. Interleukin-17 enhances the production of interferon-γ and tumour necrosis factor-α by bone marrow T lymphocytes from patients with lower risk myelodysplastic syndromes. Eur. J. Haematol. 2013, 90, 375–384. [Google Scholar] [CrossRef]
- Shao, L.L.; Zhang, L.; Hou, Y.; Yu, S.; Liu, X.G.; Huang, X.Y.; Sun, Y.X.; Tian, T.; He, N.; Ma, D.X.; et al. Th22 cells as well as Th17 cells expand differentially in patients with early-stage and late-stage myelodysplastic syndrome. PLoS ONE 2012, 7, e51339. [Google Scholar] [CrossRef]
- Sand, K.; Theorell, J.; Bruserud, Ø.; Bryceson, Y.T.; Kittang, A.O. Reduced potency of cytotoxic T lymphocytes from patients with high-risk myelodysplastic syndromes. Cancer Immunol. Immunother. 2016, 65, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- Tasis, A.; Spyropoulos, T.; Mitroulis, I. The Emerging Role of CD8(+) T Cells in Shaping Treatment Outcomes of Patients with MDS and AML. Cancers 2025, 17, 749. [Google Scholar] [CrossRef]
- Fozza, C.; Longinotti, M. Are T-cell dysfunctions the other side of the moon in the pathogenesis of myelodysplastic syndromes? Eur. J. Haematol. 2012, 88, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Shao, Z.H.; Yao, C.; He, G.S.; Liu, H.; Shi, J.; Bai, J.; Cao, Y.R.; Tu, M.F.; Wang, H.Q.; et al. Study of Th cell subsets in bone marrow of myelodysplastic syndromes patients. Zhonghua Xue Ye Xue Za Zhi 2005, 26, 743–745. [Google Scholar] [PubMed]
- Mewawalla, P.; Dasanu, C.A. Immune alterations in untreated and treated myelodysplastic syndrome. Expert. Opin. Drug Saf. 2011, 10, 351–361. [Google Scholar] [CrossRef]
- Wu, Y.; Yi, M.; Niu, M.; Mei, Q.; Wu, K. Myeloid-derived suppressor cells: An emerging target for anticancer immunotherapy. Mol. Cancer 2022, 21, 184. [Google Scholar] [CrossRef]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef]
- Schouppe, E.; Van Overmeire, E.; Laoui, D.; Keirsse, J.; Van Ginderachter, J.A. Modulation of CD8(+) T-cell activation events by monocytic and granulocytic myeloid-derived suppressor cells. Immunobiology 2013, 218, 1385–1391. [Google Scholar] [CrossRef]
- Kusmartsev, S.; Nefedova, Y.; Yoder, D.; Gabrilovich, D.I. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J. Immunol. 2004, 172, 989–999. [Google Scholar] [CrossRef]
- Tao, J.; Han, D.; Gao, S.; Zhang, W.; Yu, H.; Liu, P.; Fu, R.; Li, L.; Shao, Z. CD8+ T cells exhaustion induced by myeloid-derived suppressor cells in myelodysplastic syndromes patients might be through TIM3/Gal-9 pathway. J. Cell Mol. Med. 2020, 24, 1046–1058. [Google Scholar] [CrossRef]
- Yu, S.; Ren, X.; Meng, F.; Guo, X.; Tao, J.; Zhang, W.; Liu, Z.; Fu, R.; Li, L. TIM3/CEACAM1 pathway involves in myeloid-derived suppressor cells induced CD8(+) T cells exhaustion and bone marrow inflammatory microenvironment in myelodysplastic syndrome. Immunology 2023, 168, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Jiang, H.; Liu, P.; Xie, N.; Fu, R.; Wang, H.; Liu, C.; Zhang, T.; Wang, H.; Shao, Z. Increased myeloid-derived suppressor cells in patients with myelodysplastic syndromes suppress CD8+ T lymphocyte function through the STAT3-ARG1 pathway. Leuk. Lymphoma 2021, 62, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Li, L.; Wang, Y.; Fu, R.; Wang, H.; Shao, Z. Increased TIM3+CD8+T cells in Myelodysplastic Syndrome patients displayed less perforin and granzyme B secretion and higher CD95 expression. Leuk. Res. 2016, 51, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Asayama, T.; Tamura, H.; Ishibashi, M.; Kuribayashi-Hamada, Y.; Onodera-Kondo, A.; Okuyama, N.; Yamada, A.; Shimizu, M.; Moriya, K.; Takahashi, H.; et al. Functional expression of Tim-3 on blasts and clinical impact of its ligand galectin-9 in myelodysplastic syndromes. Oncotarget 2017, 8, 88904–88917. [Google Scholar] [CrossRef]
- Acharya, N.; Sabatos-Peyton, C.; Anderson, A.C. Tim-3 finds its place in the cancer immunotherapy landscape. J. Immunother. Cancer 2020, 8, e000911. [Google Scholar] [CrossRef]
- Fu, R.; Li, L.; Hu, J.; Wang, Y.; Tao, J.; Liu, H.; Liu, Z.; Zhang, W. Elevated TIM3 expression of T helper cells affects immune system in patients with myelodysplastic syndrome. J. Investig. Med. 2019, 67, 1125–1130. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α., and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Prima, V.; Kaliberova, L.N.; Kaliberov, S.; Curiel, D.T.; Kusmartsev, S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc. Natl. Acad. Sci. USA 2017, 114, 1117–1122. [Google Scholar] [CrossRef]
- Yang, H.; Bueso-Ramos, C.; DiNardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef]
- Geng, S.; Xu, R.; Huang, X.; Li, M.; Deng, C.; Lai, P.; Wang, Y.; Wu, P.; Chen, X.; Weng, J.; et al. Dynamics of PD-1 expression are associated with treatment efficacy and prognosis in patients with intermediate/high-risk myelodysplastic syndromes under hypomethylating treatment. Front. Immunol. 2022, 13, 950134. [Google Scholar] [CrossRef]
- Yang, X.; Ma, L.; Zhang, X.; Huang, L.; Wei, J. Targeting PD-1/PD-L1 pathway in myelodysplastic syndromes and acute myeloid leukemia. Exp. Hematol. Oncol. 2022, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Arellano-Ballestero, H.; Sabry, M.; Lowdell, M.W. A Killer Disarmed: Natural Killer Cell Impairment in Myelodysplastic Syndrome. Cells 2023, 12, 633. [Google Scholar] [CrossRef]
- Kiladjian, J.J.; Bourgeois, E.; Lobe, I.; Braun, T.; Visentin, G.; Bourhis, J.H.; Fenaux, P.; Chouaib, S.; Caignard, A. Cytolytic function and survival of natural killer cells are severely altered in myelodysplastic syndromes. Leukemia 2006, 20, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Hejazi, M.; Manser, A.R.; Fröbel, J.; Kündgen, A.; Zhao, X.; Schönberg, K.; Germing, U.; Haas, R.; Gattermann, N.; Uhrberg, M. Impaired cytotoxicity associated with defective natural killer cell differentiation in myelodysplastic syndromes. Haematologica 2015, 100, 643–652. [Google Scholar] [CrossRef]
- Epling-Burnette, P.K.; Bai, F.; Painter, J.S.; Rollison, D.E.; Salih, H.R.; Krusch, M.; Zou, J.; Ku, E.; Zhong, B.; Boulware, D.; et al. Reduced natural killer (NK) function associated with high-risk myelodysplastic syndrome (MDS) and reduced expression of activating NK receptors. Blood 2007, 109, 4816–4824. [Google Scholar] [CrossRef] [PubMed]
- Carlsten, M.; Baumann, B.C.; Simonsson, M.; Jädersten, M.; Forsblom, A.M.; Hammarstedt, C.; Bryceson, Y.T.; Ljunggren, H.G.; Hellström-Lindberg, E.; Malmberg, K.J. Reduced DNAM-1 expression on bone marrow NK cells associated with impaired killing of CD34+ blasts in myelodysplastic syndrome. Leukemia 2010, 24, 1607–1616. [Google Scholar] [CrossRef]
- Boy, M.; Bisio, V.; Zhao, L.P.; Guidez, F.; Schell, B.; Lereclus, E.; Henry, G.; Villemonteix, J.; Rodrigues-Lima, F.; Gagne, K.; et al. Myelodysplastic Syndrome associated TET2 mutations affect NK cell function and genome methylation. Nat. Commun. 2023, 14, 588. [Google Scholar] [CrossRef]
- Viel, S.; Marçais, A.; Guimaraes, F.S.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal 2016, 9, ra19. [Google Scholar] [CrossRef]
- Thangaraj, J.L.; Coffey, M.; Lopez, E.; Kaufman, D.S. Disruption of TGF-β signaling pathway is required to mediate effective killing of hepatocellular carcinoma by human iPSC-derived NK cells. Cell Stem Cell 2024, 31, 1327–1343.e5. [Google Scholar] [CrossRef]
- Shaim, H.; Shanley, M.; Basar, R.; Daher, M.; Gumin, J.; Zamler, D.B.; Uprety, N.; Wang, F.; Huang, Y.; Gabrusiewicz, K.; et al. Targeting the αv integrin/TGF-β axis improves natural killer cell function against glioblastoma stem cells. J. Clin. Investig. 2021, 131, e142116. [Google Scholar] [CrossRef]
- Regis, S.; Dondero, A.; Caliendo, F.; Bottino, C.; Castriconi, R. NK Cell Function Regulation by TGF-β-Induced Epigenetic Mechanisms. Front. Immunol. 2020, 11, 311. [Google Scholar] [CrossRef]
- Nakamura, K.; Matsunaga, K. Susceptibility of natural killer (NK) cells to reactive oxygen species (ROS) and their restoration by the mimics of superoxide dismutase (SOD). Cancer Biother. Radiopharm. 1998, 13, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Kumar, A.; Chang, C.H.; Pyaram, K. Reactive Oxygen Species Regulate the Inflammatory Function of NKT Cells through Promyelocytic Leukemia Zinc Finger. J. Immunol. 2017, 199, 3478–3487. [Google Scholar] [CrossRef]
- Akhiani, A.A.; Hallner, A.; Kiffin, R.; Aydin, E.; Werlenius, O.; Aurelius, J.; Martner, A.; Thorén, F.B.; Hellstrand, K. Idelalisib Rescues Natural Killer Cells from Monocyte-Induced Immunosuppression by Inhibiting NOX2-Derived Reactive Oxygen Species. Cancer Immunol. Res. 2020, 8, 1532–1541. [Google Scholar] [CrossRef]
- Angelucci, E.; Cianciulli, P.; Finelli, C.; Mecucci, C.; Voso, M.T.; Tura, S. Unraveling the mechanisms behind iron overload and ineffective hematopoiesis in myelodysplastic syndromes. Leuk. Res. 2017, 62, 108–115. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Van Overmeire, E.; Laoui, D.; Keirsse, J.; Van Ginderachter, J.A.; Sarukhan, A. Mechanisms driving macrophage diversity and specialization in distinct tumor microenvironments and parallelisms with other tissues. Front. Immunol. 2014, 5, 127. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Xiang, X.; Wang, J.; Lu, D.; Xu, X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 75. [Google Scholar] [CrossRef]
- Kumari, N.; Choi, S.H. Tumor-associated macrophages in cancer: Recent advancements in cancer nanoimmunotherapies. J. Exp. Clin. Cancer Res. 2022, 41, 68. [Google Scholar] [CrossRef]
- Gao, J.; Liang, Y.; Wang, L. Shaping Polarization of Tumor-Associated Macrophages in Cancer Immunotherapy. Front. Immunol. 2022, 13, 888713. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Feng, W.; Wang, R.; Yang, F.; Wang, L.; Chen, S.; Ru, Y.; Cheng, T.; Zheng, G. Repolarizing heterogeneous leukemia-associated macrophages with more M1 characteristics eliminates their pro-leukemic effects. Oncoimmunology 2018, 7, e1412910. [Google Scholar] [CrossRef]
- Yang, F.; Wu, Z.; Yang, D.; Zhang, X.; Zhang, X.; Xu, Y. Characteristics of macrophages from myelodysplastic syndrome microenvironment. Exp. Cell Res. 2021, 408, 112837. [Google Scholar] [CrossRef] [PubMed]
- Xing, T.; Yao, W.L.; Zhao, H.Y.; Wang, J.; Zhang, Y.Y.; Lv, M.; Xu, L.P.; Zhang, X.H.; Huang, X.J.; Kong, Y. Bone marrow macrophages are involved in the ineffective hematopoiesis of myelodysplastic syndromes. J. Cell Physiol. 2024, 239, e31129. [Google Scholar] [CrossRef]
- Zhang, G.; Yang, L.; Han, Y.; Niu, H.; Yan, L.; Shao, Z.; Xing, L.; Wang, H. Abnormal Macrophage Polarization in Patients with Myelodysplastic Syndrome. Mediat. Inflamm. 2021, 2021, 9913382. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Wang, H.; Shao, Z. Monocyte-Derived Macrophages Are Impaired in Myelodysplastic Syndrome. J. Immunol. Res. 2016, 2016, 5479013. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell 2010, 142, 699–713. [Google Scholar] [CrossRef]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Jan, M.; Weissman-Tsukamoto, R.; Zhao, F.; Park, C.Y.; Weissman, I.L.; Majeti, R. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res. 2011, 71, 1374–1384. [Google Scholar] [CrossRef]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef]
- Pang, W.W.; Pluvinage, J.V.; Price, E.A.; Sridhar, K.; Arber, D.A.; Greenberg, P.L.; Schrier, S.L.; Park, C.Y.; Weissman, I.L. Hematopoietic stem cell and progenitor cell mechanisms in myelodysplastic syndromes. Proc. Natl. Acad. Sci. USA 2013, 110, 3011–3016. [Google Scholar] [CrossRef]
- Van Leeuwen-Kerkhoff, N.; Westers, T.M.; Poddighe, P.J.; Povoleri, G.A.M.; Timms, J.A.; Kordasti, S.; De Gruijl, T.D.; Van de Loosdrecht, A.A. Reduced frequencies and functional impairment of dendritic cell subsets and non-classical monocytes in myelodysplastic syndromes. Haematologica 2022, 107, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Jachiet, V.; Ricard, L.; Hirsch, P.; Malard, F.; Pascal, L.; Beyne-Rauzy, O.; Peterlin, P.; Maria, A.T.J.; Vey, N.; D’Aveni, M.; et al. Reduced peripheral blood dendritic cell and monocyte subsets in MDS patients with systemic inflammatory or dysimmune diseases. Clin. Exp. Med. 2023, 23, 803–813. [Google Scholar] [CrossRef]
- Kfoury, Y.; Scadden, D.T. Mesenchymal cell contributions to the stem cell niche. Cell Stem Cell 2015, 16, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Geyh, S.; Oz, S.; Cadeddu, R.P.; Frobel, J.; Bruckner, B.; Kundgen, A.; Fenk, R.; Bruns, I.; Zilkens, C.; Hermsen, D.; et al. Insufficient stromal support in MDS results from molecular and functional deficits of mesenchymal stromal cells. Leukemia 2013, 27, 1841–1851. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, D.; Fei, C.; Guo, J.; Gu, S.; Zhu, Y.; Xu, F.; Zhang, Z.; Wu, L.; Li, X.; et al. Down-regulation of Dicer1 promotes cellular senescence and decreases the differentiation and stem cell-supporting capacities of mesenchymal stromal cells in patients with myelodysplastic syndrome. Haematologica 2014, 100, 194–204. [Google Scholar] [CrossRef]
- Ferrer, R.A.; Wobus, M.; List, C.; Wehner, R.; Schonefeldt, C.; Brocard, B.; Mohr, B.; Rauner, M.; Schmitz, M.; Stiehler, M.; et al. Mesenchymal stromal cells from patients with myelodyplastic syndrome display distinct functional alterations that are modulated by lenalidomide. Haematologica 2013, 98, 1677–1685. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.G.; Xu, W.; Yu, H.P.; Fang, B.L.; Wu, S.H.; Li, F.; Li, W.M.; Li, Q.B.; Chen, Z.C.; Zou, P. Functional characteristics of mesenchymal stem cells derived from bone marrow of patients with myelodysplastic syndromes. Cancer Lett. 2012, 317, 136–143. [Google Scholar] [CrossRef]
- Pavlaki, K.; Pontikoglou, C.G.; Demetriadou, A.; Batsali, A.K.; Damianaki, A.; Simantirakis, E.; Kontakis, M.; Galanopoulos, A.; Kotsianidis, I.; Kastrinaki, M.-C.; et al. Impaired Proliferative Potential of Bone Marrow Mesenchymal Stromal Cells in Patients with Myelodysplastic Syndromes Is Associated with Abnormal WNT Signaling Pathway. Stem Cells Dev. 2014, 23, 1568–1581. [Google Scholar] [CrossRef]
- Falconi, G.; Fabiani, E.; Fianchi, L.; Criscuolo, M.; Raffaelli, C.S.; Bellesi, S.; Hohaus, S.; Voso, M.T.; D’Alò, F.; Leone, G. Impairment of PI3K/AKT and WNT/β-catenin pathways in bone marrow mesenchymal stem cells isolated from patients with myelodysplastic syndromes. Exp. Hematol. 2016, 44, 75–83.e4. [Google Scholar] [CrossRef]
- Geyh, S.; Rodríguez-Paredes, M.; Jäger, P.; Koch, A.; Bormann, F.; Gutekunst, J.; Zilkens, C.; Germing, U.; Kobbe, G.; Lyko, F.; et al. Transforming growth factor β1-mediated functional inhibition of mesenchymal stromal cells in myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2018, 103, 1462–1471. [Google Scholar] [CrossRef]
- Ping, Z.; Chen, S.; Hermans, S.J.F.; Kenswil, K.J.G.; Feyen, J.; van Dijk, C.; Bindels, E.M.J.; Mylona, A.M.; Adisty, N.M.; Hoogenboezem, R.M.; et al. Activation of NF-κB driven inflammatory programs in mesenchymal elements attenuates hematopoiesis in low-risk myelodysplastic syndromes. Leukemia 2019, 33, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zhao, Y.S.; Fei, C.M.; Guo, J.; Jia, Y.; Wu, D.; Wu, L.Y.; Chang, C.K. Cellular senescence induced by S100A9 in mesenchymal stromal cells through NLRP3 inflammasome activation. Aging-Us 2019, 11, 9626–9642. [Google Scholar] [CrossRef]
- Santamaría, C.; Muntión, S.; Rosón, B.; Blanco, B.; López-Villar, O.; Carrancio, S.; Sánchez-Guijo, F.M.; Díez-Campelo, M.; Alvarez-Fernández, S.; Sarasquete, M.E.; et al. Impaired expression of DICER, DROSHA, SBDS and some microRNAs in mesenchymal stromal cells from myelodysplastic syndrome patients. Haematologica 2012, 97, 1218–1224. [Google Scholar] [CrossRef]
- Fei, C.M.; Guo, J.; Zhao, Y.S.; Zhao, S.D.; Zhen, Q.Q.; Shi, L.; Li, X.; Chang, C.K. Clinical significance of hyaluronan levels and its pro-osteogenic effect on mesenchymal stromal cells in myelodysplastic syndromes. J. Transl. Med. 2018, 16, 234. [Google Scholar] [CrossRef]
- Mattiucci, D.; Maurizi, G.; Leoni, P.; Poloni, A. Aging- and Senescence-associated Changes of Mesenchymal Stromal Cells in Myelodysplastic Syndromes. Cell Transplant. 2018, 27, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Maurizi, G.; Mattiucci, D.; Mariani, M.; Ciarlantini, M.; Traini, S.; Mancini, S.; Olivieri, A.; Leoni, P.; Poloni, A. DNA demethylating therapy reverts mesenchymal stromal cells derived from high risk myelodysplastic patients to a normal phenotype. Br. J. Haematol. 2017, 177, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Poon, Z.; Dighe, N.; Venkatesan, S.S.; Cheung, A.M.S.; Fan, X.; Bari, S.; Hota, M.; Ghosh, S.; Hwang, W.Y.K. Bone marrow MSCs in MDS: Contribution towards dysfunctional hematopoiesis and potential targets for disease response to hypomethylating therapy. Leukemia 2019, 33, 1487–1500. [Google Scholar] [CrossRef]
- Abe-Suzuki, S.; Kurata, M.; Abe, S.; Onishi, I.; Kirimura, S.; Nashimoto, M.; Murayama, T.; Hidaka, M.; Kitagawa, M. CXCL12+ stromal cells as bone marrow niche for CD34+ hematopoietic cells and their association with disease progression in myelodysplastic syndromes. Lab. Investig. 2014, 94, 1212–1223. [Google Scholar] [CrossRef]
- Flores-Figueroa, E.; Varma, S.; Montgomery, K.; Greenberg, P.L.; Gratzinger, D. Distinctive contact between CD34+ hematopoietic progenitors and CXCL12+ CD271+ mesenchymal stromal cells in benign and myelodysplastic bone marrow. Lab. Investig. 2012, 92, 1330–1341. [Google Scholar] [CrossRef]
- Fattizzo, B.; Giannotta, J.A.; Barcellini, W. Mesenchymal Stem Cells in Aplastic Anemia and Myelodysplastic Syndromes: The “Seed and Soil” Crosstalk. Int. J. Mol. Sci. 2020, 21, 5438. [Google Scholar] [CrossRef]
- Zheng, L.; Zhang, L.; Guo, Y.; Xu, X.; Liu, Z.; Yan, Z.; Fu, R. The immunological role of mesenchymal stromal cells in patients with myelodysplastic syndrome. Front. Immunol. 2022, 13, 1078421. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wang, Z.; Li, Q.; Li, W.; You, Y.; Zou, P. The different immunoregulatory functions of mesenchymal stem cells in patients with low-risk or high-risk myelodysplastic syndromes. PLoS ONE 2012, 7, e45675. [Google Scholar] [CrossRef]
- Sarhan, D.; Wang, J.; Sunil Arvindam, U.; Hallstrom, C.; Verneris, M.R.; Grzywacz, B.; Warlick, E.; Blazar, B.R.; Miller, J.S. Mesenchymal stromal cells shape the MDS microenvironment by inducing suppressive monocytes that dampen NK cell function. JCI Insight 2020, 5, e130155. [Google Scholar] [CrossRef]
- Liu, Z.; Guo, Y.; Huang, L.; Jia, Y.; Liu, H.; Peng, F.; Duan, L.; Zhang, H.; Fu, R. Bone marrow mesenchymal stem cells regulate the dysfunction of NK cells via the T cell immunoglobulin and ITIM domain in patients with myelodysplastic syndromes. Cell Commun. Signal 2022, 20, 169. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G. Myelodysplastic syndromes: 2023 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2023, 98, 1307–1325. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Stone, R.M.; Abaza, Y.; Al-Kali, A.; Anand, S.; Ball, B.; Bennett, J.M.; Borate, U.; Brunner, A.M.; Chai-Ho, W.; et al. NCCN Guidelines® Insights: Myelodysplastic Syndromes, Version 2.2025. J. Natl. Compr. Cancer Netw. 2025, 23, 66–75. [Google Scholar] [CrossRef]
- De Benedetti, F.; Gattorno, M.; Anton, J.; Ben-Chetrit, E.; Frenkel, J.; Hoffman, H.M.; Kone-Paut, I.; Lachmann, H.J.; Ozen, S.; Simon, A.; et al. Canakinumab for the Treatment of Autoinflammatory Recurrent Fever Syndromes. N. Engl. J. Med. 2018, 378, 1908–1919. [Google Scholar] [CrossRef] [PubMed]
- Sfriso, P.; Bindoli, S.; Doria, A.; Feist, E.; Galozzi, P. Canakinumab for the treatment of adult-onset Still’s disease. Expert. Rev. Clin. Immunol. 2020, 16, 129–138. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Tarantolo, S.; Verma, A.; Dugan, J.; Winer, E.; Giagounidis, A.; Oncology, H.; Talati, C.; Lieberman, C.; Martinez, E. A Phase 1, dose escalation trial with novel oral irak4 inhibitor ca-4948 in patients with acute myelogenous leukemia or myelodysplastic syndrome–interim report. Proc. EHA Annu. Meet. 2021, 324573, S165. [Google Scholar]
- Cheng, P.; Chen, X.; Dalton, R.; Calescibetta, A.; So, T.; Gilvary, D.; Ward, G.; Smith, V.; Eckard, S.; Fox, J.A.; et al. Immunodepletion of MDSC by AMV564, a novel bivalent, bispecific CD33/CD3 T cell engager, ex vivo in MDS and melanoma. Mol. Ther. 2022, 30, 2315–2326. [Google Scholar] [CrossRef]
- Sanford, D.; Garcia-Manero, G.; Jorgensen, J.; Konoplev, S.; Pierce, S.; Cortes, J.; Kantarjian, H.; Ravandi, F. CD33 is frequently expressed in cases of myelodysplastic syndrome and chronic myelomonocytic leukemia with elevated blast count. Leuk. Lymphoma 2016, 57, 1965–1968. [Google Scholar] [CrossRef] [PubMed]
- Eksioglu, E.A.; Chen, X.; Heider, K.H.; Rueter, B.; McGraw, K.L.; Basiorka, A.A.; Wei, M.; Burnette, A.; Cheng, P.; Lancet, J.; et al. Novel therapeutic approach to improve hematopoiesis in low risk MDS by targeting MDSCs with the Fc-engineered CD33 antibody BI 836858. Leukemia 2017, 31, 2172–2180. [Google Scholar] [CrossRef]
- Komrokji, R.S.; Carraway, H.E.; Germing, U.; Wermke, M.; Zeidan, A.M.; Fu, E.; Ruter, B.; Burkard, U.; Osswald, A.; Foran, J.M. A phase I/II multicenter, open-label, dose escalation and randomized trial of BI 836858 in patients with low- or intermediate-1-risk myelodysplastic syndrome. Haematologica 2022, 107, 2742–2747. [Google Scholar] [CrossRef] [PubMed]
- Felices, M.; Warlick, E.; Juckett, M.; Weisdorf, D.; Vallera, D.; Miller, S.; Wangen, R.; Lewis, D.; Knox, J.; Schroeder, M. 444 GTB-3550 tri-specific killer engager TriKE™ drives NK cells expansion and cytotoxicity in acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) patients. J. Immunother. Cancer 2021, 9, A473. [Google Scholar] [CrossRef]
- Jongen-Lavrencic, M.; Pabst, T.; Bories, P.; Griškevičius, L.; Huls, G.; de Leeuw, D.C.; Boettcher, S.; Pigneux, A.; Boissel, N.; Dymkowska, M.; et al. MP0533 (CD33 x CD123 x CD70 x CD3), a Tetra-Specific CD3-Engaging Darpin for the Treatment of Patients with Relapsed/Refractory AML or MDS/AML: Results of an Ongoing Phase 1/2a Study. Blood 2024, 144, 2881. [Google Scholar] [CrossRef]
- Sallman, D.A.; Elmariah, H.; Sweet, K.; Mishra, A.; Cox, C.A.; Chakaith, M.; Semnani, R.; Shehzad, S.; Anderson, A.; Sabzevari, H.; et al. Phase 1/1b Safety Study of Prgn-3006 Ultracar-T in Patients with Relapsed or Refractory CD33-Positive Acute Myeloid Leukemia and Higher Risk Myelodysplastic Syndromes. Blood 2022, 140, 10313–10315. [Google Scholar] [CrossRef]
- Sellar, R.S.; Sperling, A.S.; Słabicki, M.; Gasser, J.A.; McConkey, M.E.; Donovan, K.A.; Mageed, N.; Adams, D.N.; Zou, C.; Miller, P.G.; et al. Degradation of GSPT1 causes TP53-independent cell death in leukemia while sparing normal hematopoietic stem cells. J. Clin. InvestIG. 2022, 132, e153514. [Google Scholar] [CrossRef]
- Zhang, D.; Lin, P.; Lin, J. Molecular glues targeting GSPT1 in cancers: A potent therapy. Bioorg Chem. 2024, 143, 107000. [Google Scholar] [CrossRef]
- Brunner, A.M.; Esteve, J.; Porkka, K.; Knapper, S.; Traer, E.; Scholl, S.; Garcia-Manero, G.; Vey, N.; Wermke, M.; Janssen, J.; et al. Efficacy and Safety of Sabatolimab (MBG453) in Combination with Hypomethylating Agents (HMAs) in Patients (Pts) with Very High/High-Risk Myelodysplastic Syndrome (vHR/HR-MDS) and Acute Myeloid Leukemia (AML): Final Analysis from a Phase Ib Study. Blood 2021, 138, 244. [Google Scholar] [CrossRef]
- Chien, K.S.; Kim, K.; Nogueras-Gonzalez, G.M.; Borthakur, G.; Naqvi, K.; Daver, N.G.; Montalban-Bravo, G.; Cortes, J.E.; DiNardo, C.D.; Jabbour, E.; et al. Phase II study of azacitidine with pembrolizumab in patients with intermediate-1 or higher-risk myelodysplastic syndrome. Br. J. Haematol. 2021, 195, 378–387. [Google Scholar] [CrossRef]
- Ma, S.; Caligiuri, M.A.; Yu, J. Harnessing IL-15 signaling to potentiate NK cell-mediated cancer immunotherapy. Trends Immunol. 2022, 43, 833–847. [Google Scholar] [CrossRef]
- Yang, Y.; Lundqvist, A. Immunomodulatory Effects of IL-2 and IL-15; Implications for Cancer Immunotherapy. Cancers 2020, 12, 3586. [Google Scholar] [CrossRef] [PubMed]
- Berrien-Elliott, M.M.; Becker-Hapak, M.; Cashen, A.F.; Jacobs, M.; Wong, P.; Foster, M.; McClain, E.; Desai, S.; Pence, P.; Cooley, S.; et al. Systemic IL-15 promotes allogeneic cell rejection in patients treated with natural killer cell adoptive therapy. Blood 2022, 139, 1177–1183. [Google Scholar] [CrossRef]
- Li, W.; Wang, F.; Guo, R.; Bian, Z.; Song, Y. Targeting macrophages in hematological malignancies: Recent advances and future directions. J. Hematol. Oncol. 2022, 15, 110. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Sun, H.; Jiang, Z.; Tian, W.; Cang, S.; Yu, J. Targeting the CD47/SIRPα pathway in malignancies: Recent progress, difficulties and future perspectives. Front. Oncol. 2024, 14, 1378647. [Google Scholar] [CrossRef] [PubMed]
- Boasman, K.; Bridle, C.; Simmonds, M.; Rinaldi, C. Role of pro-phagocytic calreticulin and anti-phagocytic CD47 in MDS and MPN models treated with azacytidine or ruxolitinib. Haematologica 2017, 102, 763. [Google Scholar]
- Sallman, D.A.; Al Malki, M.M.; Asch, A.S.; Wang, E.S.; Jurcic, J.G.; Bradley, T.J.; Flinn, I.W.; Pollyea, D.A.; Kambhampati, S.; Tanaka, T.N.; et al. Magrolimab in Combination With Azacitidine in Patients With Higher-Risk Myelodysplastic Syndromes: Final Results of a Phase Ib Study. J. Clin. Oncol. 2023, 41, 2815–2826. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Erba, H.P.; Sanikommu, S.R.; Altman, J.K.; Sayar, H.; Scott, B.L.; Fong, A.P.; Guan, S.; Jin, F.; Forgie, A.J.; et al. Evorpacept (ALX148), a CD47-Blocking Myeloid Checkpoint Inhibitor, in Combination with Azacitidine: A Phase 1/2 Study in Patients with Myelodysplastic Syndrome (ASPEN-02). Blood 2021, 138, 2601. [Google Scholar] [CrossRef]
- Yang, W.; Gao, S.; Yan, X.; Guo, R.; Han, L.; Li, F.; Wang, Y.; Li, J.; Chang, C.; Yang, H.; et al. Preliminary Results of a Phase 2 Study of IMM01 Combined with Azacitidine (AZA) As the First-Line Treatment in Adult Patients with Higher Risk Myelodysplastic Syndromes (MDS). Blood 2023, 142, 320. [Google Scholar] [CrossRef]
- Boyd-Kirkup, J.; Thakkar, D.; Brauer, P.; Zhou, J.; Chng, W.-J.; Ingram, P.J. HMBD004, a Novel Anti-CD47xCD33 Bispecific Antibody Displays Potent Anti-Tumor Effects in Pre-Clinical Models of AML. Blood 2017, 130, 1378. [Google Scholar] [CrossRef]
- Chester, C.; Sanmamed, M.F.; Wang, J.; Melero, I. Immunotherapy targeting 4-1BB: Mechanistic rationale, clinical results, and future strategies. Blood 2018, 131, 49–57. [Google Scholar] [CrossRef]
- Salek-Ardakani, S.; Zajonc, D.M.; Croft, M. Agonism of 4-1BB for immune therapy: A perspective on possibilities and complications. Front. Immunol. 2023, 14, 1228486. [Google Scholar] [CrossRef] [PubMed]
- Boada, M.; Echarte, L.; Guillermo, C.; Diaz, L.; Tourino, C.; Grille, S. 5-Azacytidine restores interleukin 6-increased production in mesenchymal stromal cells from myelodysplastic patients. Hematol. Transfus. Cell Ther. 2021, 43, 35–42. [Google Scholar] [CrossRef]
- Giagounidis, A.; Arnan, M.; Chee, L.C.Y.; Cluzeau, T.; Diez-Campelo, M.; Hiwase, D.; Ross, D.M.; Sekeres, M.A.; Tan, S.; Valcarcel, D.; et al. Improvements in Hematological Parameters and Quality of Life (QOL) with Elritercept (KER-050): Results from an Ongoing Phase 2 Trial in Participants with Lower-Risk (LR) Myelodysplastic Neoplasms (MDS). Blood 2024, 144, 1825. [Google Scholar] [CrossRef]
- Hosono, N. Genetic abnormalities and pathophysiology of MDS. Int. J. Clin. Oncol. 2019, 24, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Medina, E.A.; Delma, C.R.; Yang, F.C. ASXL1/2 mutations and myeloid malignancies. J. Hematol. Oncol. 2022, 15, 127. [Google Scholar] [CrossRef]
- Köhnke, T.; Nuno, K.A.; Alder, C.C.; Gars, E.J.; Phan, P.; Fan, A.C.; Majeti, R. Human ASXL1-Mutant Hematopoiesis Is Driven by a Truncated Protein Associated with Aberrant Deubiquitination of H2AK119. Blood Cancer Discov. 2024, 5, 202–223. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. ASXL1 Mutations Promote Myeloid Transformation through Loss of PRC2-Mediated Gene Repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef]
- Inoue, D.; Fujino, T.; Sheridan, P.; Zhang, Y.Z.; Nagase, R.; Horikawa, S.; Li, Z.; Matsui, H.; Kanai, A.; Saika, M.; et al. A novel ASXL1-OGT axis plays roles in H3K4 methylation and tumor suppression in myeloid malignancies. Leukemia 2018, 32, 1327–1337. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [PubMed]
- Inoue, D.; Fujino, T.; Kitamura, T. ASXL1 as a critical regulator of epigenetic marks and therapeutic potential of mutated cells. Oncotarget 2018, 9, 35203. [Google Scholar] [CrossRef]
- Nagase, R.; Inoue, D.; Pastore, A.; Fujino, T.; Hou, H.A.; Yamasaki, N.; Goyama, S.; Saika, M.; Kanai, A.; Sera, Y.; et al. Expression of mutant Asxl1 perturbs hematopoiesis and promotes susceptibility to leukemic transformation. J. Exp. Med. 2018, 215, 1729–1747. [Google Scholar] [CrossRef] [PubMed]
- Rinke, J.; Chase, A.; Cross, N.C.P.; Hochhaus, A.; Ernst, T. EZH2 in Myeloid Malignancies. Cells 2020, 9, 1639. [Google Scholar] [CrossRef]
- Sashida, G.; Harada, H.; Matsui, H.; Oshima, M.; Yui, M.; Harada, Y.; Tanaka, S.; Mochizuki-Kashio, M.; Wang, C.; Saraya, A.; et al. Ezh2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat. Commun. 2014, 5, 4177. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627; quiz 3699. [Google Scholar] [CrossRef]
- Hasegawa, N.; Oshima, M.; Sashida, G.; Matsui, H.; Koide, S.; Saraya, A.; Wang, C.; Muto, T.; Takane, K.; Kaneda, A.; et al. Impact of combinatorial dysfunctions of Tet2 and Ezh2 on the epigenome in the pathogenesis of myelodysplastic syndrome. Leukemia 2017, 31, 861–871. [Google Scholar] [CrossRef]
- Muto, T.; Sashida, G.; Oshima, M.; Wendt, G.R.; Mochizuki-Kashio, M.; Nagata, Y.; Sanada, M.; Miyagi, S.; Saraya, A.; Kamio, A.; et al. Concurrent loss of Ezh2 and Tet2 cooperates in the pathogenesis of myelodysplastic disorders. J. Exp. Med. 2013, 210, 2627–2639. [Google Scholar] [CrossRef]
- Sood, R.; Kamikubo, Y.; Liu, P. Role of RUNX1 in hematological malignancies. Blood 2017, 129, 2070–2082. [Google Scholar] [CrossRef]
- Chuang, L.S.H.; Ito, K.; Ito, Y. RUNX family: Regulation and diversification of roles through interacting proteins. Int. J. Cancer 2013, 132, 1260–1271. [Google Scholar] [CrossRef]
- He, W.; Zhao, C.; Hu, H. Prognostic effect of RUNX1 mutations in myelodysplastic syndromes: A meta-analysis. Hematology 2020, 25, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Barreyro, L.; Sampson, A.M.; Hueneman, K.; Choi, K.; Christie, S.; Ramesh, V.; Wyder, M.; Wang, D.; Pujato, M.; Greis, K.D.; et al. Dysregulated innate immune signaling cooperates with RUNX1 mutations to transform an MDS-like disease to AML. iScience 2024, 27, 109809. [Google Scholar] [CrossRef]
- Kaisrlikova, M.; Vesela, J.; Kundrat, D.; Votavova, H.; Merkerova, M.D.; Krejcik, Z.; Divoky, V.; Jedlicka, M.; Fric, J.; Klema, J.; et al. RUNX1 mutations contribute to the progression of MDS due to disruption of antitumor cellular defense: A study on patients with lower-risk MDS. Leukemia 2022, 36, 1898–1906. [Google Scholar] [CrossRef]
- Marion, W.; Koppe, T.; Chen, C.C.; Wang, D.; Frenis, K.; Fierstein, S.; Sensharma, P.; Aumais, O.; Peters, M.; Ruiz-Torres, S.; et al. RUNX1 mutations mitigate quiescence to promote transformation of hematopoietic progenitors in Fanconi anemia. Leukemia 2023, 37, 1698–1708. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; Chen, J.Y.; Yan, M.; Davis, A.G.; Miyauchi, S.; Chen, L.; Hao, Y.; Katz, S.; Bejar, R.; Abdel-Wahab, O.; et al. RUNX1 deficiency cooperates with SRSF2 mutation to induce multilineage hematopoietic defects characteristic of MDS. Blood Adv. 2022, 6, 6078–6092. [Google Scholar] [CrossRef] [PubMed]
- Ochi, Y.; Kon, A.; Sakata, T.; Nakagawa, M.M.; Nakazawa, N.; Kakuta, M.; Kataoka, K.; Koseki, H.; Nakayama, M.; Morishita, D.; et al. Combined Cohesin-RUNX1 Deficiency Synergistically Perturbs Chromatin Looping and Causes Myelodysplastic Syndromes. Cancer Discov. 2020, 10, 836–853. [Google Scholar] [CrossRef]
- Yang, X.; Zhao, H.; Wu, H.; Guo, X.; Jia, H.; Liu, W.; Wei, Y.; Can, C.; Ma, D. Analysis of gene mutation characteristics and its correlation with prognosis in patients with myelodysplastic syndromes. Clin. Chim. Acta 2024, 554, 117789. [Google Scholar] [CrossRef]
- Guryanova, O.A.; Lieu, Y.K.; Garrett-Bakelman, F.E.; Spitzer, B.; Glass, J.L.; Shank, K.; Martinez, A.B.; Rivera, S.A.; Durham, B.H.; Rapaport, F.; et al. Dnmt3a regulates myeloproliferation and liver-specific expansion of hematopoietic stem and progenitor cells. Leukemia 2016, 30, 1133–1142. [Google Scholar] [CrossRef]
- Wu, X.; Deng, J.; Zhang, N.; Liu, X.; Zheng, X.; Yan, T.; Ye, W.; Gong, Y. Pedigree investigation, clinical characteristics, and prognosis analysis of haematological disease patients with germline TET2 mutation. BMC Cancer 2022, 22, 262. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, K.; Shen, Q.C.; Han, Y.M.; Gu, Y.; Li, X.; Zhao, D.Z.; Liu, Y.Q.; Wang, C.M.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Neves-Costa, A.; Moita, L.F. TET1 is a negative transcriptional regulator of IL-1β in the THP-1 cell line. Mol. Immunol. 2013, 54, 264–270. [Google Scholar] [CrossRef]
- Sun, J.; He, X.; Zhu, Y.; Ding, Z.; Dong, H.; Feng, Y.; Du, J.; Wang, H.; Wu, X.; Zhang, L.; et al. SIRT1 Activation Disrupts Maintenance of Myelodysplastic Syndrome Stem and Progenitor Cells by Restoring TET2 Function. Cell Stem Cell 2018, 23, 355–369 e359. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Sun, J.; Chen, W.; Zhang, L.; He, X.; Dong, H.; Wu, Y.; Wang, H.; Li, Z.; Ball, B.; et al. TET2 deficiency promotes MDS-associated leukemogenesis. Blood Cancer J. 2022, 12, 141. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Chinnaiyan, A.M. Metabolism unhinged: IDH mutations in cancer. Nat. Med. 2011, 17, 291–293. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Makishima, H.; Yoshizato, T.; Yoshida, K.; Sekeres, M.A.; Radivoyevitch, T.; Suzuki, H.; Przychodzen, B.; Nagata, Y.; Meggendorfer, M.; Sanada, M.; et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat. Genet. 2017, 49, 204–212. [Google Scholar] [CrossRef]
- Gunn, K.; Myllykoski, M.; Cao, J.Z.; Ahmed, M.; Huang, B.; Rouaisnel, B.; Diplas, B.H.; Levitt, M.M.; Looper, R.; Doench, J.G.; et al. (R)-2-Hydroxyglutarate Inhibits KDM5 Histone Lysine Demethylases to Drive Transformation in IDH-Mutant Cancers. Cancer Discov. 2023, 13, 1478–1497. [Google Scholar] [CrossRef]
- Gu, Y.; Yang, R.; Yang, Y.; Zhao, Y.; Wakeham, A.; Li, W.Y.; Tseng, A.; Leca, J.; Berger, T.; Saunders, M.; et al. IDH1 mutation contributes to myeloid dysplasia in mice by disturbing heme biosynthesis and erythropoiesis. Blood 2021, 137, 945–958. [Google Scholar] [CrossRef]
- Malcovati, L.; Stevenson, K.; Papaemmanuil, E.; Neuberg, D.; Bejar, R.; Boultwood, J.; Bowen, D.T.; Campbell, P.J.; Ebert, B.L.; Fenaux, P.; et al. SF3B1-mutant MDS as a distinct disease subtype: A proposal from the International Working Group for the Prognosis of MDS. Blood 2020, 136, 157–170. [Google Scholar] [CrossRef]
- Zhou, Z.; Gong, Q.; Wang, Y.; Li, M.; Wang, L.; Ding, H.; Li, P. The biological function and clinical significance of SF3B1 mutations in cancer. Biomark. Res. 2020, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Dalton, W.B.; Helmenstine, E.; Pieterse, L.; Li, B.; Gocke, C.D.; Donaldson, J.; Xiao, Z.J.; Gondek, L.P.; Ghiaur, G.; Gojo, I.; et al. The K666N mutation in SF3B1 is associated with increased progression of MDS and distinct RNA splicing. Blood Adv. 2020, 4, 1192–1196. [Google Scholar] [CrossRef]
- Choudhary, G.S.; Pellagatti, A.; Agianian, B.; Smith, M.A.; Bhagat, T.D.; Gordon-Mitchell, S.; Sahu, S.; Pandey, S.; Shah, N.S.; Aluri, S.; et al. Activation of targetable inflammatory immune signaling is seen in myelodysplastic syndromes with SF3B1 mutations. Elife 2022, 11, e78136. [Google Scholar] [CrossRef]
- Dolatshad, H.; Pellagatti, A.; Liberante, F.G.; Llorian, M.; Repapi, E.; Steeples, V.; Roy, S.; Scifo, L.; Armstrong, R.N.; Shaw, J.; et al. Cryptic splicing events in the iron transporter ABCB7 and other key target genes in SF3B1-mutant myelodysplastic syndromes. Leukemia 2016, 30, 2322–2331. [Google Scholar] [CrossRef]
- Bondu, S.; Alary, A.S.; Lefevre, C.; Houy, A.; Jung, G.; Lefebvre, T.; Rombaut, D.; Boussaid, I.; Bousta, A.; Guillonneau, F.; et al. A variant erythroferrone disrupts iron homeostasis in SF3B1-mutated myelodysplastic syndrome. Sci. Transl. Med. 2019, 11, eaav5467. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Ahmed, D.; Dolatshad, H.; Tatwavedi, D.; Schulze, U.; Sanchi, A.; Ryley, S.; Dhir, A.; Carpenter, L.; Watt, S.M.; et al. SF3B1 mutations induce R-loop accumulation and DNA damage in MDS and leukemia cells with therapeutic implications. Leukemia 2020, 34, 2525–2530. [Google Scholar] [CrossRef] [PubMed]
- Wagner, R.E.; Arnetzl, L.; Britto-Borges, T.; Heit-Mondrzyk, A.; Bakr, A.; Sollier, E.; Gkatza, N.A.; Panten, J.; Delaunay, S.; Sohn, D.; et al. SRSF2 safeguards efficient transcription of DNA damage and repair genes. Cell Rep. 2024, 43, 114869. [Google Scholar] [CrossRef]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef]
- Inoue, D.; Bradley, R.K.; Abdel-Wahab, O. Spliceosomal gene mutations in myelodysplasia: Molecular links to clonal abnormalities of hematopoiesis. Genes. Dev. 2016, 30, 989–1001. [Google Scholar] [CrossRef]
- Liu, X.L.; Devadiga, S.A.; Stanley, R.F.; Morrow, R.M.; Janssen, K.A.; Quesnel-Vallieres, M.; Pomp, O.; Moverley, A.A.; Li, C.C.; Skuli, N.; et al. A mitochondrial surveillance mechanism activated by SRSF2 mutations in hematologic malignancies. J. Clin. Investig. 2024, 134, e175619. [Google Scholar] [CrossRef]
- Dutta, A.; Yang, Y.; Le, B.T.; Zhang, Y.; Abdel-Wahab, O.; Zang, C.; Mohi, G. U2af1 is required for survival and function of hematopoietic stem/progenitor cells. Leukemia 2021, 35, 2382–2398. [Google Scholar] [CrossRef] [PubMed]
- Graubert, T.A.; Shen, D.; Ding, L.; Okeyo-Owuor, T.; Lunn, C.L.; Shao, J.; Krysiak, K.; Harris, C.C.; Koboldt, D.C.; Larson, D.E.; et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet. 2011, 44, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Choudhary, G.S.; Pellagatti, A.; Choi, K.; Bolanos, L.C.; Bhagat, T.D.; Gordon-Mitchell, S.; Von Ahrens, D.; Pradhan, K.; Steeples, V.; et al. U2AF1 mutations induce oncogenic IRAK4 isoforms and activate innate immune pathways in myeloid malignancies. Nat. Cell Biol. 2019, 21, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.Q.; Song, D.D.; Guo, J.; Jin, J.C.; Tao, Y.; Zhang, Z.; Xu, F.; He, Q.; Li, X.; Chang, C.K.; et al. U2AF1 mutation promotes tumorigenicity through facilitating autophagy flux mediated by FOXO3a activation in myelodysplastic syndromes. Cell Death Dis. 2021, 12, 655. [Google Scholar] [CrossRef]
- Thota, S.; Viny, A.D.; Makishima, H.; Spitzer, B.; Radivoyevitch, T.; Przychodzen, B.; Sekeres, M.A.; Levine, R.L.; Maciejewski, J.P. Genetic alterations of the cohesin complex genes in myeloid malignancies. Blood 2014, 124, 1790–1798. [Google Scholar] [CrossRef]
- Viny, A.D.; Bowman, R.L.; Liu, Y.; Lavallee, V.P.; Eisman, S.E.; Xiao, W.; Durham, B.H.; Navitski, A.; Park, J.; Braunstein, S.; et al. Cohesin Members Stag1 and Stag2 Display Distinct Roles in Chromatin Accessibility and Topological Control of HSC Self-Renewal and Differentiation. Cell Stem Cell 2019, 25, 682–696.e8. [Google Scholar] [CrossRef]
- Tothova, Z.; Valton, A.L.; Gorelov, R.A.; Vallurupalli, M.; Krill-Burger, J.M.; Holmes, A.; Landers, C.C.; Haydu, J.E.; Malolepsza, E.; Hartigan, C.; et al. Cohesin mutations alter DNA damage repair and chromatin structure and create therapeutic vulnerabilities in MDS/AML. JCI Insight 2021, 6, e142149. [Google Scholar] [CrossRef]
- Rodriguez-Sevilla, J.J.; Ganan-Gomez, I.; Ma, F.; Chien, K.; Del Rey, M.; Loghavi, S.; Montalban-Bravo, G.; Adema, V.; Wildeman, B.; Kanagal-Shamanna, R.; et al. Hematopoietic stem cells with granulo-monocytic differentiation state overcome venetoclax sensitivity in patients with myelodysplastic syndromes. Nat. Commun. 2024, 15, 2428. [Google Scholar] [CrossRef]
- Jain, A.G.; Ball, S.; Aguirre, L.; Al Ali, N.; Kaldas, D.; Tinsley-Vance, S.; Kuykendall, A.; Chan, O.; Sweet, K.; Lancet, J.E.; et al. Patterns of lower risk myelodysplastic syndrome progression: Factors predicting progression to high-risk myelodysplastic syndrome and acute myeloid leukemia. Haematologica 2024, 109, 2157–2164. [Google Scholar] [CrossRef]
- Meggendorfer, M.; de Albuquerque, A.; Nadarajah, N.; Alpermann, T.; Kern, W.; Steuer, K.; Perglerova, K.; Haferlach, C.; Schnittger, S.; Haferlach, T. Karyotype evolution and acquisition of FLT3 or RAS pathway alterations drive progression of myelodysplastic syndrome to acute myeloid leukemia. Haematologica 2015, 100, e487–e490. [Google Scholar] [CrossRef]
- Ren, Y.; Lang, W.; Mei, C.; Luo, Y.; Ye, L.; Wang, L.; Zhou, X.; Xu, G.; Ma, L.; Jin, J.; et al. Co-mutation landscape and clinical significance of RAS pathway related gene mutations in patients with myelodysplastic syndrome. Hematol. Oncol. 2023, 41, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Osswald, L.; Hamarsheh, S.; Uhl, F.M.; Andrieux, G.; Klein, C.; Dierks, C.; Duquesne, S.; Braun, L.M.; Schmitt-Graeff, A.; Duyster, J.; et al. Oncogenic Kras(G12D) Activation in the Nonhematopoietic Bone Marrow Microenvironment Causes Myelodysplastic Syndrome in Mice. Mol. Cancer Res. 2021, 19, 1596–1608. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef]
- Daver, N.G.; Maiti, A.; Kadia, T.M.; Vyas, P.; Majeti, R.; Wei, A.H.; Garcia-Manero, G.; Craddock, C.; Sallman, D.A.; Kantarjian, H.M. TP53-Mutated Myelodysplastic Syndrome and Acute Myeloid Leukemia: Biology, Current Therapy, and Future Directions. Cancer Discov. 2022, 12, 2516–2529. [Google Scholar] [CrossRef]
- Pant, V.; Quintas-Cardama, A.; Lozano, G. The p53 pathway in hematopoiesis: Lessons from mouse models, implications for humans. Blood 2012, 120, 5118–5127. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kao, Y.-R.; Sun, D.; Todorova, T.I.; Reynolds, D.; Narayanagari, S.-R.; Montagna, C.; Will, B.; Verma, A.; Steidl, U. Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nat. Med. 2019, 25, 103–110. [Google Scholar] [CrossRef]
- Sallman, D.A.; McLemore, A.F.; Aldrich, A.L.; Komrokji, R.S.; McGraw, K.L.; Dhawan, A.; Geyer, S.; Hou, H.A.; Eksioglu, E.A.; Sullivan, A.; et al. TP53 mutations in myelodysplastic syndromes and secondary AML confer an immunosuppressive phenotype. Blood 2020, 136, 2812–2823. [Google Scholar] [CrossRef]
- Stoddart, A.; Fernald, A.A.; Wang, J.; Davis, E.M.; Karrison, T.; Anastasi, J.; Le Beau, M.M. Haploinsufficiency of del(5q) genes, Egr1 and Apc, cooperate with Tp53 loss to induce acute myeloid leukemia in mice. Blood 2014, 123, 1069–1078. [Google Scholar] [CrossRef]
- Hsu, J.; Reilly, A.; Hayes, B.J.; Clough, C.A.; Konnick, E.Q.; Torok-Storb, B.; Gulsuner, S.; Wu, D.; Becker, P.S.; Keel, S.B.; et al. Reprogramming identifies functionally distinct stages of clonal evolution in myelodysplastic syndromes. Blood 2019, 134, 186–198. [Google Scholar] [CrossRef]
- Feurstein, S.; Churpek, J.E.; Walsh, T.; Keel, S.; Hakkarainen, M.; Schroeder, T.; Germing, U.; Geyh, S.; Heuser, M.; Thol, F.; et al. Germline variants drive myelodysplastic syndrome in young adults. Leukemia 2021, 35, 2439–2444. [Google Scholar] [CrossRef]
- Keel, S.B.; Scott, A.; Sanchez-Bonilla, M.; Ho, P.A.; Gulsuner, S.; Pritchard, C.C.; Abkowitz, J.L.; King, M.C.; Walsh, T.; Shimamura, A. Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients. Haematologica 2016, 101, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Attardi, E.; Tiberi, L.; Mattiuz, G.; Formicola, D.; Dirupo, E.; Raddi, M.G.; Consagra, A.; Vergani, D.; Artuso, R.; Santini, V. Prospective genetic germline evaluation in a consecutive group of adult patients aged <60 years with myelodysplastic syndromes. Hemasphere 2024, 8, e112. [Google Scholar] [CrossRef]
- Chlon, T.M.; Stepanchick, E.; Hershberger, C.E.; Daniels, N.J.; Hueneman, K.M.; Kuenzi Davis, A.; Choi, K.; Zheng, Y.; Gurnari, C.; Haferlach, T.; et al. Germline DDX41 mutations cause ineffective hematopoiesis and myelodysplasia. Cell Stem Cell 2021, 28, 1966–1981 e6. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, J.T.; Ghazale, N.; Pradhan, K.; Gupta, V.; Potts, K.S.; Tricomi, B.; Daniels, N.J.; Padgett, R.A.; De Oliveira, S.; Verma, A.; et al. Excessive R-loops trigger an inflammatory cascade leading to increased HSPC production. Dev. Cell 2021, 56, 627–640 e625. [Google Scholar] [CrossRef]
- Nagata, Y.; Narumi, S.; Guan, Y.; Przychodzen, B.P.; Hirsch, C.M.; Makishima, H.; Shima, H.; Aly, M.; Pastor, V.; Kuzmanovic, T.; et al. Germline loss-of-function SAMD9 and SAMD9L alterations in adult myelodysplastic syndromes. Blood 2018, 132, 2309–2313. [Google Scholar] [CrossRef]
- Chong, C.E.; Venugopal, P.; Stokes, P.H.; Lee, Y.K.; Brautigan, P.J.; Yeung, D.T.O.; Babic, M.; Engler, G.A.; Lane, S.W.; Klingler-Hoffmann, M.; et al. Differential effects on gene transcription and hematopoietic differentiation correlate with GATA2 mutant disease phenotypes. Leukemia 2018, 32, 194–202. [Google Scholar] [CrossRef]
- Homan, C.C.; Venugopal, P.; Arts, P.; Shahrin, N.H.; Feurstein, S.; Rawlings, L.; Lawrence, D.M.; Andrews, J.; King-Smith, S.L.; Harvey, N.L.; et al. GATA2 deficiency syndrome: A decade of discovery. Hum. Mutat. 2021, 42, 1399–1421. [Google Scholar] [CrossRef] [PubMed]
- Giudice, V.; Wu, Z.; Kajigaya, S.; Fernandez Ibanez, M.D.P.; Rios, O.; Cheung, F.; Ito, S.; Young, N.S. Circulating S100A8 and S100A9 protein levels in plasma of patients with acquired aplastic anemia and myelodysplastic syndromes. Cytokine 2019, 113, 462–465. [Google Scholar] [CrossRef]
- Wang, Y.H.; Lin, C.C.; Yao, C.Y.; Amaral, F.M.R.; Yu, S.C.; Kao, C.J.; Shih, P.T.; Hou, H.A.; Chou, W.C.; Tien, H.F. High BM plasma S100A8/A9 is associated with a perturbed microenvironment and poor prognosis in myelodysplastic syndromes. Blood Adv. 2023, 7, 2528–2533. [Google Scholar] [CrossRef]
- Orhan, B.; Öztürk Nazlıoğlu, H.; Dik, O.; Gürbüz, B.; Özkocaman, V.; Ersal, T.; Pınar, İ.E.; Yalçın, C.; Çubukçu, S.; Güllü Koca, T.; et al. Predictive value of TGF-β1 and SMAD-7 expression at diagnosis for treatment response in low-risk myelodysplastic syndrome. Biomol. Biomed. 2025, 25, 1175–1183. [Google Scholar] [CrossRef]
- Han, D.; Tao, J.; Fu, R.; Shao, Z. Myeloid-derived suppressor cell cytokine secretion as prognostic factor in myelodysplastic syndromes. Innate Immun. 2020, 26, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Finke, C.; Lasho, T.L.; Al-Kali, A.; Begna, K.H.; Hanson, C.A.; Tefferi, A. IPSS-independent prognostic value of plasma CXCL10, IL-7 and IL-6 levels in myelodysplastic syndromes. Leukemia 2012, 26, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, A.B.; Hansen, J.W.; Ørskov, A.D.; Dimopoulos, K.; Salem, M.; Grigorian, M.; Bruunsgaard, H.; Grønbæk, K. Inflammatory Cytokine Profiles Do Not Differ Between Patients With Idiopathic Cytopenias of Undetermined Significance and Myelodysplastic Syndromes. Hemasphere 2022, 6, e0713. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.M.; Bewersdorf, J.P.; Hasle, V.; Shallis, R.M.; Thompson, E.; de Menezes, D.L.; Rose, S.; Boss, I.; Halene, S.; Haferlach, T.; et al. Integrated genetic, epigenetic, and immune landscape of TP53 mutant AML and higher risk MDS treated with azacitidine. Ther. Adv. Hematol. 2024, 15, 20406207241257904. [Google Scholar] [CrossRef]
| Gene | Wide-Type’s Main Biological Functions | Mutational Frequencies and Clinical Significance | Mechanisms of Mutational Effects |
|---|---|---|---|
| ASXL1 | Cooperates with PRC2 to facilitate H3K27me3; cooperates with PRC1 to facilitate H2AK119Ub; downregulates the HOXA cluster via PRC2. The OGT-ASXL1 axis mediates methylation of H3K4 [156,157,158,159]. | 15–20%. Some studies report poorer OS in ASXL1-mutated MDS patients [160,161]. | ASXL1 mutation or knockdown causes loss of PRC2-mediated H3K27me3 and upregulation of HOXA genes, leading to impaired hematopoiesis and MDS-like phenotypes [158,162]. ASXL1 mutation reduces the expression of genes related to erythroid differentiation and/or maturation, and reduction in H2AK119Ub is related to leukemic transformation [158,162,163]. |
| EZH2 | Catalyzes the recruitment of PRC2 by interacting with ASXL1; maintains normal H3K27me3 levels [158,164]. | 5–10%. EZH2 mutations often co-mutate with TET2 or RUNX1; but the prognostic values of mutant-EZH2 remain uncertain [165,166,167]. | In MDS, loss-of-function EZH2 mutations are more frequent [164]. Loss of EZH2 function with TET2KD/KD induces aberrant DNA hypermethylation and promotes the pathogenesis of MDS [168,169]. |
| RUNX1 | Key transcription factor for hematopoiesis. Involved in the epigenetic regulation [170,171]. | 10–15%. Mutant-RUNX1 correlates with worse OS and LFS in MDS [172]. | RUNX1 mutation with deletion of miR-146a can drive the transformation of normal HSPCs to MDS, and subsequent progression to AML [173]. RUNX1 mutations lead to the elimination of the DDR-mediated senescence barrier and promote the progression of MDS [174]. RUNX1-mutated HPCs from edited Fanconi anemia iPSC have higher expression of IRAK1 and activated NF-κB pathway and show MDS-like phenotypes [175]. RUNX1 deficiency with SRSF2 mutation induces MDS phenotype by causing mis-splicing of genes in the DDR and cell cycle checkpoint pathways [176]. Co-deficiency of STAG2/RUNX1 induces MDS-like phenotypes by disrupting enhancer-promoter looping dynamics and downregulating genes with high basal transcriptional pausing [177]. |
| Gene | Wide-Type’s Main Biological Functions | Mutational Frequencies and Clinical Significance | Mechanisms of Mutational Effects |
|---|---|---|---|
| DNMT3A | Encoding enzymes for initiating de novo DNA methylation, catalyzing the conversion of unmethylated cytosine to methylated status at CpG sites [6,155]. | 10–15%. DNMT3A mutation is an independent risk factor for death in patients with MDS [178]. | Dnmt3a-KO mice show MDS-like phenotypes and hepatomegaly. The Dnmt3a-null progenitor cells show global hypomethylation and reactivation of fetal liver hematopoiesis transcriptional programs [179]. |
| TET2 | Cooperates with α-KG to demethylate DNA by hydroxylating 5-methylcytosine [6]. | 20–30%. The prognostic value of TET2 mutations remains uncertain [178,180]. | The absence of TET2 leads to increased expression of IL-6 and IL-1β in response to inflammatory stimuli, enhancing innate immune responses in MDS [181,182]. Reduced expression of SIRT1 in MDS HSPCs leads to TET2 hyperacetylation, enhanced self-renewal and maintenance of MDS HSPCs [183]. TET2 deletion in MDS HSPCs results in a reduced global level of 5hmC; the deficiency of TET2 activity increases the risk of MDS transforming to AML by a higher occurrence of secondary malignant mutations [184]. |
| IDH1 or IDH2 | Converts isocitrate to α-KG; α-KG with TET2 hydroxylates 5-methylcytosine [185,186]. | 2–5%. IDH2 mutations are more prevalent in high-risk MDS than low-risk MDS [187]. | Abnormal IDH encoded by mutant IDH1/2 catalyzes α-KG to R-2-HG, which promotes the occurrence and progression of AML by reducing global levels of 5hmC and inhibiting KDM5 histone lysine demethylases [188]. In murine models, R-2-HG inhibits oxoglutarate dehydrogenase activity and leads to reduced production of CoA, then the insufficiency of succinyl-CoA attenuates the biosynthesis of heme in IDH1-mutant hematopoietic cells and induces abnormal erythropoiesis [189]. |
| Gene | Wide-Type’s Main Biological Functions | Mutational Frequencies and Clinical Significance | Mechanisms of Mutational Effects |
|---|---|---|---|
| SF3B1 | SF3B1 is the core component of the spliceosome [190]. | 20–30%. Notably, SF3B1 mutations are more frequent in patients with MDS with ring sideroblasts and are associated with a relatively better prognosis [166,191]. However, SF3B1K666N mutation may correlate with poorer prognosis compared to other SF3B1 mutations in MDS patients [192]. | Mutated SF3B1 induces aberrant splicing of IRAK4, resulting in a long IRAK isoform that leads to hyperactivation of the NF-κB pathway [193]. Mutated SF3B1 induces aberrant splicing of the iron transporter ABCB7, leading to reduced ABCB7 expression and iron accumulation in the mitochondria in erythroid progenitors [194]. Mis-splicing of ERFE, a key regulator of iron homeostasis, further exacerbates iron dysregulation in SF3B1-mutant MDS [195]. Mutated SF3B1 induces the accumulation of R-loops in MDS and leukemia cells, contributing to DNA damage and genomic instability [196]. |
| SRSF2 | Regulates pre-mRNA splicing in the nucleus [197]. | 10–15%. SRSF2 mutations often co-mutate with IDH2 mutations and are associated with a shorter leukemia-free survival in MDS [167]. | Conditional expression of the SRSF2P95H mutation in murine models recapitulates MDS phenotypes, driven by mutant SRSF2’s altered preference for specific exonic splicing enhancer motifs [198]. The mis-splicing results in the aberrant isoforms of some key hematopoietic regulators and degradation of the EZH2, impairing hematopoietic differentiation and increasing leukemic risk [198,199]. SRSF2P95H/+ impairs the splicing of mitochondrial mRNAs, increases mitophagy, and elevates the expression of PINK1 (which is vital for the survival of SRSF2-mutant cells) [200]. |
| U2AF1 | In pre-mRNA splicing, U2AF1 participates in the recognition of the 3’ splice site, and is essential for the maintenance and normal function of HSPCs [201]. | 5–10%. MDS patients harboring U2AF1 mutations generally present with a poorer prognosis [178,202]. | Mutant U2AF1 leads to high-activity isoform long IRAK4, amplifying downstream innate immune responses [203]. SKM-1 and K562 cells with U2AF1S34F mutation show reduced proliferation and increased apoptosis, and the U2AF1S34F SKM-1 cells show elevated mRNA of FOXO3a; the dysregulation of FOXO3a restores autophagy flux and activates the NLRP3 inflammasome [204]. |
| Drugs | Phase | MDS Types and Gene Abnormalities | Main Mechanisms | Register No. |
|---|---|---|---|---|
| Luspatercept | Phase 2 | Lower-risk MDS with splicing mutation (SRSF2, U2AF1, ZRSR2), or with SF3B1 mutation and received HMA and/or lenalidomide prior treatments | Binding to TGF-β and reducing SMAD2/3 | NCT05732961 |
| Emavusertib (CA-4948) | Phase 1/2 | Refractory/relapse (R/R) higher-risk MDS with spliceosome mutations of SF3B1 or U2AF1 | Inhibitor of IRAK4 and FLT3 | NCT04278768 |
| Eltrombopag | Phase 2 | Lower-risk MDS with TET2 mutations | Thrombopoietin receptor agonist and inhibiting the growth of TET2-mutated cells | NCT06630221 |
| Ivosidenib-based therapies | - | MDS with IDH1 mutation (The specific types of MDS depend on the study designs) | Inhibitor of mutant IDH1 | NCT02074839; NCT04250051; NCT03471260; NCT03839771 |
| Olutasidenib-based therapies | - | MDS with IDH1 mutation (The specific types of MDS depend on the study designs) | Inhibitor of mutant IDH1 | NCT06543381; NCT06597734 |
| Enasidenib-based therapies | - | MDS with IDH2 mutation (The specific types of MDS depend on the study designs) | Inhibitor of mutant IDH2 | NCT03744390; NCT06577441; NCT03839771; |
| Oral Arsenic Trioxide | Phase 2 | MDS with TP53 mutation | Rescuing structural p53 mutations | NCT06778187 |
| Quizartinib | Phase 1/2 | MDS with FLT3-ITD mutation, or presence of CBL exon 8 or 9 deletions or point mutations | Inhibitor of FLT3 | NCT04493138 |
| Gilteritinib-based therapies | - | MDS with FLT3 mutations (The specific types of MDS depend on the study designs) | Inhibitor of FLT3 | NCT04027309; NCT05010122 |
| Components | Prognostic Correlations | References |
|---|---|---|
| S100A8/A9 heterodimer | High concentration of S100A8/A9 heterodimer in bone marrow plasma (cutoff: 7093 ng/mL) is correlated with worse LFS and OS. | [229] |
| Hyaluronan | Higher concentration of hyaluronan in bone marrow serum (>100 μg/L) is correlated with worse LFS and OS. | [114] |
| IL-6, IL-7, and CXCL10 | Patients with normal plasma levels of IL-6, IL-7, and CXCL10 have better OS than those with elevated levels of at least one of the three cytokines; elevated level of IL-6 correlates with worse LFS. | [232] |
| High inflammatory load | High inflammatory load (IL-6, TNF-α, IL-10, and CXCL10) in blood plasma correlates with shorter OS in clonal cytopenias of undetermined significance (CCUS) and MDS. | [233] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Zou, C.; Xiang, X.; Zhao, L.; Chen, M.; Yang, C.; Wu, Y. Myelodysplastic Neoplasms (MDS): Pathogenesis and Therapeutic Prospects. Biomolecules 2025, 15, 761. https://doi.org/10.3390/biom15060761
Li X, Zou C, Xiang X, Zhao L, Chen M, Yang C, Wu Y. Myelodysplastic Neoplasms (MDS): Pathogenesis and Therapeutic Prospects. Biomolecules. 2025; 15(6):761. https://doi.org/10.3390/biom15060761
Chicago/Turabian StyleLi, Xuefeng, Chaoyu Zou, Xinrong Xiang, Lei Zhao, Mengran Chen, Chenlu Yang, and Yu Wu. 2025. "Myelodysplastic Neoplasms (MDS): Pathogenesis and Therapeutic Prospects" Biomolecules 15, no. 6: 761. https://doi.org/10.3390/biom15060761
APA StyleLi, X., Zou, C., Xiang, X., Zhao, L., Chen, M., Yang, C., & Wu, Y. (2025). Myelodysplastic Neoplasms (MDS): Pathogenesis and Therapeutic Prospects. Biomolecules, 15(6), 761. https://doi.org/10.3390/biom15060761