




Neuroprotective Roles of the Biliverdin Reductase-A/Bilirubin Axis in the Brain


Abstract
1. Introduction
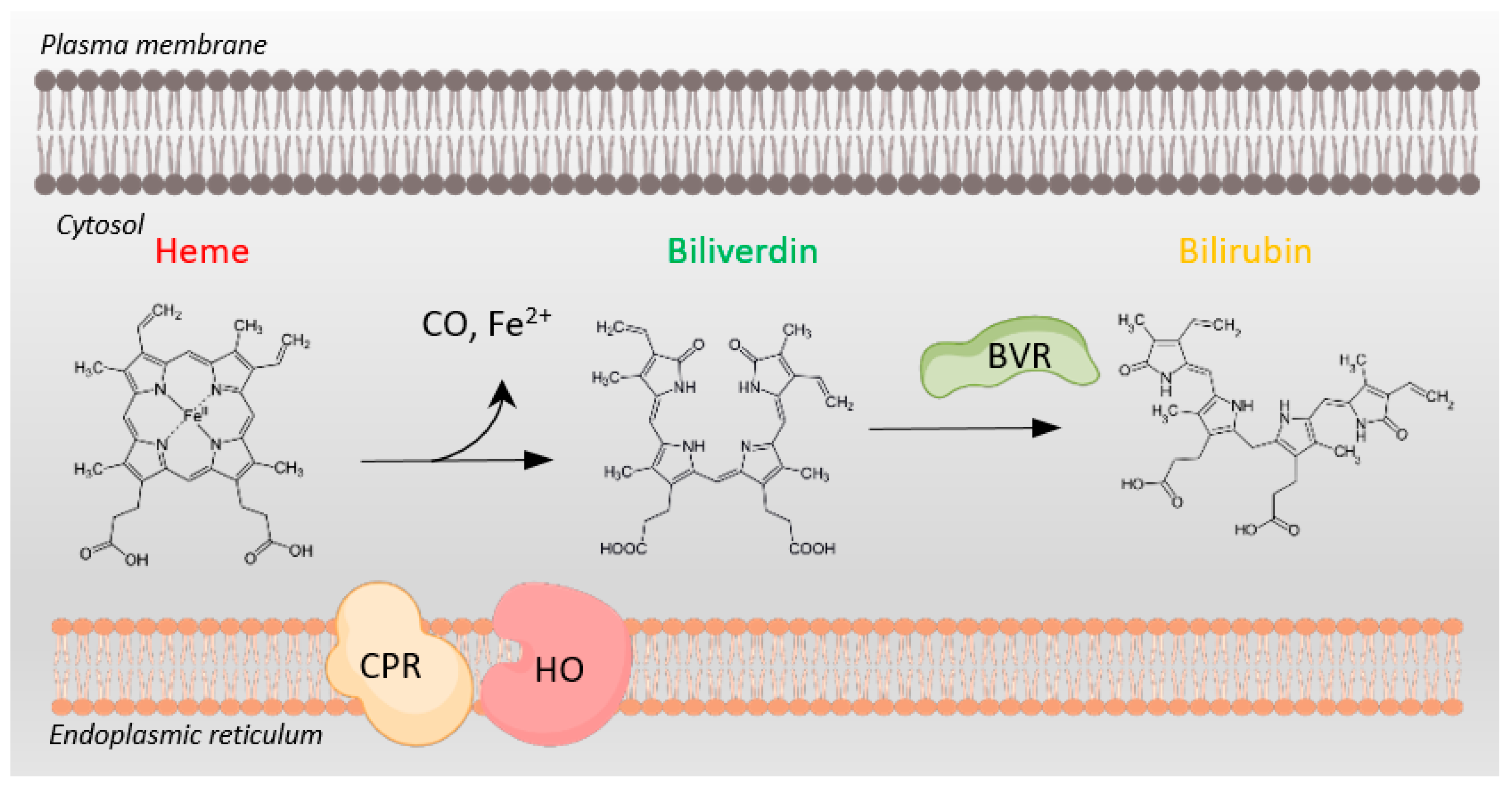
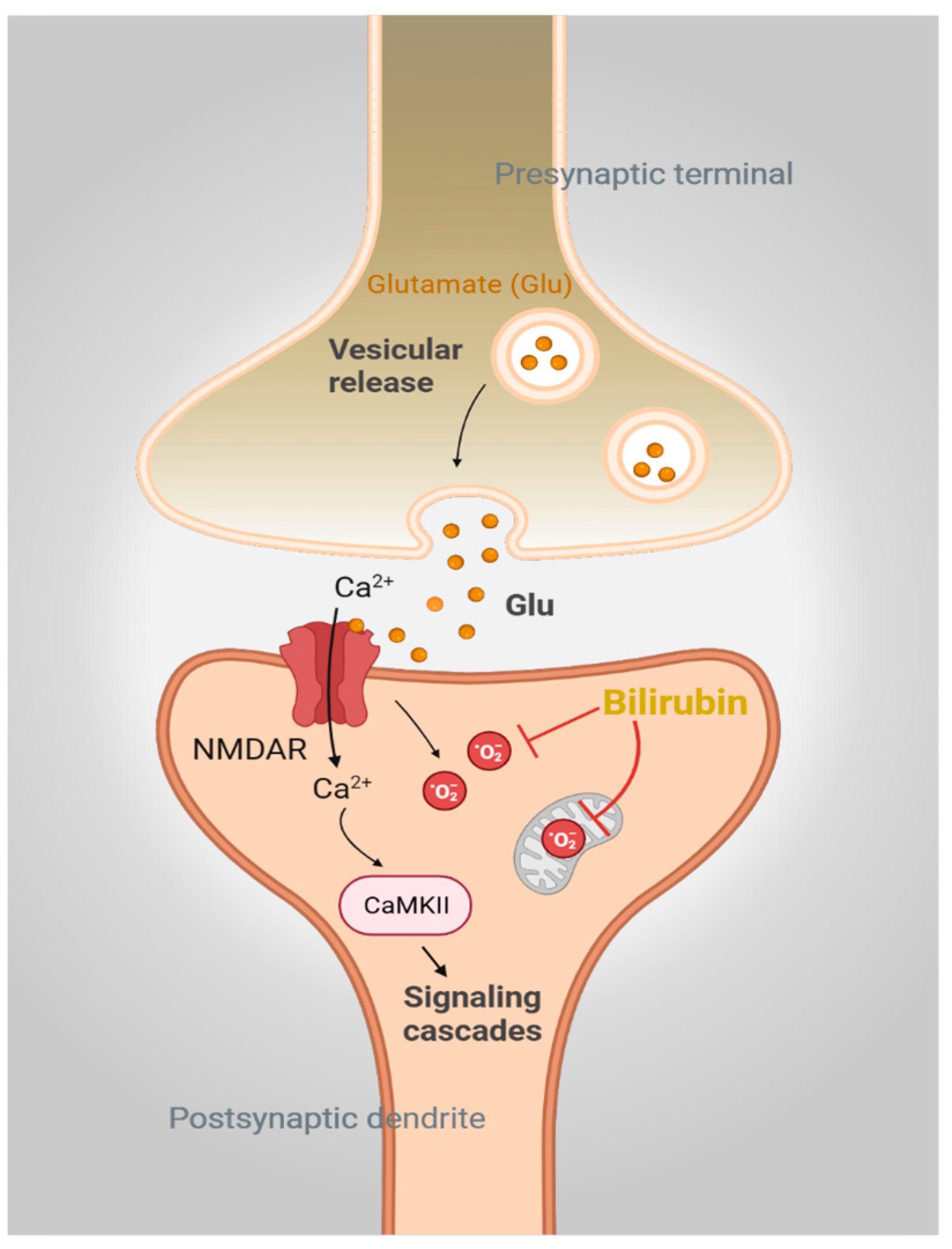
2. Bilirubin in the Brain: The Good, the Bad, and the Ugly
3. Biliverdin Reductase-A in the Brain: More Than an Enzyme for Bilirubin Synthesis
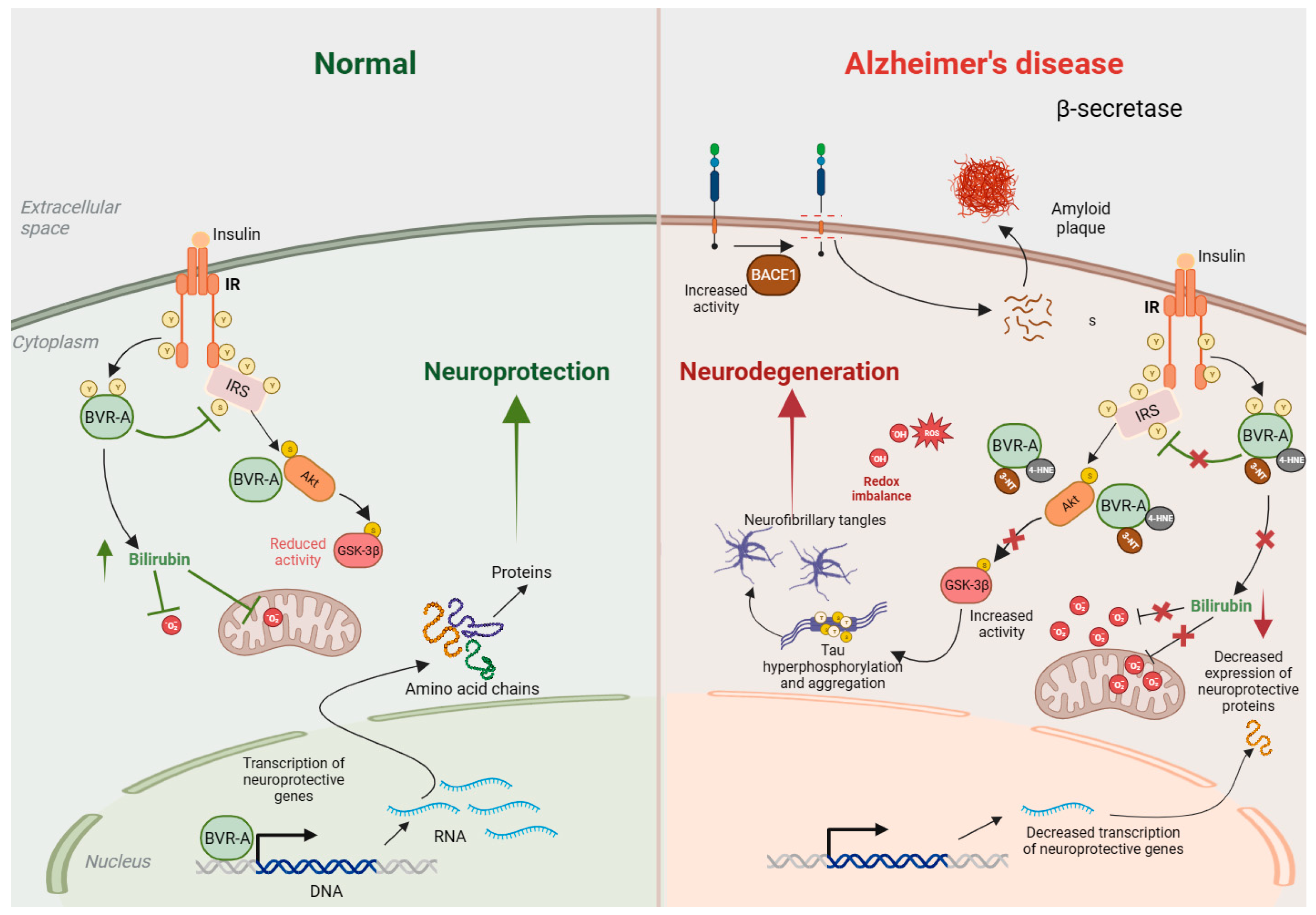
3.1. BVRA and Bilirubin in Alzheimer’s Disease
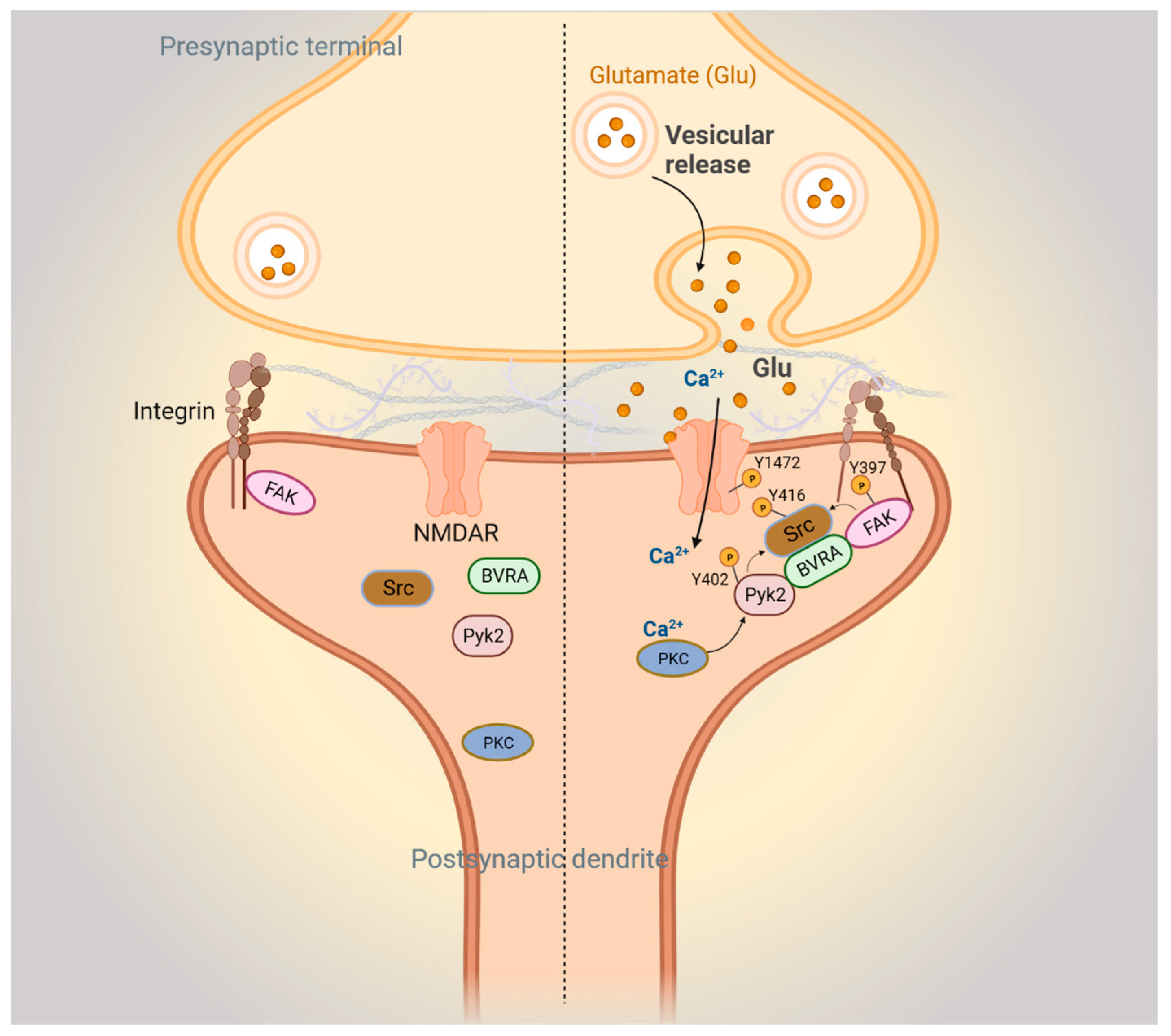
3.2. BVRA and Synaptic Plasticity
4. Therapeutic Opportunities
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Lemberg, R.; Wyndham, R.A. Reduction of biliverdin to bilirubin in tissues. Biochem. J. 1936, 30, 1147–1170. [Google Scholar] [CrossRef] [PubMed]
- Singleton, J.W.; Laster, L. Biliverdin reductase of guinea pig liver. J. Biol. Chem. 1965, 240, 4780–4789. [Google Scholar] [CrossRef] [PubMed]
- Kutty, R.K.; Maines, M.D. Purification and characterization of biliverdin reductase from rat liver. J. Biol. Chem. 1981, 256, 3956–3962. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D.; Trakshel, G.M. Purification and characterization of human biliverdin reductase. Arch. Biochem. Biophys. 1993, 300, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Kapitulnik, J.; Maines, M.D. Pleiotropic functions of biliverdin reductase: Cellular signaling and generation of cytoprotective and cytotoxic bilirubin. Trends Pharmacol. Sci. 2009, 30, 129–137. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, L.; Hosick, P.A.; John, K.; Stec, D.E.; Hinds, T.D., Jr. Biliverdin reductase isozymes in metabolism. Trends Endocrinol. Metab. 2015, 26, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, O.; Gore, M.G.; Mantle, T.J. Initial-rate kinetics of the flavin reductase reaction catalysed by human biliverdin-IXbeta reductase (BVR-B). Biochem. J. 2000, 345 Pt 2, 393–399. [Google Scholar] [CrossRef]
- Vasavda, C.; Kothari, R.; Malla, A.P.; Tokhunts, R.; Lin, A.; Ji, M.; Ricco, C.; Xu, R.; Saavedra, H.G.; Sbodio, J.I.; et al. Bilirubin Links Heme Metabolism to Neuroprotection by Scavenging Superoxide. Cell Chem. Biol. 2019, 26, 1450–1460.e7. [Google Scholar] [CrossRef]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef]
- Mireles, L.C.; Lum, M.A.; Dennery, P.A. Antioxidant and cytotoxic effects of bilirubin on neonatal erythrocytes. Pediatr. Res. 1999, 45, 355–362. [Google Scholar] [CrossRef]
- McDonagh, A.F. Is bilirubin good for you? Clin. Perinatol. 1990, 17, 359–369. [Google Scholar] [CrossRef]
- Mancuso, C.; Barone, E.; Guido, P.; Miceli, F.; Di Domenico, F.; Perluigi, M.; Santangelo, R.; Preziosi, P. Inhibition of lipid peroxidation and protein oxidation by endogenous and exogenous antioxidants in rat brain microsomes in vitro. Neurosci. Lett. 2012, 518, 101–105. [Google Scholar] [CrossRef]
- Mancuso, C. Biliverdin as a disease-modifying agent: An integrated viewpoint. Free Radic. Biol. Med. 2023, 207, 133–143. [Google Scholar] [CrossRef]
- Sedlak, T.W.; Saleh, M.; Higginson, D.S.; Paul, B.D.; Juluri, K.R.; Snyder, S.H. Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc. Natl. Acad. Sci. USA 2009, 106, 5171–5176. [Google Scholar] [CrossRef]
- Baranano, D.E.; Rao, M.; Ferris, C.D.; Snyder, S.H. Biliverdin reductase: A major physiologic cytoprotectant. Proc. Natl. Acad. Sci. USA 2002, 99, 16093–16098. [Google Scholar] [CrossRef] [PubMed]
- Kaasik, K.; Lee, C.C. Reciprocal regulation of haem biosynthesis and the circadian clock in mammals. Nature 2004, 430, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Falchuk, K.H.; Contin, J.M.; Dziedzic, T.S.; Feng, Z.; French, T.C.; Heffron, G.J.; Montorzi, M. A role for biliverdin IXalpha in dorsal axis development of Xenopus laevis embryos. Proc. Natl. Acad. Sci. USA 2002, 99, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Tukey, R.H.; Strassburg, C.P. Human UDP-glucuronosyltransferases: Metabolism, expression, and disease. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 581–616. [Google Scholar] [CrossRef] [PubMed]
- Abu-Bakar, A.; Arthur, D.M.; Aganovic, S.; Ng, J.C.; Lang, M.A. Inducible bilirubin oxidase: A novel function for the mouse cytochrome P450 2A5. Toxicol. Appl. Pharmacol. 2011, 257, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Vitek, L.; Ostrow, J.D. Bilirubin chemistry and metabolism; harmful and protective aspects. Curr. Pharm. Des. 2009, 15, 2869–2883. [Google Scholar] [CrossRef] [PubMed]
- Boon, A.C.; Bulmer, A.C.; Coombes, J.S.; Fassett, R.G. Circulating bilirubin and defense against kidney disease and cardiovascular mortality: Mechanisms contributing to protection in clinical investigations. Am. J. Physiol. Renal Physiol. 2014, 307, F123–F136. [Google Scholar] [CrossRef]
- Wagner, K.H.; Wallner, M.; Molzer, C.; Gazzin, S.; Bulmer, A.C.; Tiribelli, C.; Vitek, L. Looking to the horizon: The role of bilirubin in the development and prevention of age-related chronic diseases. Clin. Sci. 2015, 129, 1–25. [Google Scholar] [CrossRef]
- Gazzin, S.; Masutti, F.; Vitek, L.; Tiribelli, C. The molecular basis of jaundice: An old symptom revisited. Liver Int. 2017, 37, 1094–1102. [Google Scholar] [CrossRef]
- Wagner, K.H.; Khoei, N.S.; Hana, C.A.; Doberer, D.; Marculescu, R.; Bulmer, A.C.; Hormann-Wallner, M.; Molzer, C. Oxidative Stress and Related Biomarkers in Gilbert’s Syndrome: A Secondary Analysis of Two Case-Control Studies. Antioxidants 2021, 10, 1474. [Google Scholar] [CrossRef]
- Canu, G.; Minucci, A.; Zuppi, C.; Capoluongo, E. Gilbert and Crigler Najjar syndromes: An update of the UDP-glucuronosyltransferase 1A1 (UGT1A1) gene mutation database. Blood Cells Mol. Dis. 2013, 50, 273–280. [Google Scholar] [CrossRef]
- Gantla, S.; Bakker, C.T.; Deocharan, B.; Thummala, N.R.; Zweiner, J.; Sinaasappel, M.; Roy Chowdhury, J.; Bosma, P.J.; Roy Chowdhury, N. Splice-site mutations: A novel genetic mechanism of Crigler-Najjar syndrome type 1. Am. J. Hum. Genet. 1998, 62, 585–592. [Google Scholar] [CrossRef]
- Ritter, J.K.; Yeatman, M.T.; Ferreira, P.; Owens, I.S. Identification of a genetic alteration in the code for bilirubin UDP-glucuronosyltransferase in the UGT1 gene complex of a Crigler-Najjar type I patient. J. Clin. Investig. 1992, 90, 150–155. [Google Scholar] [CrossRef]
- Crigler, J.F., Jr.; Najjar, V.A. Congenital familial nonhemolytic jaundice with kernicterus. Pediatrics 1952, 10, 169–180. [Google Scholar] [PubMed]
- Reyes, R.C.; Brennan, A.M.; Shen, Y.; Baldwin, Y.; Swanson, R.A. Activation of neuronal NMDA receptors induces superoxide-mediated oxidative stress in neighboring neurons and astrocytes. J. Neurosci. 2012, 32, 12973–12978. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Holley, A.E.; Flitter, W.D.; Slater, T.F.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Jenner, P.; Marsden, C.D. Increased levels of lipid hydroperoxides in the parkinsonian substantia nigra: An HPLC and ESR study. Mov. Disord. 1994, 9, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H., Jr.; Beal, M.F. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 1997, 69, 2064–2074. [Google Scholar] [CrossRef]
- Subbarao, K.V.; Richardson, J.S.; Ang, L.C. Autopsy samples of Alzheimer’s cortex show increased peroxidation in vitro. J. Neurochem. 1990, 55, 342–345. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Vasavda, C.; Snyder, S.H.; Paul, B.D. Quantitative measurement of reactive oxygen species in ex vivo mouse brain slices. STAR Protoc. 2021, 2, 100332. [Google Scholar] [CrossRef]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kontos, C.D.; Wei, E.P.; Williams, J.I.; Kontos, H.A.; Povlishock, J.T. Cytochemical detection of superoxide in cerebral inflammation and ischemia in vivo. Am. J. Physiol. 1992, 263, H1234–H1242. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D. Multiple forms of biliverdin reductase: Age-related change in pattern of expression in rat liver and brain. Mol. Pharmacol. 1990, 38, 481–485. [Google Scholar]
- Panahian, N.; Huang, T.; Maines, M.D. Enhanced neuronal expression of the oxidoreductase--biliverdin reductase--after permanent focal cerebral ischemia. Brain Res. 1999, 850, 1–13. [Google Scholar] [CrossRef]
- Gibbs, P.E.; Tudor, C.; Maines, M.D. Biliverdin reductase: More than a namesake-the reductase, its Peptide fragments, and biliverdin regulate activity of the three classes of protein kinase C. Front. Pharmacol. 2012, 3, 31. [Google Scholar] [CrossRef]
- Cimini, F.A.; Perluigi, M.; Barchetta, I.; Cavallo, M.G.; Barone, E. Role of Biliverdin Reductase A in the Regulation of Insulin Signaling in Metabolic and Neurodegenerative Diseases: An Update. Int. J. Mol. Sci. 2022, 23, 5574. [Google Scholar] [CrossRef] [PubMed]
- Lerner-Marmarosh, N.; Shen, J.; Torno, M.D.; Kravets, A.; Hu, Z.; Maines, M.D. Human biliverdin reductase: A member of the insulin receptor substrate family with serine/threonine/tyrosine kinase activity. Proc. Natl. Acad. Sci. USA 2005, 102, 7109–7114. [Google Scholar] [CrossRef] [PubMed]
- Lerner-Marmarosh, N.; Miralem, T.; Gibbs, P.E.; Maines, M.D. Regulation of TNF-alpha-activated PKC-zeta signaling by the human biliverdin reductase: Identification of activating and inhibitory domains of the reductase. FASEB J. 2007, 21, 3949–3962. [Google Scholar] [CrossRef] [PubMed]
- Lerner-Marmarosh, N.; Miralem, T.; Gibbs, P.E.; Maines, M.D. Human biliverdin reductase is an ERK activator; hBVR is an ERK nuclear transporter and is required for MAPK signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 6870–6875. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D. Biliverdin reductase: PKC interaction at the cross-talk of MAPK and PI3K signaling pathways. Antioxid. Redox Signal. 2007, 9, 2187–2195. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Burns, K.A.; Hosick, P.A.; McBeth, L.; Nestor-Kalinoski, A.; Drummond, H.A.; AlAmodi, A.A.; Hankins, M.W.; Vanden Heuvel, J.P.; Stec, D.E. Biliverdin Reductase A Attenuates Hepatic Steatosis by Inhibition of Glycogen Synthase Kinase (GSK) 3beta Phosphorylation of Serine 73 of Peroxisome Proliferator-activated Receptor (PPAR) alpha. J. Biol. Chem. 2016, 291, 25179–25191. [Google Scholar] [CrossRef]
- Imazu, M.; Strickland, W.G.; Chrisman, T.D.; Exton, J.H. Phosphorylation and inactivation of liver glycogen synthase by liver protein kinases. J. Biol. Chem. 1984, 259, 1813–1821. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Orena, S.J.; Torchia, A.J.; Garofalo, R.S. Inhibition of glycogen-synthase kinase 3 stimulates glycogen synthase and glucose transport by distinct mechanisms in 3T3-L1 adipocytes. J. Biol. Chem. 2000, 275, 15765–15772. [Google Scholar] [CrossRef] [PubMed]
- Miralem, T.; Lerner-Marmarosh, N.; Gibbs, P.E.; Jenkins, J.L.; Heimiller, C.; Maines, M.D. Interaction of human biliverdin reductase with Akt/protein kinase B and phosphatidylinositol-dependent kinase 1 regulates glycogen synthase kinase 3 activity: A novel mechanism of Akt activation. FASEB J. 2016, 30, 2926–2944. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, P.E.; Lerner-Marmarosh, N.; Poulin, A.; Farah, E.; Maines, M.D. Human biliverdin reductase-based peptides activate and inhibit glucose uptake through direct interaction with the kinase domain of insulin receptor. FASEB J. 2014, 28, 2478–2491. [Google Scholar] [CrossRef]
- Song, S.; Wang, S.; Ma, J.; Yao, L.; Xing, H.; Zhang, L.; Liao, L.; Zhu, D. Biliverdin reductase/bilirubin mediates the anti-apoptotic effect of hypoxia in pulmonary arterial smooth muscle cells through ERK1/2 pathway. Exp. Cell Res. 2013, 319, 1973–1987. [Google Scholar] [CrossRef]
- Wegiel, B.; Gallo, D.; Csizmadia, E.; Roger, T.; Kaczmarek, E.; Harris, C.; Zuckerbraun, B.S.; Otterbein, L.E. Biliverdin inhibits Toll-like receptor-4 (TLR4) expression through nitric oxide-dependent nuclear translocation of biliverdin reductase. Proc. Natl. Acad. Sci. USA 2011, 108, 18849–18854. [Google Scholar] [CrossRef]
- Gustavsson, A.; Norton, N.; Fast, T.; Frolich, L.; Georges, J.; Holzapfel, D.; Kirabali, T.; Krolak-Salmon, P.; Rossini, P.M.; Ferretti, M.T.; et al. Global estimates on the number of persons across the Alzheimer’s disease continuum. Alzheimers Dement. 2023, 19, 658–670. [Google Scholar] [CrossRef]
- Karch, C.M.; Goate, A.M. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Limorenko, G.; Lashuel, H.A. Revisiting the grammar of Tau aggregation and pathology formation: How new insights from brain pathology are shaping how we study and target Tauopathies. Chem. Soc. Rev. 2022, 51, 513–565. [Google Scholar] [CrossRef] [PubMed]
- Santarella, R.A.; Skiniotis, G.; Goldie, K.N.; Tittmann, P.; Gross, H.; Mandelkow, E.M.; Mandelkow, E.; Hoenger, A. Surface-decoration of microtubules by human tau. J. Mol. Biol. 2004, 339, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Kopke, E.; Tung, Y.C.; Shaikh, S.; Alonso, A.C.; Iqbal, K.; Grundke-Iqbal, I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 1993, 268, 24374–24384. [Google Scholar] [CrossRef] [PubMed]
- Davidson, R.; Krider, R.I.; Borsellino, P.; Noorda, K.; Alhwayek, G.; Vida, T.A. Untangling Tau: Molecular Insights into Neuroinflammation, Pathophysiology, and Emerging Immunotherapies. Curr. Issues Mol. Biol. 2023, 45, 8816–8839. [Google Scholar] [CrossRef]
- Tracy, T.; Claiborn, K.C.; Gan, L. Regulation of Tau Homeostasis and Toxicity by Acetylation. Adv. Exp. Med. Biol. 2019, 1184, 47–55. [Google Scholar] [CrossRef]
- Shin, M.K.; Vazquez-Rosa, E.; Koh, Y.; Dhar, M.; Chaubey, K.; Cintron-Perez, C.J.; Barker, S.; Miller, E.; Franke, K.; Noterman, M.F.; et al. Reducing acetylated tau is neuroprotective in brain injury. Cell 2021, 184, 2715–2732.e23. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-beta Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Lindholm, K.; Yang, L.B.; Yue, X.; Citron, M.; Yan, R.; Beach, T.; Sue, L.; Sabbagh, M.; Cai, H.; et al. Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2004, 101, 3632–3637. [Google Scholar] [CrossRef] [PubMed]
- Granzotto, A.; Sensi, S.L. Once upon a time, the Amyloid Cascade Hypothesis. Ageing Res. Rev. 2024, 93, 102161. [Google Scholar] [CrossRef] [PubMed]
- Kimpara, T.; Takeda, A.; Yamaguchi, T.; Arai, H.; Okita, N.; Takase, S.; Sasaki, H.; Itoyama, Y. Increased bilirubins and their derivatives in cerebrospinal fluid in Alzheimer’s disease. Neurobiol. Aging 2000, 21, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.S.; Pae, C.U.; Yoon, S.J.; Jang, W.Y.; Lee, N.J.; Kim, J.J.; Lee, S.J.; Lee, C.; Paik, I.H.; Lee, C.U. Decreased plasma antioxidants in patients with Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2006, 21, 344–348. [Google Scholar] [CrossRef]
- Takahashi, M.; Dore, S.; Ferris, C.D.; Tomita, T.; Sawa, A.; Wolosker, H.; Borchelt, D.R.; Iwatsubo, T.; Kim, S.H.; Thinakaran, G.; et al. Amyloid precursor proteins inhibit heme oxygenase activity and augment neurotoxicity in Alzheimer’s disease. Neuron 2000, 28, 461–473. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Cenini, G.; Sultana, R.; Coccia, R.; Preziosi, P.; Perluigi, M.; Mancuso, C.; Butterfield, D.A. Oxidative and nitrosative modifications of biliverdin reductase-A in the brain of subjects with Alzheimer’s disease and amnestic mild cognitive impairment. J. Alzheimers Dis. 2011, 25, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Di Domenico, F.; Cenini, G.; Sultana, R.; Cini, C.; Preziosi, P.; Perluigi, M.; Mancuso, C.; Butterfield, D.A. Biliverdin reductase—A protein levels and activity in the brains of subjects with Alzheimer disease and mild cognitive impairment. Biochim. Biophys. Acta 2011, 1812, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Di Domenico, F.; Mancuso, C.; Butterfield, D.A. The Janus face of the heme oxygenase/biliverdin reductase system in Alzheimer disease: It’s time for reconciliation. Neurobiol. Dis. 2014, 62, 144–159. [Google Scholar] [CrossRef]
- Di Domenico, F.; Barone, E.; Mancuso, C.; Perluigi, M.; Cocciolo, A.; Mecocci, P.; Butterfield, D.A.; Coccia, R. HO-1/BVR-a system analysis in plasma from probable Alzheimer’s disease and mild cognitive impairment subjects: A potential biochemical marker for the prediction of the disease. J. Alzheimers Dis. 2012, 32, 277–289. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Cassano, T.; Arena, A.; Tramutola, A.; Lavecchia, M.A.; Coccia, R.; Butterfield, D.A.; Perluigi, M. Impairment of biliverdin reductase-A promotes brain insulin resistance in Alzheimer disease: A new paradigm. Free Radic. Biol. Med. 2016, 91, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Lanzillotta, C.; Zuliani, I.; Vasavda, C.; Snyder, S.H.; Paul, B.D.; Perluigi, M.; Di Domenico, F.; Barone, E. BVR-A Deficiency Leads to Autophagy Impairment through the Dysregulation of AMPK/mTOR Axis in the Brain-Implications for Neurodegeneration. Antioxidants 2020, 9, 671. [Google Scholar] [CrossRef]
- Triani, F.; Tramutola, A.; Di Domenico, F.; Sharma, N.; Butterfield, D.A.; Head, E.; Perluigi, M.; Barone, E. Biliverdin reductase-A impairment links brain insulin resistance with increased Abeta production in an animal model of aging: Implications for Alzheimer disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3181–3194. [Google Scholar] [CrossRef]
- Barone, E.; Mancuso, C.; Di Domenico, F.; Sultana, R.; Murphy, M.P.; Head, E.; Butterfield, D.A. Biliverdin reductase-A: A novel drug target for atorvastatin in a dog pre-clinical model of Alzheimer disease. J. Neurochem. 2012, 120, 135–146. [Google Scholar] [CrossRef]
- Liu, J.; Dong, H.; Zhang, Y.; Cao, M.; Song, L.; Pan, Q.; Bulmer, A.; Adams, D.B.; Dong, X.; Wang, H. Bilirubin Increases Insulin Sensitivity by Regulating Cholesterol Metabolism, Adipokines and PPARgamma Levels. Sci. Rep. 2015, 5, 9886. [Google Scholar] [CrossRef]
- Kipp, Z.A.; Xu, M.; Bates, E.A.; Lee, W.H.; Kern, P.A.; Hinds, T.D., Jr. Bilirubin Levels Are Negatively Correlated with Adiposity in Obese Men and Women, and Its Catabolized Product, Urobilin, Is Positively Associated with Insulin Resistance. Antioxidants 2023, 12, 170. [Google Scholar] [CrossRef]
- Cimini, F.A.; Arena, A.; Barchetta, I.; Tramutola, A.; Ceccarelli, V.; Lanzillotta, C.; Fontana, M.; Bertoccini, L.; Leonetti, F.; Capoccia, D.; et al. Reduced biliverdin reductase-A levels are associated with early alterations of insulin signaling in obesity. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1490–1501. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Tramutola, A.; Lanzillotta, C.; Arena, A.; Blarzino, C.; Cassano, T.; Butterfield, D.A.; Di Domenico, F.; Perluigi, M.; Barone, E. Loss of biliverdin reductase-A favors Tau hyper-phosphorylation in Alzheimer’s disease. Neurobiol. Dis. 2019, 125, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, L.; Li, Z.; Song, Y.; Yuan, Y.; Liu, T.; Hong, J.; Wang, Q.; Chang, H.; Kuang, Z.; et al. Potential Serum Biomarkers for Postoperative Neurocognitive Disorders Based on Proteomic Analysis of Cognitive-Related Brain Regions. Front. Aging Neurosci. 2021, 13, 741263. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Li, Y.; Li, T.; Xu, F.; Zeng, D.; Shi, Y.; Zhao, N.; Zhang, L.; Ma, Y.Z.; Wang, Q.; et al. Sex differences between serum total bilirubin levels and cognition in patients with schizophrenia. BMC Psychiatry 2021, 21, 396. [Google Scholar] [CrossRef] [PubMed]
- Becklen, M.; Orhan, F.; Piehl, F.; Cervenka, S.; Sellgren, C.M.; Flyckt, L.; Erhardt, S.; Fatouros-Bergman, H. Plasma bilirubin levels are reduced in first-episode psychosis patients and associates to working memory and duration of untreated psychosis. Sci. Rep. 2021, 11, 7527. [Google Scholar] [CrossRef] [PubMed]
- Moccia, M.; Picillo, M.; Erro, R.; Longo, K.; Amboni, M.; Santangelo, G.; Palladino, R.; Allocca, R.; Caporale, O.; Triassi, M.; et al. Increased bilirubin levels in de novo Parkinson’s disease. Eur. J. Neurol. 2015, 22, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef]
- Barone, E.; Tramutola, A.; Triani, F.; Calcagnini, S.; Di Domenico, F.; Ripoli, C.; Gaetani, S.; Grassi, C.; Butterfield, D.A.; Cassano, T.; et al. Biliverdin Reductase-A Mediates the Beneficial Effects of Intranasal Insulin in Alzheimer Disease. Mol. Neurobiol. 2019, 56, 2922–2943. [Google Scholar] [CrossRef]
- Tramutola, A.; Triplett, J.C.; Di Domenico, F.; Niedowicz, D.M.; Murphy, M.P.; Coccia, R.; Perluigi, M.; Butterfield, D.A. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): Analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J. Neurochem. 2015, 133, 739–749. [Google Scholar] [CrossRef]
- Lanzillotta, C.; Tramutola, A.; Di Giacomo, G.; Marini, F.; Butterfield, D.A.; Di Domenico, F.; Perluigi, M.; Barone, E. Insulin resistance, oxidative stress and mitochondrial defects in Ts65dn mice brain: A harmful synergistic path in down syndrome. Free Radic. Biol. Med. 2021, 165, 152–170. [Google Scholar] [CrossRef]
- Hatano, T.; Saiki, S.; Okuzumi, A.; Mohney, R.P.; Hattori, N. Identification of novel biomarkers for Parkinson’s disease by metabolomic technologies. J. Neurol. Neurosurg. Psychiatry 2016, 87, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Albillos, S.M.; Montero, O.; Calvo, S.; Solano-Vila, B.; Trejo, J.M.; Cubo, E. Plasma acyl-carnitines, bilirubin, tyramine and tetrahydro-21-deoxycortisol in Parkinson’s disease and essential tremor. A case control biomarker study. Park. Relat. Disord. 2021, 91, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, D.; Surucu, S.; Basak, A.N.; Ulusu, N.N. Evaluation of the Hematological and Serum Biochemistry Parameters in the Pre-Symptomatic and Symptomatic Stages of ALS Disease to Support Early Diagnosis and Prognosis. Cells 2022, 11, 3569. [Google Scholar] [CrossRef] [PubMed]
- Ilzecka, J.; Stelmasiak, Z. Serum bilirubin concentration in patients with amyotrophic lateral sclerosis. Clin. Neurol. Neurosurg. 2003, 105, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Xue, L.; Luo, D.M. Lower serum bilirubin concentration in patients with migraine. Int. J. Clin. Exp. Med. 2015, 8, 13398–13402. [Google Scholar] [PubMed]
- Peng, F.; Deng, X.; Yu, Y.; Chen, X.; Shen, L.; Zhong, X.; Qiu, W.; Jiang, Y.; Zhang, J.; Hu, X. Serum bilirubin concentrations and multiple sclerosis. J. Clin. Neurosci. 2011, 18, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Ljubisavljevic, S.; Stojanovic, I.; Vojinovic, S.; Milojkovic, M.; Dunjic, O.; Stojanov, D.; Pavlovic, D. Association of serum bilirubin and uric acid levels changes during neuroinflammation in patients with initial and relapsed demyelination attacks. Metab. Brain Dis. 2013, 28, 629–638. [Google Scholar] [CrossRef]
- Vasavda, C.; Semenza, E.R.; Liew, J.; Kothari, R.; Dhindsa, R.S.; Shanmukha, S.; Lin, A.; Tokhunts, R.; Ricco, C.; Snowman, A.M.; et al. Biliverdin reductase bridges focal adhesion kinase to Src to modulate synaptic signaling. Sci. Signal. 2022, 15, eabh3066. [Google Scholar] [CrossRef]
- Cingolani, L.A.; Goda, Y. Actin in action: The interplay between the actin cytoskeleton and synaptic efficacy. Nat. Rev. Neurosci. 2008, 9, 344–356. [Google Scholar] [CrossRef]
- Huganir, R.L.; Nicoll, R.A. AMPARs and synaptic plasticity: The last 25 years. Neuron 2013, 80, 704–717. [Google Scholar] [CrossRef]
- Sutton, M.A.; Schuman, E.M. Dendritic protein synthesis, synaptic plasticity, and memory. Cell 2006, 127, 49–58. [Google Scholar] [CrossRef]
- Mayford, M.; Siegelbaum, S.A.; Kandel, E.R. Synapses and memory storage. Cold Spring Harb. Perspect. Biol. 2012, 4, a005751. [Google Scholar] [CrossRef]
- Luscher, C.; Malenka, R.C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012, 4, a005710. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 2005, 6, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, D.D.; Hanks, S.K.; Hunter, T.; van der Geer, P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature 1994, 372, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Husi, H.; Ward, M.A.; Choudhary, J.S.; Blackstock, W.P.; Grant, S.G. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat. Neurosci. 2000, 3, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Ma, Y.L.; Chen, S.K.; Wang, C.W.; Lee, E.H. Focal adhesion kinase is required, but not sufficient, for the induction of long-term potentiation in dentate gyrus neurons in vivo. J. Neurosci. 2003, 23, 4072–4080. [Google Scholar] [CrossRef] [PubMed]
- Babayan, A.H.; Kramar, E.A.; Barrett, R.M.; Jafari, M.; Haettig, J.; Chen, L.Y.; Rex, C.S.; Lauterborn, J.C.; Wood, M.A.; Gall, C.M.; et al. Integrin dynamics produce a delayed stage of long-term potentiation and memory consolidation. J. Neurosci. 2012, 32, 12854–12861. [Google Scholar] [CrossRef] [PubMed]
- Cimini, F.A.; Tramutola, A.; Barchetta, I.; Ceccarelli, V.; Gangitano, E.; Lanzillotta, S.; Lanzillotta, C.; Cavallo, M.G.; Barone, E. Dynamic Changes of BVRA Protein Levels Occur in Response to Insulin: A Pilot Study in Humans. Int. J. Mol. Sci. 2023, 24, 7282. [Google Scholar] [CrossRef]
- Xu, J.; Ji, J.; Yan, X.H. Cross-talk between AMPK and mTOR in regulating energy balance. Crit. Rev. Food Sci. Nutr. 2012, 52, 373–381. [Google Scholar] [CrossRef]
- Mancuso, C. Biliverdin reductase as a target in drug research and development: Facts and hypotheses. Free Radic. Biol. Med. 2021, 172, 521–529. [Google Scholar] [CrossRef]
- Gibbs, P.E.; Miralem, T.; Maines, M.D. Biliverdin reductase: A target for cancer therapy? Front. Pharmacol. 2015, 6, 119. [Google Scholar] [CrossRef]
- Ai, W.; Bae, S.; Ke, Q.; Su, S.; Li, R.; Chen, Y.; Yoo, D.; Lee, E.; Jon, S.; Kang, P.M. Bilirubin Nanoparticles Protect Against Cardiac Ischemia/Reperfusion Injury in Mice. J. Am. Heart Assoc. 2021, 10, e021212. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Creeden, J.F.; Gordon, D.M.; Stec, D.F.; Donald, M.C.; Stec, D.E. Bilirubin Nanoparticles Reduce Diet-Induced Hepatic Steatosis, Improve Fat Utilization, and Increase Plasma beta-Hydroxybutyrate. Front. Pharmacol. 2020, 11, 594574. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, H.; Kang, S.; Lee, J.; Park, J.; Jon, S. Bilirubin Nanoparticles as a Nanomedicine for Anti-inflammation Therapy. Angew. Chem. Int. Ed. Engl. 2016, 55, 7460–7463. [Google Scholar] [CrossRef]
- Sundararaghavan, V.L.; Sindhwani, P.; Hinds, T.D., Jr. Glucuronidation and UGT isozymes in bladder: New targets for the treatment of uroepithelial carcinomas? Oncotarget 2017, 8, 3640–3648. [Google Scholar] [CrossRef] [PubMed]
- Kataura, T.; Saiki, S.; Ishikawa, K.I.; Akamatsu, W.; Sasazawa, Y.; Hattori, N.; Imoto, M. BRUP-1, an intracellular bilirubin modulator, exerts neuroprotective activity in a cellular Parkinson’s disease model. J. Neurochem. 2020, 155, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Calabrese, V.; Mancuso, C. Ferulic acid and its therapeutic potential as a hormetin for age-related diseases. Biogerontology 2009, 10, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Catino, S.; Paciello, F.; Miceli, F.; Rolesi, R.; Troiani, D.; Calabrese, V.; Santangelo, R.; Mancuso, C. Ferulic Acid Regulates the Nrf2/Heme Oxygenase-1 System and Counteracts Trimethyltin-Induced Neuronal Damage in the Human Neuroblastoma Cell Line SH-SY5Y. Front. Pharmacol. 2015, 6, 305. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Garagnani, P.; Marquis, J.; Delledonne, M.; Pirazzini, C.; Marasco, E.; Kwiatkowska, K.M.; Iannuzzi, V.; Bacalini, M.G.; Valsesia, A.; Carayol, J.; et al. Whole-genome sequencing analysis of semi-supercentenarians. elife 2021, 10, e57849. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Status of Bilirubin and BVRA | Reference |
|---|---|---|
| Alzheimer’s disease (3xTg-AD mouse model) | Decreased BVRA levels and activity, decreased Tyr-phosphorylation. Increased 3-NT modification of BVRA. | [75,87,88] |
| Alzheimer’s disease and MCI patients | Reduced BVR-A Tyr-phosphorylation and elevated 3-NT modification of BVR-A in the hippocampus. | [71,72,89] |
| Down Syndrome (Ts65dn mouse model) | Reduced BVR-A Tyr-phosphorylation in the frontal cortex (9 months). | [90] |
| Aging (C57BL/6J mice) | Decreased BVRA levels and phosphorylation (12 months) and increased 3-NT modification of BVRA (18 months) in the hippocampus. | [75] |
| Aging (Canine model) | Decreased BVRA Tyr-phosphorylation (4–12 months), elevated 3-NT modification of BVRA (10–12 months) in the parietal cortex. | [77] |
| Parkinson’s disease patients (without dementia) | Low bilirubin levels in serum of PD patients. | [91] |
| Parkinson’s disease (without dementia) | Higher bilirubin levels in plasma of PD patients. | [92] |
| Parkinson’s disease patients (de novo, drug-naïve) | Higher levels of bilirubin in serum, which correlated with better outcomes in a 2-year longitudinal study. | [86] |
| Amyotrophic lateral sclerosis (G93A SOD1 rat model) | Higher serum bilirubin levels in the symptomatic stage of ALS. | [93] |
| ALS patients | Lower serum bilirubin levels in patients with long duration ALS. | [94] |
| Migraine | Lower serum bilirubin in people with migraine. | [95] |
| Multiple sclerosis | Lower serum bilirubin in people with MS. | [96] |
| Multiple sclerosis (relapsing, remitting, or RRMS) | Lower serum bilirubin in people with MS | [97] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paul, B.D.; Pieper, A.A. Neuroprotective Roles of the Biliverdin Reductase-A/Bilirubin Axis in the Brain. Biomolecules 2024, 14, 155. https://doi.org/10.3390/biom14020155
Paul BD, Pieper AA. Neuroprotective Roles of the Biliverdin Reductase-A/Bilirubin Axis in the Brain. Biomolecules. 2024; 14(2):155. https://doi.org/10.3390/biom14020155
Chicago/Turabian StylePaul, Bindu D., and Andrew A. Pieper. 2024. "Neuroprotective Roles of the Biliverdin Reductase-A/Bilirubin Axis in the Brain" Biomolecules 14, no. 2: 155. https://doi.org/10.3390/biom14020155
APA StylePaul, B. D., & Pieper, A. A. (2024). Neuroprotective Roles of the Biliverdin Reductase-A/Bilirubin Axis in the Brain. Biomolecules, 14(2), 155. https://doi.org/10.3390/biom14020155