The Dichotomous Role of Inflammation in the CNS: A Mitochondrial Point of View

 ,
,  ,
,  ,
, 

Abstract
1. Introduction: The Cellular Players of Neuroinflammation
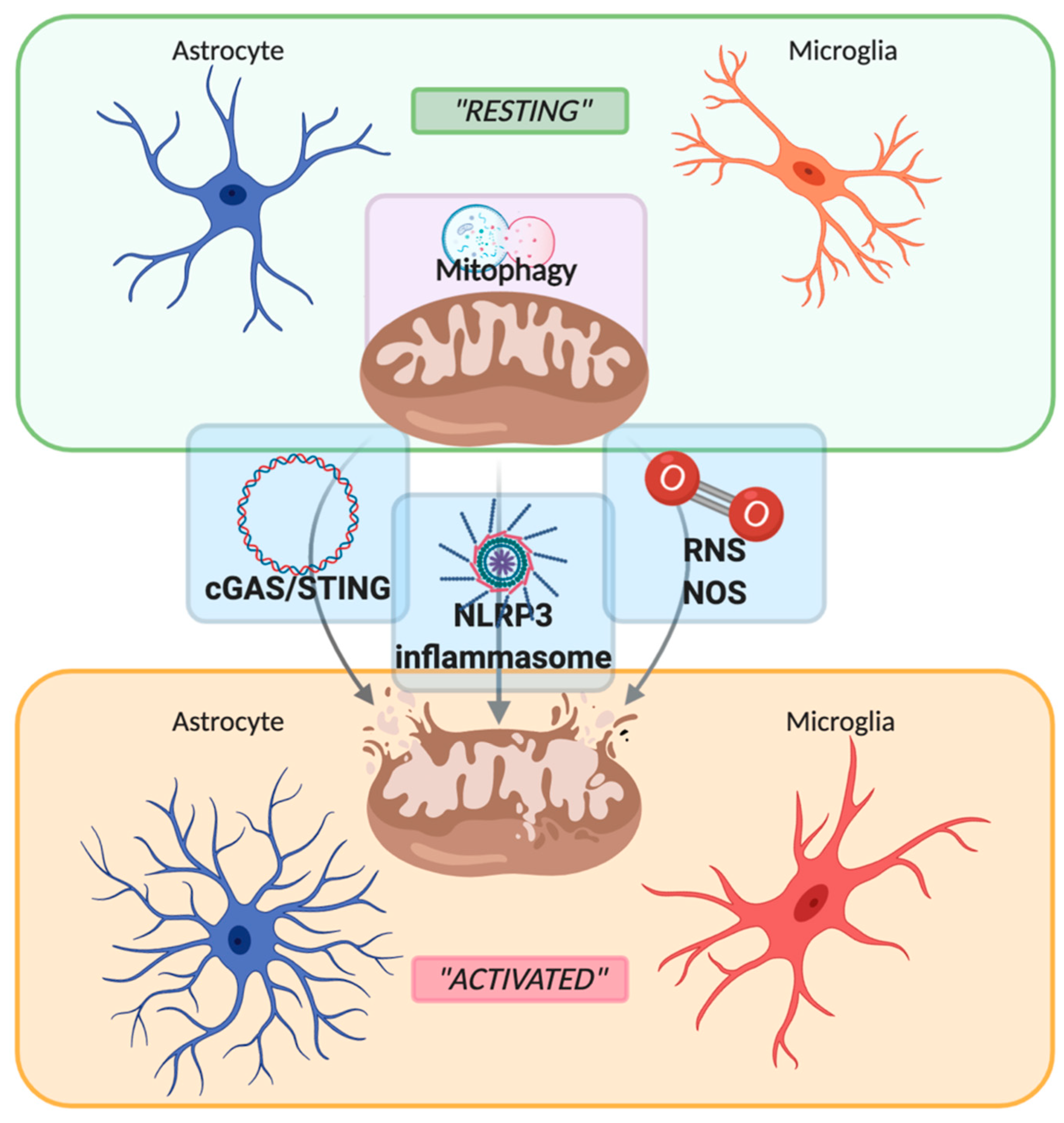
2. Role of Mitochondria in Neuroinflammation
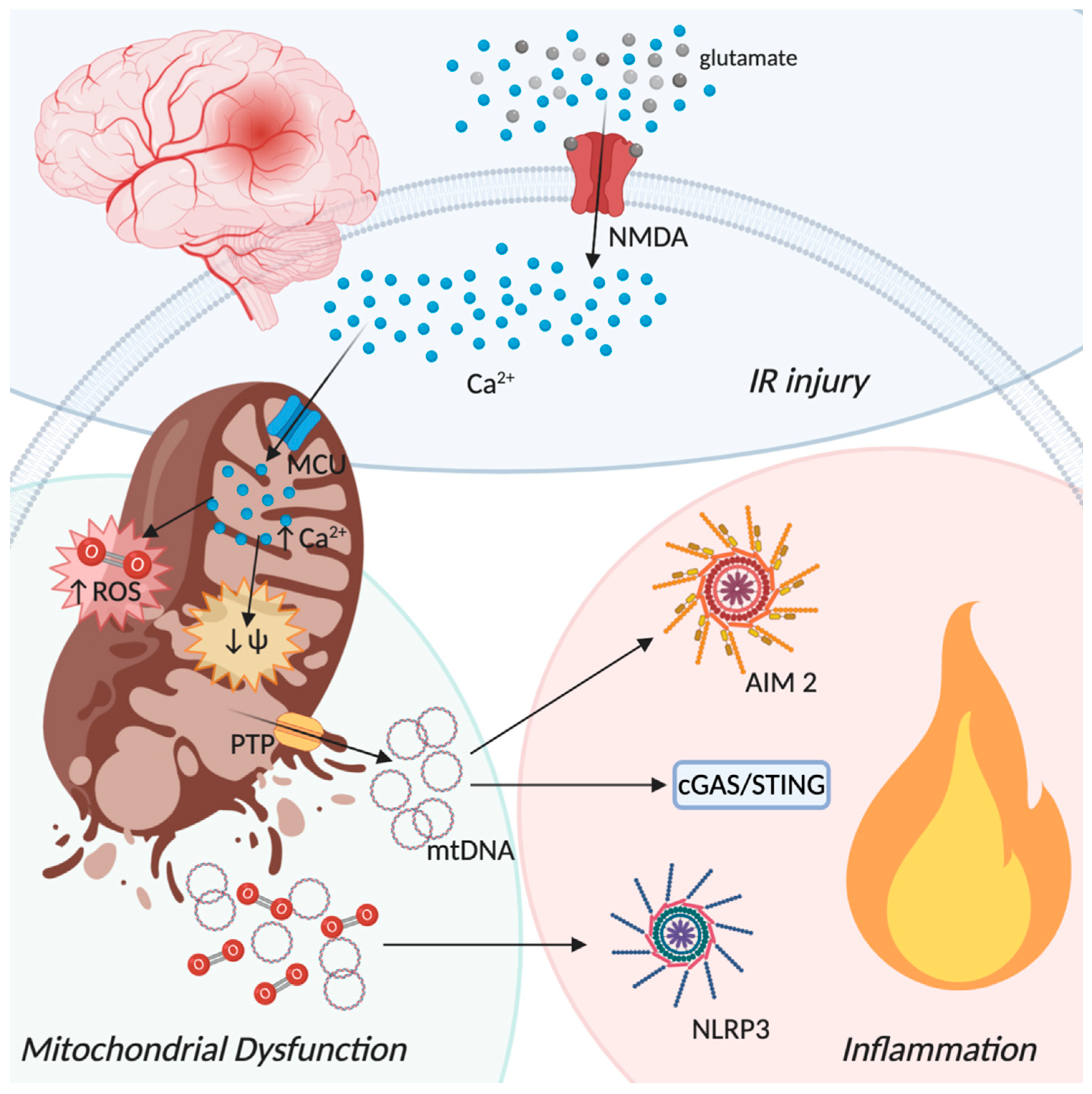
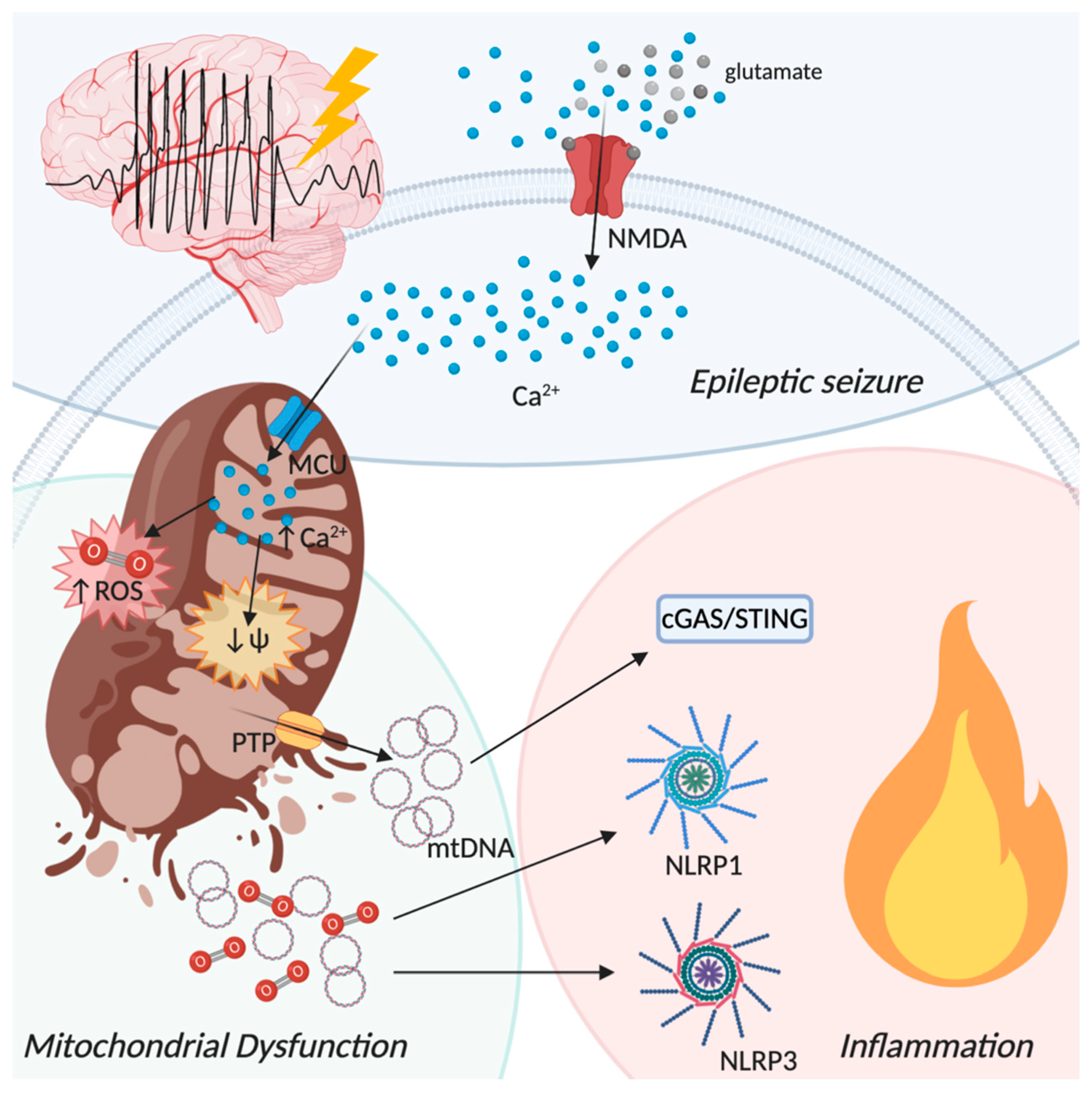
2.1. PRRs Signaling: Focus on cGAS-STING Pathway
2.2. PRRs Signaling: Focus on NLRP3 Inflammasome
2.3. Reactive Oxygen Species
2.4. Mitophagy
3. From Chronic Neuroinflammation to Neurodegeneration: Multiple Sclerosis, Parkinson’s, and Alzheimer’s Disease
3.1. Multiple Sclerosis
3.2. Parkinson’s Disease
3.3. Alzheimer’s Disease
4. Ischemic Stroke and Mitochondrial Induced Neuroinflammation
5. The Neuroinflammatory Process in Epilepsy: The Involvement of Mitochondria
6. Current Therapies Targeting Neuroinflammation
6.1. Targeting Neuroinflammation in Multiple Sclerosis
6.2. Targeting Neuroinflammation in Parkinson’s Disease
6.3. Targeting Neuroinflammation in Alzheimer’s Disease
6.4. Targeting Neuroinflammation in Ischemic Stroke
6.5. Targeting Neuroinflammation in Epilepsy
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| AIM2 | absent in melanoma 2 |
| ALRs | AIM2-like receptors |
| ALS | amyotrophic lateral sclerosis |
| ASC | apoptosis-associated speck-like protein containing CARD |
| ATP | adenosine triphosphate |
| Aβ | β-amyloid protein |
| BBB | blood–brain barrier |
| BNIP3 | B-cell lymphoma 2 nineteen kilodalton interacting protein 3 |
| BNIP3L | BNIP3-like |
| cGAMP | 2′,3′-cyclic GMP-AMP |
| cGAS | cyclic GMP-AMP synthetase |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| CXC3CR1 | CX3C chemokine receptor 1 |
| DAMPs | damage associated molecular patterns |
| dsDNA | double strand DNA |
| EAE | experimental autoimmune encephalomyelitis |
| ER | endoplasmic reticulum |
| ETC | electron transport chain |
| GTP | guanosine triphosphate |
| GLP1 | glucagon-like peptide 1 |
| IFN | interferon |
| IL-10 | interleukin-10 |
| IL-18 | interleukin-18 |
| IL-1β | interleukin-1β |
| IL-6 | interleukin-6 |
| IR | ischemic reperfusion |
| IRF3 | interferon regulatory factor 3 |
| IS | ischemic stroke |
| LIR | LC3-interacting region |
| LRR | leucine-rich repeats |
| MAMs | mitochondria associated membranes |
| MCU | mitochondria calcium uniporter |
| MICU1 | mitochondrial calcium uptake 1 |
| MPPs | matrix metalloproteinases |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MS | multiple sclerosis |
| mtDNA | mitochondrial DNA |
| NACHT | nucleotide and oligomerization domain |
| NBR1 | neighbor of Brca1 |
| NDDs | neurodegenerative diseases |
| NDP52 | nuclear dot protein 52 |
| NF-ĸB | nuclear factor-ĸB |
| NLRP3 | nucleotide-binding domain and leucine-rich repeat containing protein 3 |
| NLRs | nucleotide-binding oligomerization domain-like (NOD) receptors |
| NMDA | N-methyl-D-aspartate |
| NO | nitric oxide |
| NSAIDs | non steroidal anti-inflammatory drugs |
| OGD | oxygen and glucose deprivation |
| OMM | outer mitochondrial membrane |
| OPTN | optineurin |
| OXPHOS | oxidative phosphorylation |
| p62/SQSTM1 | p62/sequestosome 1 |
| PAMPs | pathogen-associated molecular patterns |
| PBMCs | peripheral blood mononuclear cells |
| PD | Parkinson’s disease |
| PINK1 | PTEN-induced putative kinase 1 |
| PRRs | pattern recognition receptors |
| PTP | permeability transition pore |
| PYD | pyrine |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SE | status epilepticus |
| STING | stimulator of interferon genes |
| TACE | TNF-a-converting enzyme |
| TAX1BP1 | tax 1 binding protein 1 |
| TBK1 | TANK-binding kinase 1 |
| TGF-β | transforming growth factor β |
| TLE | temporal lobe epilepsy |
| TLR | Toll-like receptor |
| TNF-α | tumor necrosis factor α |
| TREM2 | triggering receptor expressed on myeloid cells-2 |
| α-Syn | α-synuclein |
| Ψm | mitochondrial membrane potential |
References
- Barker, C.F.; Billingham, R.E. Immunologically privileged sites. Adv. Immunol. 1977, 25, 1–54. [Google Scholar] [PubMed]
- Medawar, P.B. Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br. J. Exp. Pathol. 1948, 29, 58–69. [Google Scholar] [PubMed]
- Louveau, A.; Harris, T.H.; Kipnis, J. Revisiting the Mechanisms of CNS Immune Privilege. Trends Immunol. 2015, 36, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Papaneophytou, C.; Georgiou, E.; Kleopa, K.A. The role of oligodendrocyte gap junctions in neuroinflammation. Channels 2019, 13, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Biernacki, K.; Wosik, K.; Antel, J.P. Glial cell influence on the human blood-brain barrier. Glia 2001, 36, 145–155. [Google Scholar] [CrossRef]
- Koehler, R.C.; Gebremedhin, D.; Harder, D.R. Role of astrocytes in cerebrovascular regulation. J. Appl. Physiol. 2006, 100, 307–317. [Google Scholar] [CrossRef][Green Version]
- Gimenez, M.A.; Sim, J.; Archambault, A.S.; Klein, R.S.; Russell, J.H. A tumor necrosis factor receptor 1-dependent conversation between central nervous system-specific T cells and the central nervous system is required for inflammatory infiltration of the spinal cord. Am. J. Pathol. 2006, 168, 1200–1209. [Google Scholar] [CrossRef]
- Carson, M.J.; Thrash, J.C.; Walter, B. The cellular response in neuroinflammation: The role of leukocytes, microglia and astrocytes in neuronal death and survival. Clin. Neurosci. Res. 2006, 6, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Del Río Hortega, P. Noticia de un nuevo y fácil método para la coloración de la neuroglia y el tejido conjuntivo. Trab. Lab. Investig. Biol. 1918, 15, 367–378. [Google Scholar]
- Ransohoff, R.M.; El Khoury, J. Microglia in Health and Disease. Cold Spring Harb. Perspect. Biol. 2015, 8, a020560. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S.T. Microglia and neuroinflammation: A pathological perspective. J. Neuroinflamm. 2004, 1, 14. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Da Mesquita, S.; Fu, Z.; Kipnis, J. The Meningeal Lymphatic System: A New Player in Neurophysiology. Neuron 2018, 100, 375–388. [Google Scholar] [CrossRef]
- McGavern, D.B.; Homann, D.; Oldstone, M.B.A. T cells in the central nervous system: The delicate balance between viral clearance and disease. J. Infect. Dis. 2002, 186 (Suppl. S2), S145–S151. [Google Scholar] [CrossRef]
- Sabatino, J.J.; Pröbstel, A.-K.; Zamvil, S.S. B cells in autoimmune and neurodegenerative central nervous system diseases. Nat. Rev. Neurosci. 2019, 20, 728–745. [Google Scholar] [CrossRef]
- Heneka, M.T.; O’Banion, M.K. Inflammatory processes in Alzheimer’s disease. J. Neuroimmunol. 2007, 184, 69–91. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Björkqvist, M.; Wild, E.J.; Tabrizi, S.J. Harnessing immune alterations in neurodegenerative diseases. Neuron 2009, 64, 21–24. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Di Filippo, M.; Chiasserini, D.; Tozzi, A.; Picconi, B.; Calabresi, P. Mitochondria and the link between neuroinflammation and neurodegeneration. J. Alzheimer’s Dis. 2010, 20 (Suppl. S2), S369–S379. [Google Scholar] [CrossRef] [PubMed]
- Aktas, O.; Ullrich, O.; Infante-Duarte, C.; Nitsch, R.; Zipp, F. Neuronal damage in brain inflammation. Arch. Neurol. 2007, 64, 185–189. [Google Scholar] [CrossRef] [PubMed]
- de Barcelos, I.P.; Troxell, R.M.; Graves, J.S. Mitochondrial Dysfunction and Multiple Sclerosis. Biology 2019, 8, 37. [Google Scholar] [CrossRef]
- Abou-Sleiman, P.M.; Muqit, M.M.K.; Wood, N.W. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat. Rev. Neurosci. 2006, 7, 207–219. [Google Scholar] [CrossRef]
- Silva, D.F.; Selfridge, J.E.; Lu, J.; Lezi, E.; Cardoso, S.M.; Swerdlow, R.H. Mitochondrial abnormalities in Alzheimer’s disease: Possible targets for therapeutic intervention. Adv. Pharmacol. 2012, 64, 83–126. [Google Scholar] [CrossRef]
- Antico Arciuch, V.G.; Elguero, M.E.; Poderoso, J.J.; Carreras, M.C. Mitochondrial regulation of cell cycle and proliferation. Antioxid. Redox Signal. 2012, 16, 1150–1180. [Google Scholar] [CrossRef]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Are “resting” microglia more “m2”? Front. Immunol. 2014, 5, 594. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef]
- Craft, J.M.; Watterson, D.M.; Van Eldik, L.J. Neuroinflammation: A potential therapeutic target. Expert Opin. Ther. Targets 2005, 9, 887–900. [Google Scholar] [CrossRef] [PubMed]
- ElAli, A.; Rivest, S. Microglia Ontology and Signaling. Front. Cell Dev. Biol. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, T.; Weiner, H.L. CNS inflammation and neurodegeneration. J. Clin. Investig. 2017, 127, 3577–3587. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.-K.; Chen, Z.J. Identification and Characterization of MAVS, a Mitochondrial Antiviral Signaling Protein that Activates NF-κB and IRF3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Feng, M.; Guan, W. Mitochondrial DNA sensing by STING signaling participates in inflammation, cancer and beyond. Int. J. Cancer 2016, 139, 736–741. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef]
- Janeway, C.A. The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol. Today 1992, 13, 11–16. [Google Scholar] [CrossRef]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11. [Google Scholar] [CrossRef]
- Matzinger, P. The danger model: A renewed sense of self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef]
- Newton, K.; Dixit, V.M. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Sagan, L. On the origin of mitosing cells. J. Theor. Biol. 1967, 14, 255–274. [Google Scholar] [CrossRef]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Patrushev, M.; Kasymov, V.; Patrusheva, V.; Ushakova, T.; Gogvadze, V.; Gaziev, A. Mitochondrial permeability transition triggers the release of mtDNA fragments. Cell. Mol. Life Sci. 2004, 61, 3100–3103. [Google Scholar] [CrossRef] [PubMed]
- Bader, V.; Winklhofer, K. Mitochondria at the interface between neurodegeneration and neuroinflammation. Semin. Cell Dev. Biol. 2020, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16. [Google Scholar] [CrossRef]
- Adamczak, S.E.; De Rivero Vaccari, J.P.; Dale, G.; Brand, F.J.; Nonner, D.; Bullock, M.; Dahl, G.P.; Dietrich, W.D.; Keane, R.W. Pyroptotic neuronal cell death mediated by the AIM2 inflammasome. J. Cereb. Blood Flow Metab. 2014, 34, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Cameron, J.S.; Alexopoulou, L.; Sloane, J.A.; DiBernardo, A.B.; Ma, Y.; Kosaras, B.; Flavell, R.; Strittmatter, S.M.; Volpe, J.; Sidman, R.; et al. Toll-like receptor 3 is a potent negative regulator of axonal growth in mammals. J. Neurosci. 2007, 27, 13033–13041. [Google Scholar] [CrossRef]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Röhl, I.; Hopfner, K.-P.; Ludwig, J.; Hornung, V. cGAS produces a 2’-5’-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef]
- Shang, G.; Zhang, C.; Chen, Z.J.; Bai, X.-C.; Zhang, X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature 2019, 567, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.-T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [PubMed]
- Gui, X.; Yang, H.; Li, T.; Tan, X.; Shi, P.; Li, M.; Du, F.; Chen, Z.J. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 2019, 567, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Chin, A.C. Neuroinflammation and the cGAS-STING pathway. J. Neurophysiol. 2019, 121, 1087–1091. [Google Scholar] [CrossRef]
- Srinivasula, S.M.; Poyet, J.-L.; Razmara, M.; Datta, P.; Zhang, Z.; Alnemri, E.S. The PYRIN-CARD Protein ASC Is an Activating Adaptor for Caspase-1. J. Biol. Chem. 2002, 277, 21119–21122. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.-D. Regulation of inflammasome activation. Immunol. Rev. 2015, 265, 6–21. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Lu, M.; Sun, X.-L.; Qiao, C.; Liu, Y.; Ding, J.-H.; Hu, G. Uncoupling protein 2 deficiency aggravates astrocytic endoplasmic reticulum stress and nod-like receptor protein 3 inflammasome activation. Neurobiol. Aging 2014, 35, 421–430. [Google Scholar] [CrossRef]
- Gustin, A.; Kirchmeyer, M.; Koncina, E.; Felten, P.; Losciuto, S.; Heurtaux, T.; Tardivel, A.; Heuschling, P.; Dostert, C. NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes. PLoS ONE 2015, 10, e0130624. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Singhal, G.; Jaehne, E.J.; Corrigan, F.; Toben, C.; Baune, B.T. Inflammasomes in neuroinflammation and changes in brain function: A focused review. Front. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Deora, V.; Lee, J.D.; Albornoz, E.A.; McAlary, L.; Jagaraj, C.J.; Robertson, A.A.B.; Atkin, J.D.; Cooper, M.A.; Schroder, K.; Yerbury, J.J.; et al. The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia 2020, 68, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Hwang, I.; Park, S.; Hong, S.; Hwang, B.; Cho, Y.; Son, J.; Yu, J.-W. MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ. 2019, 26, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Scheiblich, H.; Schlütter, A.; Golenbock, D.T.; Latz, E.; Martinez-Martinez, P.; Heneka, M.T. Activation of the NLRP3 inflammasome in microglia: The role of ceramide. J. Neurochem. 2017, 143, 534–550. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Hu, D.; Wu, C.; Feng, Y.; Li, A.; Liu, W.; Wang, Y.; Chen, K.; Tian, M.; Xiao, F.; et al. STING promotes NLRP3 localization in ER and facilitates NLRP3 deubiquitination to activate the inflammasome upon HSV-1 infection. PLoS Pathog. 2020, 16, e1008335. [Google Scholar] [CrossRef]
- Colton, C.A.; Gilbert, D.L. Production of superoxide anions by a CNS macrophage, the microglia. FEBS Lett. 1987, 223, 284–288. [Google Scholar] [CrossRef]
- Handy, D.E.; Loscalzo, J. Redox regulation of mitochondrial function. Antioxid. Redox Signal. 2012, 16, 1323–1367. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Kobylarek, D.; Iwanowski, P.; Lewandowska, Z.; Limphaibool, N.; Szafranek, S.; Labrzycka, A.; Kozubski, W. Advances in the Potential Biomarkers of Epilepsy. Front. Neurol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Barker, J.E.; Bolaños, J.P.; Land, J.M.; Clark, J.B.; Heales, S.J. Glutathione protects astrocytes from peroxynitrite-mediated mitochondrial damage: Implications for neuronal/astrocytic trafficking and neurodegeneration. Dev. Neurosci. 1996, 18, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Eliasson, M.J.; Huang, Z.; Ferrante, R.J.; Sasamata, M.; Molliver, M.E.; Snyder, S.H.; Moskowitz, M.A. Neuronal nitric oxide synthase activation and peroxynitrite formation in ischemic stroke linked to neural damage. J. Neurosci. 1999, 19, 5910–5918. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.; Brian, C.; Woods, J.; Pappa, A.; Panayiotidis, M.I.; Powers, R.; Franco, R. Mitochondrial dysfunction in glial cells: Implications for neuronal homeostasis and survival. Toxicology 2017, 391, 109–115. [Google Scholar] [CrossRef]
- Yin, F.; Sancheti, H.; Patil, I.; Cadenas, E. Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic. Biol. Med. 2016, 100, 108–122. [Google Scholar] [CrossRef]
- Fernandez-Fernandez, S.; Almeida, A.; Bolaños, J.P. Antioxidant and bioenergetic coupling between neurons and astrocytes. Biochem. J. 2012, 443, 3–11. [Google Scholar] [CrossRef]
- Herrero-Mendez, A.; Almeida, A.; Fernández, E.; Maestre, C.; Moncada, S.; Bolaños, J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 2009, 11, 747–752. [Google Scholar] [CrossRef]
- van Horssen, J.; van Schaik, P.; Witte, M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci. Lett. 2019, 710, 132931. [Google Scholar] [CrossRef]
- Martinez-Vicente, M. Neuronal Mitophagy in Neurodegenerative Diseases. Front. Mol. Neurosci. 2017, 10, 64. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Tavernarakis, N. Mitophagy in neurodegeneration and aging. Front. Genet. 2012, 3, 297. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Pinton, P. Mitophagy and Mitochondrial Balance. Methods Mol Biol. 2015, 1241, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, N.; Lu, B. Mechanisms and roles of mitophagy in neurodegenerative diseases. CNS Neurosci. Ther. 2019, 25, 859–875. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sorensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef]
- Lublin, F.D. New multiple sclerosis phenotypic classification. Eur. Neurol. 2014, 72 (Suppl. S1), 1–5. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Fossati, V.; Bonora, M.; Giorgi, C.; Marchi, S.; Missiroli, S.; Rusielewicz, T.; Wieckowski, M.R.; Pinton, P. Mitochondria in Multiple Sclerosis: Molecular Mechanisms of Pathogenesis. Int Rev Cell Mol Biol. 2017, 382, 49–103. [Google Scholar] [CrossRef]
- Ciccarelli, O. Multiple sclerosis in 2018: New therapies and biomarkers. Lancet Neurol. 2019, 18, 10–12. [Google Scholar] [CrossRef]
- Khaibullin, T.; Ivanova, V.; Martynova, E.; Cherepnev, G.; Khabirov, F.; Granatov, E.; Rizvanov, A.; Khaiboullina, S. Elevated Levels of Proinflammatory Cytokines in Cerebrospinal Fluid of Multiple Sclerosis Patients. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Patergnani, S.; Castellazzi, M.; Bonora, M.; Marchi, S.; Casetta, I.; Pugliatti, M.; Giorgi, C.; Granieri, E.; Pinton, P. Autophagy and mitophagy elements are increased in body fluids of multiple sclerosis-affected individuals. J. Neurol. Neurosurg. Psychiatry 2018, 89, 439–441. [Google Scholar] [CrossRef]
- Seifert, T.; Kieseier, B.C.; Ropele, S.; Strasser-Fuchs, S.; Quehenberger, F.; Fazekas, F.; Hartung, H.-P. TACE mRNA expression in peripheral mononuclear cells precedes new lesions on MRI in multiple sclerosis. Mult. Scler. J. 2002, 8, 447–451. [Google Scholar] [CrossRef]
- Leppert, D.; Ford, J.; Stabler, G.; Grygar, C.; Lienert, C.; Huber, S.; Miller, K.M.; Hauser, S.L.; Kappos, L. Matrix metalloproteinase-9 (gelatinase B) is selectively elevated in CSF during relapses and stable phases of multiple sclerosis. Brain 1998, 121 Pt 1, 2327–2334. [Google Scholar] [CrossRef]
- Leppert, D.; Waubant, E.; Bürk, M.R.; Oksenberg, J.R.; Hauser, S.L. Interferon beta-1b inhibits gelatinase secretion and in vitro migration of human T cells: A possible mechanism for treatment efficacy in multiple sclerosis. Ann. Neurol. 1996, 40, 846–852. [Google Scholar] [CrossRef]
- Libbey, J.E.; Cusick, M.F.; Fujinami, R.S. Role of pathogens in multiple sclerosis. Int. Rev. Immunol. 2014, 33, 266–283. [Google Scholar] [CrossRef]
- Ding, Z.; Mathur, V.; Ho, P.P.; James, M.L.; Lucin, K.M.; Hoehne, A.; Alabsi, H.; Gambhir, S.S.; Steinman, L.; Luo, J.; et al. Antiviral drug ganciclovir is a potent inhibitor of microglial proliferation and neuroinflammation. J. Exp. Med. 2014, 211, 189–198. [Google Scholar] [CrossRef]
- Mathur, V.; Burai, R.; Vest, R.T.; Bonanno, L.N.; Lehallier, B.; Zardeneta, M.E.; Mistry, K.N.; Do, D.; Marsh, S.E.; Abud, E.M.; et al. Activation of the STING-Dependent Type I Interferon Response Reduces Microglial Reactivity and Neuroinflammation. Neuron 2017, 96, 1290–1302.e6. [Google Scholar] [CrossRef] [PubMed]
- Ming, X.; Li, W.; Maeda, Y.; Blumberg, B.; Raval, S.; Cook, S.D.; Dowling, P.C. Caspase-1 expression in multiple sclerosis plaques and cultured glial cells. J. Neurol. Sci. 2002, 197, 9–18. [Google Scholar] [CrossRef]
- Cao, Y.; Goods, B.A.; Raddassi, K.; Nepom, G.T.; Kwok, W.W.; Love, J.C.; Hafler, D.A. Functional inflammatory profiles distinguish myelin-reactive T cells from patients with multiple sclerosis. Sci. Transl. Med. 2015, 7, 287ra74. [Google Scholar] [CrossRef]
- Huang, W.-X.; Huang, P.; Hillert, J. Increased expression of caspase-1 and interleukin-18 in peripheral blood mononuclear cells in patients with multiple sclerosis. Mult. Scler. J. 2004, 10, 482–487. [Google Scholar] [CrossRef]
- Barclay, W.; Shinohara, M.L. Inflammasome activation in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Brain Pathol. 2017, 27, 213–219. [Google Scholar] [CrossRef]
- Gris, D.; Ye, Z.; Iocca, H.A.; Wen, H.; Craven, R.R.; Gris, P.; Huang, M.; Schneider, M.; Miller, S.D.; Ting, J.P.-Y. NLRP3 Plays a Critical Role in the Development of Experimental Autoimmune Encephalomyelitis by Mediating Th1 and Th17 Responses. J. Immunol. 2010, 185, 974–981. [Google Scholar] [CrossRef]
- Shaw, P.J.; Lukens, J.R.; Burns, S.; Chi, H.; McGargill, M.A.; Kanneganti, T.-D. Cutting edge: Critical role for PYCARD/ASC in the development of experimental autoimmune encephalomyelitis. J. Immunol. 2010, 184, 4610–4614. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, S.; Río, J.; Urcelay, E.; Nurtdinov, R.; Bustamante, M.F.; Fernández, O.; Oliver, B.; Zettl, U.; Brassat, D.; Killestein, J.; et al. NLRP3 inflammasome is associated with the response to IFN-β in patients with multiple sclerosis. Brain 2015, 138, 644–652. [Google Scholar] [CrossRef]
- Mahad, D.; Ziabreva, I.; Lassmann, H.; Turnbull, D. Mitochondrial defects in acute multiple sclerosis lesions. Brain 2008, 131, 1722–1735. [Google Scholar] [CrossRef]
- Iñarrea, P.; Alarcia, R.; Alava, M.A.; Capablo, J.L.; Casanova, A.; Iñiguez, C.; Iturralde, M.; Larrodé, P.; Martín, J.; Mostacero, E.; et al. Mitochondrial complex enzyme activities and cytochrome C expression changes in multiple sclerosis. Mol. Neurobiol. 2014, 49, 1–9. [Google Scholar] [CrossRef]
- Bonora, M.; De Marchi, E.; Patergnani, S.; Suski, J.M.; Celsi, F.; Bononi, A.; Giorgi, C.; Marchi, S.; Rimessi, A.; Duszyński, J.; et al. Tumor necrosis factor-α impairs oligodendroglial differentiation through a mitochondria-dependent process. Cell Death Differ. 2014, 21, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Bros, H.; Millward, J.M.; Paul, F.; Niesner, R.; Infante-Duarte, C. Oxidative damage to mitochondria at the nodes of Ranvier precedes axon degeneration in ex vivo transected axons. Exp. Neurol. 2014, 261, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, K.G.; Santos, G.D.; dos Santos, N.B.; Munhoz, C.D.; Azzi-Nogueira, D.; Campos, A.C.; Pagano, R.L.; Britto, L.R.; Hernandes, M.S. Nox2-dependent neuroinflammation in an EAE model of multiple sclerosis. Transl. Neurosci. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jansen, E.H.; Ruskovska, T. Comparative Analysis of Serum (Anti)oxidative Status Parameters in Healthy Persons. Int. J. Mol. Sci. 2013, 14, 6106–6115. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk, B.; Adamczyk-Sowa, M. New Insights into the Role of Oxidative Stress Mechanisms in the Pathophysiology and Treatment of Multiple Sclerosis. Oxid. Med. Cell. Longev. 2016, 2016, 1973834. [Google Scholar] [CrossRef]
- Tranah, G.J.; Santaniello, A.; Caillier, S.J.; D’Alfonso, S.; Martinelli Boneschi, F.; Hauser, S.L.; Oksenberg, J.R. Mitochondrial DNA sequence variation in multiple sclerosis. Neurology 2015, 85, 325–330. [Google Scholar] [CrossRef]
- Andalib, S.; Talebi, M.; Sakhinia, E.; Farhoudi, M.; Sadeghi-Bazargani, H.; Motavallian, A.; Pilehvar-Soltanahmadi, Y. Multiple sclerosis and mitochondrial gene variations: A review. J. Neurol. Sci. 2013, 330, 10–15. [Google Scholar] [CrossRef]
- Campbell, G.R.; Ziabreva, I.; Reeve, A.K.; Krishnan, K.J.; Reynolds, R.; Howell, O.; Lassmann, H.; Turnbull, D.M.; Mahad, D.J. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann. Neurol. 2011, 69, 481–492. [Google Scholar] [CrossRef]
- Castellazzi, M.; Patergnani, S.; Donadio, M.; Giorgi, C.; Bonora, M.; Fainardi, E.; Casetta, I.; Granieri, E.; Pugliatti, M.; Pinton, P. Correlation between auto/mitophagic processes and magnetic resonance imaging activity in multiple sclerosis patients. J. Neuroinflamm. 2019, 16, 131. [Google Scholar] [CrossRef]
- Nussbaum, R.L. The Identification of Alpha-Synuclein as the First Parkinson Disease Gene. J. Parkinsons Dis. 2017, 7, S43–S49. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, C.; Yin, J.; Li, X.; Cheng, F.; Li, Y.; Yang, H.; Uéda, K.; Chan, P.; Yu, S. α-Synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neurosci. Lett. 2009, 454, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial Import and Accumulation of α-Synuclein Impair Complex I in Human Dopaminergic Neuronal Cultures and Parkinson Disease Brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Duan, C.; Lü, L.; Gao, H.; Zhao, C.; Yu, S.; Uéda, K.; Chan, P.; Yang, H. α-Synuclein overexpression impairs mitochondrial function by associating with adenylate translocator. Int. J. Biochem. Cell Biol. 2011, 43, 732–741. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L. The role of parkin in familial and sporadic Parkinson’s disease. Mov. Disord. 2010, 25, S32–S39. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Thomas, R.R.; Keeney, P.M.; Bennett, J.P. Impaired complex-I mitochondrial biogenesis in Parkinson disease frontal cortex. J. Parkinsons Dis. 2012, 2, 67–76. [Google Scholar] [CrossRef]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518–520. [Google Scholar] [CrossRef]
- Fan, Z.; Pan, Y.-T.; Zhang, Z.-Y.; Yang, H.; Yu, S.-Y.; Zheng, Y.; Ma, J.-H.; Wang, X.-M. Systemic activation of NLRP3 inflammasome and plasma α-synuclein levels are correlated with motor severity and progression in Parkinson’s disease. J. Neuroinflamm. 2020, 17, 11. [Google Scholar] [CrossRef]
- Wang, X.; Chi, J.; Huang, D.; Ding, L.; Zhao, X.; Jiang, L.; Yu, Y.; Gao, F. α-synuclein promotes progression of Parkinson’s disease by upregulating autophagy signaling pathway to activate NLRP3 inflammasome. Exp. Ther. Med. 2019, 9, 931–938. [Google Scholar] [CrossRef]
- Sarkar, S.; Malovic, E.; Harishchandra, D.S.; Ghaisas, S.; Panicker, N.; Charli, A.; Palanisamy, B.N.; Rokad, D.; Jin, H.; Anantharam, V.; et al. Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. NPJ Parkinson’s Dis. 2017, 3, 30. [Google Scholar] [CrossRef]
- Fan, Z.; Liang, Z.; Yang, H.; Pan, Y.; Zheng, Y.; Wang, X. Tenuigenin protects dopaminergic neurons from inflammation via suppressing NLRP3 inflammasome activation in microglia. J. Neuroinflamm. 2017, 14, 256. [Google Scholar] [CrossRef]
- Mouton-Liger, F.; Rosazza, T.; Sepulveda-Diaz, J.; Ieang, A.; Hassoun, S.-M.; Claire, E.; Mangone, G.; Brice, A.; Michel, P.P.; Corvol, J.-C.; et al. Parkin deficiency modulates NLRP3 inflammasome activation by attenuating an A20-dependent negative feedback loop. Glia 2018, 66, 1736–1751. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, eaah4066. [Google Scholar] [CrossRef] [PubMed]
- Thakur, P.; Nehru, B. Inhibition of Neuroinflammation and Mitochondrial Dysfunctions by Carbenoxolone in the Rotenone Model of Parkinson’s Disease. Mol. Neurobiol. 2015, 51, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Rakovic, A.; Grünewald, A.; Voges, L.; Hofmann, S.; Orolicki, S.; Lohmann, K.; Klein, C. PINK1-Interacting Proteins: Proteomic Analysis of Overexpressed PINK1. Parkinsons Dis. 2011, 2011, 1–9. [Google Scholar] [CrossRef]
- Rakovic, A.; Grunewald, A.; Seibler, P.; Ramirez, A.; Kock, N.; Orolicki, S.; Lohmann, K.; Klein, C. Effect of endogenous mutant and wild-type PINK1 on Parkin in fibroblasts from Parkinson disease patients. Hum. Mol. Genet. 2010, 19, 3124–3137. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.-F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S. PINK1 protein in normal human brain and Parkinson’s disease. Brain 2006, 129, 1720–1731. [Google Scholar] [CrossRef]
- Jagmag, S.A.; Tripathi, N.; Shukla, S.D.; Maiti, S.; Khurana, S. Evaluation of Models of Parkinson’s Disease. Front. Neurosci. 2015, 9, 503. [Google Scholar] [CrossRef] [PubMed]
- Giaime, E.; Shen, J. Regulation of mitochondrial function by PINK1, Parkin and DJ-1. Mol. Neurodegener. 2013, 8, O13. [Google Scholar] [CrossRef]
- Giaime, E.; Yamaguchi, H.; Gautier, C.A.; Kitada, T.; Shen, J. Loss of DJ-1 Does Not Affect Mitochondrial Respiration but Increases ROS Production and Mitochondrial Permeability Transition Pore Opening. PLoS ONE 2012, 7, e40501. [Google Scholar] [CrossRef]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.J.; Wood, N.W.; et al. PINK1-Associated Parkinson’s Disease Is Caused by Neuronal Vulnerability to Calcium-Induced Cell Death. Mol. Cell 2009, 33, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Du, H.; Yan, S.S.; Fang, F.; Wang, C.; Lue, L.-F.; Guo, L.; Chen, D.; Stern, D.M.; Gunn Moore, F.J.; et al. Inhibition of Amyloid-(A) Peptide-Binding Alcohol Dehydrogenase-A Interaction Reduces A Accumulation and Improves Mitochondrial Function in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2011, 31, 2313–2320. [Google Scholar] [CrossRef] [PubMed]
- Tillement, L.; Lecanu, L.; Yao, W.; Greeson, J.; Papadopoulos, V. The spirostenol (22R, 25R)-20α-spirost-5-en-3β-yl hexanoate blocks mitochondrial uptake of Aβ in neuronal cells and prevents Aβ-induced impairment of mitochondrial function. Steroids 2006, 71, 725–735. [Google Scholar] [CrossRef]
- Walls, K.C.; Coskun, P.; Gallegos-Perez, J.L.; Zadourian, N.; Freude, K.; Rasool, S.; Blurton-Jones, M.; Green, K.N.; LaFerla, F.M. Swedish Alzheimer mutation induces mitochondrial dysfunction mediated by HSP60 mislocalization of amyloid precursor protein (APP) and beta-amyloid. J. Biol. Chem. 2012, 287, 30317–30327. [Google Scholar] [CrossRef]
- Wang, J.; Markesbery, W.R.; Lovell, M.A. Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J. Neurochem. 2006, 96, 825–832. [Google Scholar] [CrossRef]
- Bonda, D.J.; Wang, X.; Lee, H.-G.; Smith, M.A.; Perry, G.; Zhu, X. Neuronal failure in Alzheimer’s disease: A view through the oxidative stress looking-glass. Neurosci. Bull. 2014, 30, 243–252. [Google Scholar] [CrossRef]
- Coskun, P.E.; Beal, M.F.; Wallace, D.C. Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc. Natl. Acad. Sci. USA 2004, 101, 10726–10731. [Google Scholar] [CrossRef] [PubMed]
- Keogh, M.J.; Chinnery, P.F. Mitochondrial DNA mutations in neurodegeneration. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Stancu, I.-C.; Cremers, N.; Vanrusselt, H.; Couturier, J.; Vanoosthuyse, A.; Kessels, S.; Lodder, C.; Brône, B.; Huaux, F.; Octave, J.-N.; et al. Aggregated Tau activates NLRP3–ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019, 137, 599–617. [Google Scholar] [CrossRef]
- Shaftel, S.S.; Griffin, W.S.T.; O’Banion, M.K. The role of interleukin-1 in neuroinflammation and Alzheimer disease: An evolving perspective. J. Neuroinflamm. 2008, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10. [Google Scholar] [CrossRef]
- Griffin, W.; Liu, L.; Li, Y.; Mrak, R.; Barger, S. Interleukin-1 mediates Alzheimer and Lewy body pathologies. J Neuroinflamm. 2006, 3, 1–9. [Google Scholar] [CrossRef]
- Sheng, J.G.; Ito, K.; Skinner, R.D.; Mrak, R.E.; Rovnaghi, C.R.; Van Eldik, L.J.; Griffin, W.S. In vivo and in vitro evidence supporting a role for the inflammatory cytokine interleukin-1 as a driving force in Alzheimer pathogenesis. Neurobiol. Aging 1996, 17, 761–766. [Google Scholar] [CrossRef]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 Signaling Rescues Cognition, Attenuates Tau Pathology, and Restores Neuronal β-Catenin Pathway Function in an Alzheimer’s Disease Model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef]
- Bossu, P.; Ciaramella, A.; Moro, M.L.; Bellincampi, L.; Bernardini, S.; Federici, G.; Trequattrini, A.; Macciardi, F.; Spoletini, I.; Di Iulio, F.; et al. Interleukin 18 gene polymorphisms predict risk and outcome of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2007, 78, 807–811. [Google Scholar] [CrossRef]
- Bossù, P.; Ciaramella, A.; Salani, F.; Bizzoni, F.; Varsi, E.; Di Iulio, F.; Giubilei, F.; Gianni, W.; Trequattrini, A.; Moro, M.L.; et al. Interleukin-18 produced by peripheral blood cells is increased in Alzheimer’s disease and correlates with cognitive impairment. Brain Behav. Immun. 2008, 22, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, M.; Dell’Ombra, N.; Zambito, A.M.; Malaguarnera, M.; Nicoletti, F.; Malaguarnera, L. Chitotriosidase and inflammatory mediator levels in Alzheimer’s disease and cerebrovascular dementia. Eur. J. Neurosci. 2006, 23, 2648–2656. [Google Scholar] [CrossRef] [PubMed]
- Ojala, J.O.; Sutinen, E.M.; Salminen, A.; Pirttilä, T. Interleukin-18 increases expression of kinases involved in tau phosphorylation in SH-SY5Y neuroblastoma cells. J. Neuroimmunol. 2008, 205, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Knight, A.G.; Gupta, S.; Keller, J.N.; Bruce-Keller, A.J. Saturated long-chain fatty acids activate inflammatory signaling in astrocytes. J. Neurochem. 2012, 120, 1060–1071. [Google Scholar] [CrossRef]
- Du, F.; Yu, Q.; Yan, S.; Hu, G.; Lue, L.-F.; Walker, D.G.; Wu, L.; Yan, S.F.; Tieu, K.; Yan, S.S. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain 2017, 140, 3233–3251. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Castellazzi, M.; Patergnani, S.; Donadio, M.; Giorgi, C.; Bonora, M.; Bosi, C.; Brombo, G.; Pugliatti, M.; Seripa, D.; Zuliani, G.; et al. Autophagy and mitophagy biomarkers are reduced in sera of patients with Alzheimer’s disease and mild cognitive impairment. Sci. Rep. 2019, 9, 20009. [Google Scholar] [CrossRef]
- Veitch, D.P.; Weiner, M.W.; Aisen, P.S.; Beckett, L.A.; Cairns, N.J.; Green, R.C.; Harvey, D.; Jack, C.R.; Jagust, W.; Morris, J.C.; et al. Understanding disease progression and improving Alzheimer’s disease clinical trials: Recent highlights from the Alzheimer’s Disease Neuroimaging Initiative. Alzheimer’s Dement. 2019, 15, 106–152. [Google Scholar] [CrossRef]
- Toledo, J.B.; Arnold, M.; Kastenmüller, G.; Chang, R.; Baillie, R.A.; Han, X.; Thambisetty, M.; Tenenbaum, J.D.; Suhre, K.; Thompson, J.W.; et al. Metabolic network failures in Alzheimer’s disease: A biochemical road map. Alzheimer’s Dement. 2017, 13, 965–984. [Google Scholar] [CrossRef]
- Hatano, S. Experience from a multicentre stroke register: A preliminary report. Bull. World Health Organ. 1976, 54, 541–553. [Google Scholar]
- Hui, C.; Tadi, P.; Patti, L. Ischemic Stroke; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Katan, M.; Luft, A. Global Burden of Stroke. Semin. Neurol. 2018, 38, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Hossmann, K.-A. Pathophysiology and Therapy of Experimental Stroke. Cell. Mol. Neurobiol. 2006, 26, 1055–1081. [Google Scholar] [CrossRef] [PubMed]
- Khoshnam, S.E.; Winlow, W.; Farzaneh, M.; Farbood, Y.; Moghaddam, H.F. Pathogenic mechanisms following ischemic stroke. Neurol. Sci. 2017, 38, 1167–1186. [Google Scholar] [CrossRef] [PubMed]
- Ames, A. CNS energy metabolism as related to function. Brain Res. Rev. 2000, 34, 42–68. [Google Scholar] [CrossRef]
- Bandera, E.; Botteri, M.; Minelli, C.; Sutton, A.; Abrams, K.R.; Latronico, N. Cerebral Blood Flow Threshold of Ischemic Penumbra and Infarct Core in Acute Ischemic Stroke. Stroke 2006, 37, 1334–1339. [Google Scholar] [CrossRef] [PubMed]
- Siket, M.S. Treatment of Acute Ischemic Stroke. Emerg. Med. Clin. N. Am. 2016, 34, 861–882. [Google Scholar] [CrossRef]
- Jung, S.; Gilgen, M.; Slotboom, J.; El-Koussy, M.; Zubler, C.; Kiefer, C.; Luedi, R.; Mono, M.-L.; Heldner, M.R.; Weck, A.; et al. Factors that determine penumbral tissue loss in acute ischaemic stroke. Brain 2013, 136, 3554–3560. [Google Scholar] [CrossRef]
- Murphy, B.D.; Fox, A.J.; Lee, D.H.; Sahlas, D.J.; Black, S.E.; Hogan, M.J.; Coutts, S.B.; Demchuk, A.M.; Goyal, M.; Aviv, R.I.; et al. White matter thresholds for ischemic penumbra and infarct core in patients with acute stroke: CT perfusion study. Radiology 2008, 247, 818–825. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell Biology of Ischemia/Reperfusion Injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [CrossRef]
- Li, Q.; Cao, Y.; Dang, C.; Han, B.; Han, R.; Ma, H.; Hao, J.; Wang, L. Inhibition of double-strand DNA-sensing cGAS ameliorates brain injury after ischemic stroke. EMBO Mol. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Lammerding, L.; Slowik, A.; Johann, S.; Beyer, C.; Zendedel, A. Poststroke Inflammasome Expression and Regulation in the Peri-Infarct Area by Gonadal Steroids after Transient Focal Ischemia in the Rat Brain. Neuroendocrinology 2016, 103, 460–475. [Google Scholar] [CrossRef]
- Kim, H.; Seo, J.S.; Lee, S.-Y.; Ha, K.-T.; Choi, B.T.; Shin, Y.-I.; Ju Yun, Y.; Shin, H.K. AIM2 inflammasome contributes to brain injury and chronic post-stroke cognitive impairment in mice. Brain Behav. Immun. 2020. [Google Scholar] [CrossRef] [PubMed]
- Fann, D.Y.-W.; Lee, S.-Y.; Manzanero, S.; Chunduri, P.; Sobey, C.G.; Arumugam, T.V. Pathogenesis of acute stroke and the role of inflammasomes. Ageing Res. Rev. 2013, 12, 941–966. [Google Scholar] [CrossRef] [PubMed]
- Ismael, S.; Zhao, L.; Nasoohi, S.; Ishrat, T. Inhibition of the NLRP3-inflammasome as a potential approach for neuroprotection after stroke. Sci. Rep. 2018, 8, 5971. [Google Scholar] [CrossRef] [PubMed]
- Su, S.-H.; Wu, Y.-F.; Lin, Q.; Wang, D.-P.; Hai, J. URB597 protects against NLRP3 inflammasome activation by inhibiting autophagy dysfunction in a rat model of chronic cerebral hypoperfusion. J. Neuroinflamm. 2019, 16, 260. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; Tang, C.; Li, H.; Lei, J.; Zhu, L.; Kou, L.; Li, H.; Luo, S.; Li, C.; Chen, W.; et al. Eicosapentaenoic acid prevents inflammation induced by acute cerebral infarction through inhibition of NLRP3 inflammasome activation. Life Sci. 2020, 242, 117133. [Google Scholar] [CrossRef] [PubMed]
- Yang-Wei Fann, D.; Lee, S.-Y.; Manzanero, S.; Tang, S.-C.; Gelderblom, M.; Chunduri, P.; Bernreuther, C.; Glatzel, M.; Cheng, Y.-L.; Thundyil, J.; et al. Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome-mediated neuronal death in ischemic stroke. Cell Death Dis. 2013, 4, e790. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Pan, J.; Shen, Q.; Li, M.; Peng, Y. Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J. Neuroinflamm. 2018, 15, 242. [Google Scholar] [CrossRef]
- Gurung, P.; Lukens, J.R.; Kanneganti, T.-D. Mitochondria: Diversity in the regulation of the NLRP3 inflammasome. Trends Mol. Med. 2015, 21, 193–201. [Google Scholar] [CrossRef]
- Mongin, A.A. Disruption of ionic and cell volume homeostasis in cerebral ischemia: The perfect storm. Pathophysiology 2007, 14, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Epstein, F.H.; Lipton, S.A.; Rosenberg, P.A. Excitatory Amino Acids as a Final Common Pathway for Neurologic Disorders. N. Engl. J. Med. 1994, 330, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Schipke, C.G.; Ohlemeyer, C.; Matyash, M.; Nolte, C.; Kettenmann, H.; Kirchhoff, F. Astrocytes of the mouse neocortex express functional N-methyl-D-aspartate receptors. FASEB J. 2001, 15, 1270–1272. [Google Scholar] [CrossRef] [PubMed]
- Kaindl, A.M.; Degos, V.; Peineau, S.; Gouadon, E.; Chhor, V.; Loron, G.; Le Charpentier, T.; Josserand, J.; Ali, C.; Vivien, D.; et al. Activation of microglial N-methyl-D-aspartate receptors triggers inflammation and neuronal cell death in the developing and mature brain. Ann. Neurol. 2012, 72, 536–549. [Google Scholar] [CrossRef] [PubMed]
- White, R.; Reynolds, I. Mitochondrial depolarization in glutamate-stimulated neurons: An early signal specific to excitotoxin exposure. J. Neurosci. 1996, 16, 5688–5697. [Google Scholar] [CrossRef] [PubMed]
- Mody, I. NMDA receptor-dependent excitotoxicity: The role of intracellular Ca2+ release. Trends Pharmacol. Sci. 1995, 16, 356–359. [Google Scholar] [CrossRef]
- Smith, J.P.; Cunningham, L.A.; Partridge, L.D. Coupling of AMPA receptors with the Na+/Ca2+ exchanger in cultured rat astrocytes. Brain Res. 2000, 887, 98–109. [Google Scholar] [CrossRef]
- Chinopoulos, C.; Adam-Vizi, V. Mitochondrial Ca2+ sequestration and precipitation revisited. FEBS J. 2010, 277, 3637–3651. [Google Scholar] [CrossRef]
- Triantafilou, K.; Hughes, T.R.; Triantafilou, M.; Morgan, B.P. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J. Cell Sci. 2013, 126, 2903–2913. [Google Scholar] [CrossRef]
- Qiu, J.; Tan, Y.-W.; Hagenston, A.M.; Martel, M.-A.; Kneisel, N.; Skehel, P.A.; Wyllie, D.J.A.; Bading, H.; Hardingham, G.E. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun. 2013, 4, 2034. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Yan, J.; Wang, L.; Tian, X.; Zhang, T.; Li, B.; Wang, W.; Guo, L.; Liu, X. Expression of MICU1 after experimental focal cerebral ischemia in adult rats. Chin. Neurosurg. J. 2017, 3, 13. [Google Scholar] [CrossRef]
- Hill, J.K.; Gunion-Rinker, L.; Kulhanek, D.; Lessov, N.; Kim, S.; Clark, W.M.; Dixon, M.P.; Nishi, R.; Stenzel-Poore, M.P.; Eckenstein, F.P. Temporal modulation of cytokine expression following focal cerebral ischemia in mice. Brain Res. 1999, 820, 45–54. [Google Scholar] [CrossRef]
- Lambertsen, K.L.; Meldgaard, M.; Ladeby, R.; Finsen, B. A Quantitative Study of Microglial—Macrophage Synthesis of Tumor Necrosis Factor during Acute and Late Focal Cerebral Ischemia in Mice. J. Cereb. Blood Flow Metab. 2005, 25, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Clausen, B.H.; Lambertsen, K.L.; Babcock, A.A.; Holm, T.H.; Dagnaes-Hansen, F.; Finsen, B. Interleukin-1beta and tumor necrosis factor-alpha are expressed by different subsets of microglia and macrophages after ischemic stroke in mice. J. Neuroinflamm. 2008, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [PubMed]
- Pétrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007, 14, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, T.H.; Reynolds, C.A.; Kumar, R.; Przyklenk, K.; Hüttemann, M. Molecular Mechanisms of Ischemia–Reperfusion Injury in Brain: Pivotal Role of the Mitochondrial Membrane Potential in Reactive Oxygen Species Generation. Mol. Neurobiol. 2013, 47, 9–23. [Google Scholar] [CrossRef]
- Cruz, C.M.; Rinna, A.; Forman, H.J.; Ventura, A.L.M.; Persechini, P.M.; Ojcius, D.M. ATP Activates a Reactive Oxygen Species-dependent Oxidative Stress Response and Secretion of Proinflammatory Cytokines in Macrophages. J. Biol. Chem. 2007, 282, 2871–2879. [Google Scholar] [CrossRef] [PubMed]
- Loor, G.; Kondapalli, J.; Iwase, H.; Chandel, N.S.; Waypa, G.B.; Guzy, R.D.; Vanden Hoek, T.L.; Schumacker, P.T. Mitochondrial oxidant stress triggers cell death in simulated ischemia–reperfusion. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1382–1394. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kussmaul, L.; Hirst, J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7607–7612. [Google Scholar] [CrossRef] [PubMed]
- Bleier, L.; Dröse, S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochim. Biophys. Acta Bioenerg. 2013, 1827, 1320–1331. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial Complex I Inhibitor Rotenone Induces Apoptosis through Enhancing Mitochondrial Reactive Oxygen Species Production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar] [CrossRef]
- Horng, T. Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends Immunol. 2014, 35, 253–261. [Google Scholar] [CrossRef]
- Zhuang, Y.; Yasinta, M.; Hu, C.; Zhao, M.; Ding, G.; Bai, M.; Yang, L.; Ni, J.; Wang, R.; Jia, Z.; et al. Mitochondrial dysfunction confers albumin-induced NLRP3 inflammasome activation and renal tubular injury. Am. J. Physiol. Renal Physiol. 2015, 308, F857–F866. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Schinzel, A.C.; Takeuchi, O.; Huang, Z.; Fisher, J.K.; Zhou, Z.; Rubens, J.; Hetz, C.; Danial, N.N.; Moskowitz, M.A.; Korsmeyer, S.J. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 2005, 102, 12005–12010. [Google Scholar] [CrossRef]
- Sun, J.; Li, Y.; Ding, Y.; Wang, J.; Geng, J.; Yang, H.; Ren, J.; Tang, J.; Gao, J. Neuroprotective effects of gallic acid against hypoxia/reoxygenation-induced mitochondrial dysfunctions in vitro and cerebral ischemia/reperfusion injury in vivo. Brain Res. 2014, 1589, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Murozono, M.; Kanazawa, M.; Nara, T.; Ozawa, T.; Watanabe, Y. Edaravone and cyclosporine A as neuroprotective agents for acute ischemic stroke. Acute Med. Surg. 2018, 5, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Chen, S.; Hu, Q.; Feng, H.; Zhang, J.H.; Tang, J. NLRP3 inflammasome contributes to inflammation after intracerebral hemorrhage. Ann. Neurol. 2014, 75, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nartiss, Y.; Steipe, B.; McQuibban, G.A.; Kim, P.K. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 2012, 8, 1462–1476. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S.P.; Qian, T.; Grissom, S.F.; Lemasters, J.J. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001, 15, 1–17. [Google Scholar] [CrossRef]
- Lyamzaev, K.G.; Tokarchuk, A.V.; Panteleeva, A.A.; Mulkidjanian, A.Y.; Skulachev, V.P.; Chernyak, B.V. Induction of autophagy by depolarization of mitochondria. Autophagy 2018, 14, 921–924. [Google Scholar] [CrossRef]
- Cao, S.; Shrestha, S.; Li, J.; Yu, X.; Chen, J.; Yan, F.; Ying, G.; Gu, C.; Wang, L.; Chen, G. Melatonin-mediated mitophagy protects against early brain injury after subarachnoid hemorrhage through inhibition of NLRP3 inflammasome activation. Sci. Rep. 2017, 7, 2417. [Google Scholar] [CrossRef]
- Di, Y.; He, Y.-L.; Zhao, T.; Huang, X.; Wu, K.-W.; Liu, S.-H.; Zhao, Y.-Q.; Fan, M.; Wu, L.-Y.; Zhu, L.-L. Methylene Blue Reduces Acute Cerebral Ischemic Injury via the Induction of Mitophagy. Mol. Med. 2015, 21, 420–429. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, T.; Wang, J.; Zhang, Z.; Zhai, Y.; Yang, G.-Y.; Sun, X. Rapamycin attenuates mitochondrial dysfunction via activation of mitophagy in experimental ischemic stroke. Biochem. Biophys. Res. Commun. 2014, 444, 182–188. [Google Scholar] [CrossRef]
- He, Q.; Li, Z.; Meng, C.; Wu, J.; Zhao, Y.; Zhao, J. Parkin-Dependent Mitophagy is Required for the Inhibition of ATF4 on NLRP3 Inflammasome Activation in Cerebral Ischemia-Reperfusion Injury in Rats. Cells 2019, 8, 897. [Google Scholar] [CrossRef]
- Yuan, Y.; Zheng, Y.; Zhang, X.; Chen, Y.; Wu, X.; Wu, J.; Shen, Z.; Jiang, L.; Wang, L.; Yang, W.; et al. BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy 2017, 13, 1754–1766. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.-Y.; Zhu, S.-H.; Li, V.; Gibson, S.B.; Xu, X.-S.; Kong, J.-M. BNIP3 Interacting with LC3 Triggers Excessive Mitophagy in Delayed Neuronal Death in Stroke. CNS Neurosci. Ther. 2014, 20, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Chen, X.; Guan, B.; Li, C.; Qiu, J.; Shen, J. Inhibition of Peroxynitrite-Induced Mitophagy Activation Attenuates Cerebral Ischemia-Reperfusion Injury. Mol. Neurobiol. 2018, 55, 6369–6386. [Google Scholar] [CrossRef] [PubMed]
- Leach, J.P. Treatment of epilepsy—Towards precision. F1000Research 2018, 7. [Google Scholar] [CrossRef]
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE official report: A practical clinical definition of epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef]
- Laxer, K.D.; Trinka, E.; Hirsch, L.J.; Cendes, F.; Langfitt, J.; Delanty, N.; Resnick, T.; Benbadis, S.R. The consequences of refractory epilepsy and its treatment. Epilepsy Behav. 2014, 37, 59–70. [Google Scholar] [CrossRef]
- Rana, A.; Musto, A.E. The role of inflammation in the development of epilepsy. J. Neuroinflamm. 2018, 15, 144. [Google Scholar] [CrossRef]
- Vezzani, A.; Conti, M.; De Luigi, A.; Ravizza, T.; Moneta, D.; Marchesi, F.; De Simoni, M.G. Interleukin-1beta immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: Functional evidence for enhancement of electrographic seizures. J. Neurosci. 1999, 19, 5054–5065. [Google Scholar] [CrossRef]
- McElroy, P.B.; Liang, L.-P.; Day, B.J.; Patel, M. Scavenging reactive oxygen species inhibits status epilepticus-induced neuroinflammation. Exp. Neurol. 2017, 298, 13–22. [Google Scholar] [CrossRef]
- Pauletti, A.; Terrone, G.; Shekh-Ahmad, T.; Salamone, A.; Ravizza, T.; Rizzi, M.; Pastore, A.; Pascente, R.; Liang, L.-P.; Villa, B.R.; et al. Targeting oxidative stress improves disease outcomes in a rat model of acquired epilepsy. Brain 2017, 140, 1885–1899. [Google Scholar] [CrossRef]
- Terrone, G.; Balosso, S.; Pauletti, A.; Ravizza, T.; Vezzani, A. Inflammation and reactive oxygen species as disease modifiers in epilepsy. Neuropharmacology 2020, 167, 107742. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S. Pathophysiology of mitochondrial disease causing epilepsy and status epilepticus. Epilepsy Behav. 2015, 49, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Téllez-Zenteno, J.F.; Hernández-Ronquillo, L. A review of the epidemiology of temporal lobe epilepsy. Epilepsy Res. Treat. 2012, 2012, 630853. [Google Scholar] [CrossRef] [PubMed]
- Pitkänen, A.; Lukasiuk, K. Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol. 2011, 10, 173–186. [Google Scholar] [CrossRef]
- Shen, K.; Mao, Q.; Yin, X.; Zhang, C.; Jin, Y.; Deng, A.; Gu, Z.; Chen, B. NLRP3 Inflammasome Activation Leads to Epileptic Neuronal Apoptosis. Curr. Neurovasc. Res. 2018, 15, 276–281. [Google Scholar] [CrossRef]
- Meng, X.-F.; Tan, L.; Tan, M.-S.; Jiang, T.; Tan, C.-C.; Li, M.-M.; Wang, H.-F.; Yu, J.-T. Inhibition of the NLRP3 inflammasome provides neuroprotection in rats following amygdala kindling-induced status epilepticus. J. Neuroinflamm. 2014, 11, 212. [Google Scholar] [CrossRef]
- Rong, S.; Wan, D.; Fan, Y.; Liu, S.; Sun, K.; Huo, J.; Zhang, P.; Li, X.; Xie, X.; Wang, F.; et al. Amentoflavone Affects Epileptogenesis and Exerts Neuroprotective Effects by Inhibiting NLRP3 Inflammasome. Front. Pharmacol. 2019, 10, 856. [Google Scholar] [CrossRef]
- Sun, Y.; Ma, J.; Li, D.; Li, P.; Zhou, X.; Li, Y.; He, Z.; Qin, L.; Liang, L.; Luo, X. Interleukin-10 inhibits interleukin-1β production and inflammasome activation of microglia in epileptic seizures. J. Neuroinflamm. 2019, 16, 66. [Google Scholar] [CrossRef]
- Liu, Z.; Xian, H.; Ye, X.; Chen, J.; Ma, Y.; Huang, W. Increased levels of NLRP3 in children with febrile seizures. Brain Dev. 2020, 42, 336–341. [Google Scholar] [CrossRef]
- Wang, H.; Xu, P.; Liao, D.; Dang, R.; He, X.; Guo, Y.; Jiang, P. Association between NLPR1, NLPR3, and P2X7R Gene Polymorphisms with Partial Seizures. BioMed Res. Int. 2017, 2017, 9547902. [Google Scholar] [CrossRef]
- Tan, C.-C.; Zhang, J.-G.; Tan, M.-S.; Chen, H.; Meng, D.-W.; Jiang, T.; Meng, X.-F.; Li, Y.; Sun, Z.; Li, M.-M.; et al. NLRP1 inflammasome is activated in patients with medial temporal lobe epilepsy and contributes to neuronal pyroptosis in amygdala kindling-induced rat model. J. Neuroinflamm. 2015, 12, 18. [Google Scholar] [CrossRef] [PubMed]
- Dixit, A.B.; Banerjee, J.; Srivastava, A.; Tripathi, M.; Sarkar, C.; Kakkar, A.; Jain, M.; Chandra, P.S. RNA-seq analysis of hippocampal tissues reveals novel candidate genes for drug refractory epilepsy in patients with MTLE-HS. Genomics 2016, 107, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.; Backos, D.S.; Reigan, P.; Patel, M. Post-translational oxidative modification and inactivation of mitochondrial complex I in epileptogenesis. J. Neurosci. 2012, 32, 11250–11258. [Google Scholar] [CrossRef]
- Waldbaum, S.; Liang, L.-P.; Patel, M. Persistent impairment of mitochondrial and tissue redox status during lithium-pilocarpine-induced epileptogenesis. J. Neurochem. 2010, 115, 1172–1182. [Google Scholar] [CrossRef]
- Rowley, S.; Liang, L.-P.; Fulton, R.; Shimizu, T.; Day, B.; Patel, M. Mitochondrial respiration deficits driven by reactive oxygen species in experimental temporal lobe epilepsy. Neurobiol. Dis. 2015, 75, 151–158. [Google Scholar] [CrossRef]
- Liang, L.P.; Ho, Y.S.; Patel, M. Mitochondrial superoxide production in kainate-induced hippocampal damage. Neuroscience 2000, 101, 563–570. [Google Scholar] [CrossRef]
- Pearson, J.N.; Rowley, S.; Liang, L.-P.; White, A.M.; Day, B.J.; Patel, M. Reactive oxygen species mediate cognitive deficits in experimental temporal lobe epilepsy. Neurobiol. Dis. 2015, 82, 289–297. [Google Scholar] [CrossRef]
- Walker, M.C. Pathophysiology of status epilepticus. Neurosci. Lett. 2018, 667, 84–91. [Google Scholar] [CrossRef]
- Kovac, S.; Domijan, A.-M.; Walker, M.C.; Abramov, A.Y. Seizure activity results in calcium- and mitochondria-independent ROS production via NADPH and xanthine oxidase activation. Cell Death Dis. 2014, 5, e1442. [Google Scholar] [CrossRef]
- Ding, S.; Fellin, T.; Zhu, Y.; Lee, S.-Y.; Auberson, Y.P.; Meaney, D.F.; Coulter, D.A.; Carmignoto, G.; Haydon, P.G. Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J. Neurosci. 2007, 27, 10674–10684. [Google Scholar] [CrossRef] [PubMed]
- Volmering, E.; Niehusmann, P.; Peeva, V.; Grote, A.; Zsurka, G.; Altmüller, J.; Nürnberg, P.; Becker, A.J.; Schoch, S.; Elger, C.E.; et al. Neuropathological signs of inflammation correlate with mitochondrial DNA deletions in mesial temporal lobe epilepsy. Acta Neuropathol. 2016, 132, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Shefa, U.; Jeong, N.Y.; Song, I.O.; Chung, H.-J.; Kim, D.; Jung, J.; Huh, Y. Mitophagy links oxidative stress conditions and neurodegenerative diseases. Neural Regen. Res. 2019, 14, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Liu, X.; Chi, X.; Zhang, L.; Xiong, W.; Chiang, S.M.V.; Zhou, D.; Li, J. Mitophagy in Refractory Temporal Lobe Epilepsy Patients with Hippocampal Sclerosis. Cell. Mol. Neurobiol. 2018, 38, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.-J.; Wang, R.-F.; Hölscher, C.; Mi, R.-L.; Yuan, Z.-Y.; Li, D.-F.; Xue, G.-F. The novel GLP-1/GIP dual receptor agonist DA3-CH is neuroprotective in the pilocarpine-induced epileptogenesis rat model. Epilepsy Res. 2019, 154, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, M.; Zhu, W.; Yu, J.; Wang, Q.; Zhang, J.; Cui, Y.; Pan, X.; Gao, X.; Sun, H. Succinate accumulation induces mitochondrial reactive oxygen species generation and promotes status epilepticus in the kainic acid rat model. Redox Biol. 2020, 28, 101365. [Google Scholar] [CrossRef] [PubMed]
- Redman, B.G.; Abubakr, Y.; Chou, T.; Esper, P.; Flaherty, L.E. Phase II trial of recombinant interleukin-1 beta in patients with metastatic renal cell carcinoma. J. Immunother. Emphas. Tumor Immunol. 1994, 16, 211–215. [Google Scholar] [CrossRef]
- Chari, D.M. Remyelination in multiple sclerosis. Int. Rev. Neurobiol. 2007, 79, 589–620. [Google Scholar] [CrossRef]
- Lampron, A.; Larochelle, A.; Laflamme, N.; Préfontaine, P.; Plante, M.-M.; Sánchez, M.G.; Yong, V.W.; Stys, P.K.; Tremblay, M.-È.; Rivest, S. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J. Exp. Med. 2015, 212, 481–495. [Google Scholar] [CrossRef]
- Kocur, M.; Schneider, R.; Pulm, A.-K.; Bauer, J.; Kropp, S.; Gliem, M.; Ingwersen, J.; Goebels, N.; Alferink, J.; Prozorovski, T.; et al. IFNβ secreted by microglia mediates clearance of myelin debris in CNS autoimmunity. Acta Neuropathol. Commun. 2015, 3, 20. [Google Scholar] [CrossRef]
- De Mercanti, S.; Rolla, S.; Cucci, A.; Bardina, V.; Cocco, E.; Vladic, A.; Soldo-Butkovic, S.; Habek, M.; Adamec, I.; Horakova, D.; et al. Alemtuzumab long-term immunologic effect: Treg suppressor function increases up to 24 months. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e194. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.; Herrod, S.S.; Alvarez-Gonzalez, C.; Giovannoni, G.; Schmierer, K. Interpreting Lymphocyte Reconstitution Data From the Pivotal Phase 3 Trials of Alemtuzumab. JAMA Neurol. 2017, 74, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Pinschewer, D.D.; Ochsenbein, A.F.; Odermatt, B.; Brinkmann, V.; Hengartner, H.; Zinkernagel, R.M. FTY720 immunosuppression impairs effector T cell peripheral homing without affecting induction, expansion, and memory. J. Immunol. 2000, 164, 5761–5770. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Antel, J.; Comi, G.; Montalban, X.; O’Connor, P.; Polman, C.H.; Haas, T.; Korn, A.A.; Karlsson, G.; Radue, E.W.; et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N. Engl. J. Med. 2006, 355, 1124–1140. [Google Scholar] [CrossRef]
- Cohen, J.A.; Barkhof, F.; Comi, G.; Hartung, H.-P.; Khatri, B.O.; Montalban, X.; Pelletier, J.; Capra, R.; Gallo, P.; Izquierdo, G.; et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 402–415. [Google Scholar] [CrossRef]
- Nagatsu, T.; Mogi, M.; Ichinose, H.; Togari, A. Changes in cytokines and neurotrophins in Parkinson’s disease. J. Neural Transm. Suppl. 2000, 277–290. [Google Scholar] [CrossRef]
- Campolo, M.; Paterniti, I.; Siracusa, R.; Filippone, A.; Esposito, E.; Cuzzocrea, S. TLR4 absence reduces neuroinflammation and inflammasome activation in Parkinson’s diseases in vivo model. Brain Behav. Immun. 2019, 76, 236–247. [Google Scholar] [CrossRef]
- Muthuraman, M.; Koirala, N.; Ciolac, D.; Pintea, B.; Glaser, M.; Groppa, S.; Tamás, G.; Groppa, S. Deep Brain Stimulation and L-DOPA Therapy: Concepts of Action and Clinical Applications in Parkinson’s Disease. Front. Neurol. 2018, 9, 711. [Google Scholar] [CrossRef] [PubMed]
- Tomás-Camardiel, M.; Rite, I.; Herrera, A.J.; de Pablos, R.M.; Cano, J.; Machado, A.; Venero, J.L. Minocycline reduces the lipopolysaccharide-induced inflammatory reaction, peroxynitrite-mediated nitration of proteins, disruption of the blood-brain barrier, and damage in the nigral dopaminergic system. Neurobiol. Dis. 2004, 16, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Diguet, E.; Fernagut, P.-O.; Wei, X.; Du, Y.; Rouland, R.; Gross, C.; Bezard, E.; Tison, F. Deleterious effects of minocycline in animal models of Parkinson’s disease and Huntington’s disease. Eur. J. Neurosci. 2004, 19, 3266–3276. [Google Scholar] [CrossRef]
- Castaño, A.; Herrera, A.J.; Cano, J.; Machado, A. The degenerative effect of a single intranigral injection of LPS on the dopaminergic system is prevented by dexamethasone, and not mimicked by rh-TNF-alpha, IL-1beta and IFN-gamma. J. Neurochem. 2002, 81, 150–157. [Google Scholar] [CrossRef]
- Liu, B.; Du, L.; Hong, J.S. Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J. Pharmacol. Exp. Ther. 2000, 293, 607–617. [Google Scholar]
- Li, Y.; Hu, X.; Liu, Y.; Bao, Y.; An, L. Nimodipine protects dopaminergic neurons against inflammation-mediated degeneration through inhibition of microglial activation. Neuropharmacology 2009, 56, 580–589. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, L.; Li, L.; Hölscher, C. Semaglutide is Neuroprotective and Reduces α-Synuclein Levels in the Chronic MPTP Mouse Model of Parkinson’s Disease. J. Parkinsons Dis. 2019, 9, 157–171. [Google Scholar] [CrossRef]
- Sanz, J.M.; Chiozzi, P.; Colaianna, M.; Zotti, M.; Ferrari, D.; Trabace, L.; Zuliani, G.; Di Virgilio, F. Nimodipine inhibits IL-1β release stimulated by amyloid β from microglia. Br. J. Pharmacol. 2012, 167, 1702–1711. [Google Scholar] [CrossRef]
- Vlad, S.C.; Miller, D.R.; Kowall, N.W.; Felson, D.T. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 2008, 70, 1672–1677. [Google Scholar] [CrossRef]
- Jaturapatporn, D.; Isaac, M.G.E.K.N.; McCleery, J.; Tabet, N. Aspirin, steroidal and non-steroidal anti-inflammatory drugs for the treatment of Alzheimer’s disease. Cochrane Database Syst. Rev. 2012, CD006378. [Google Scholar] [CrossRef]
- Daniels, M.J.D.; Rivers-Auty, J.; Schilling, T.; Spencer, N.G.; Watremez, W.; Fasolino, V.; Booth, S.J.; White, C.S.; Baldwin, A.G.; Freeman, S.; et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat. Commun. 2016, 7, 12504. [Google Scholar] [CrossRef]
- Sato, T.; Hanyu, H.; Hirao, K.; Kanetaka, H.; Sakurai, H.; Iwamoto, T. Efficacy of PPAR-γ agonist pioglitazone in mild Alzheimer disease. Neurobiol. Aging 2011, 32, 1626–1633. [Google Scholar] [CrossRef]
- Hall, H.; Iulita, M.F.; Gubert, P.; Flores Aguilar, L.; Ducatenzeiler, A.; Fisher, A.; Cuello, A.C. AF710B, an M1/sigma-1 receptor agonist with long-lasting disease-modifying properties in a transgenic rat model of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 811–823. [Google Scholar] [CrossRef]
- Woodling, N.S.; Colas, D.; Wang, Q.; Minhas, P.; Panchal, M.; Liang, X.; Mhatre, S.D.; Brown, H.; Ko, N.; Zagol-Ikapitte, I.; et al. Cyclooxygenase inhibition targets neurons to prevent early behavioural decline in Alzheimer’s disease model mice. Brain 2016, 139, 2063–2081. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhang, N.; Ren, L.; Yan, Y.; Sun, N.; Li, Y.-J.; Han, W.; Xue, R.; Liu, Q.; Hao, J.; et al. Impact of an immune modulator fingolimod on acute ischemic stroke. Proc. Natl. Acad. Sci. USA 2014, 111, 18315–18320. [Google Scholar] [CrossRef] [PubMed]
- Lampl, Y.; Boaz, M.; Gilad, R.; Lorberboym, M.; Dabby, R.; Rapoport, A.; Anca-Hershkowitz, M.; Sadeh, M. Minocycline treatment in acute stroke: An open-label, evaluator-blinded study. Neurology 2007, 69, 1404–1410. [Google Scholar] [CrossRef] [PubMed]
- Kohler, E.; Prentice, D.A.; Bates, T.R.; Hankey, G.J.; Claxton, A.; van Heerden, J.; Blacker, D. Intravenous minocycline in acute stroke: A randomized, controlled pilot study and meta-analysis. Stroke 2013, 44, 2493–2499. [Google Scholar] [CrossRef] [PubMed]
- Emsley, H.C.A.; Smith, C.J.; Georgiou, R.F.; Vail, A.; Hopkins, S.J.; Rothwell, N.J.; Tyrrell, P.J.; Acute Stroke Investigators. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1366–1372. [Google Scholar] [CrossRef]
- Smith, C.J.; Hulme, S.; Vail, A.; Heal, C.; Parry-Jones, A.R.; Scarth, S.; Hopkins, K.; Hoadley, M.; Allan, S.M.; Rothwell, N.J.; et al. SCIL-STROKE (Subcutaneous Interleukin-1 Receptor Antagonist in Ischemic Stroke): A Randomized Controlled Phase 2 Trial. Stroke 2018, 49, 1210–1216. [Google Scholar] [CrossRef]
- Hill, M.D.; Goyal, M.; Menon, B.K.; Nogueira, R.G.; McTaggart, R.A.; Demchuk, A.M.; Poppe, A.Y.; Buck, B.H.; Field, T.S.; Dowlatshahi, D.; et al. Efficacy and safety of nerinetide for the treatment of acute ischaemic stroke (ESCAPE-NA1): A multicentre, double-blind, randomised controlled trial. Lancet 2020, 395, 878–887. [Google Scholar] [CrossRef]
- Vezzani, A.; Moneta, D.; Conti, M.; Richichi, C.; Ravizza, T.; De Luigi, A.; De Simoni, M.G.; Sperk, G.; Andell-Jonsson, S.; Lundkvist, J.; et al. Powerful anticonvulsant action of IL-1 receptor antagonist on intracerebral injection and astrocytic overexpression in mice. Proc. Natl. Acad. Sci. USA 2000, 97, 11534–11539. [Google Scholar] [CrossRef]
- Vezzani, A.; Maroso, M.; Balosso, S.; Sanchez, M.-A.; Bartfai, T. IL-1 receptor/Toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav. Immun. 2011, 25, 1281–1289. [Google Scholar] [CrossRef]
- Kenney-Jung, D.L.; Vezzani, A.; Kahoud, R.J.; LaFrance-Corey, R.G.; Ho, M.-L.; Muskardin, T.W.; Wirrell, E.C.; Howe, C.L.; Payne, E.T. Febrile infection-related epilepsy syndrome treated with anakinra. Ann. Neurol. 2016, 80, 939–945. [Google Scholar] [CrossRef]
- Stienen, M.N.; Haghikia, A.; Dambach, H.; Thöne, J.; Wiemann, M.; Gold, R.; Chan, A.; Dermietzel, R.; Faustmann, P.M.; Hinkerohe, D.; et al. Anti-inflammatory effects of the anticonvulsant drug levetiracetam on electrophysiological properties of astroglia are mediated via TGFβ1 regulation. Br. J. Pharmacol. 2011, 162, 491–507. [Google Scholar] [CrossRef]
- Silvestro, S.; Mammana, S.; Cavalli, E.; Bramanti, P.; Mazzon, E. Use of Cannabidiol in the Treatment of Epilepsy: Efficacy and Security in Clinical Trials. Molecules 2019, 24, 1459. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapy | Disease | Effects | Clinically Approved | Reference |
|---|---|---|---|---|
| Alemtuzumab | MS | Controversial | No | [272,273] |
| ANAVEX2-73 | AD | Anti-inflammatory; Antioxidant | No | NCT03790709 |
| ANAVEX3-71 | AD | Anti-inflammatory | No | [291]; NCT04442945 |
| Cannabidiol | Epilepsy | Anti-inflammatory; Antioxidant | Yes | [303]; NCT02224690 |
| Dexamethasone | PD | Neuroprotectant | No | [282] |
| Exenatide | PD | Neuroprotectant | No | NCT04232969 |
| Fenamate NSAIDs | AD | Anti-inflammatory | No | [289] |
| Fenebrutinib | MS | Anti-inflammatory | No | NCT04544449 |
| Fingolimod | IS | Neuroprotectant | No | [293] |
| Fingolimod | MS | Anti-inflammatory | No | [274,275,276] |
| H2M | IS | Antioxidant; neuroprotectant. | No | [287,288] |
| Ibuprofen | AD | Controversial | No | [287,288] |
| Idebenone | PD | Antioxidant | No | [299,300] |
| IL-1Ra | Epilepsy | Anticonvulsant | No | [299,300] |
| IL-1Ra | IS | Controversial | No | [296,297] |
| Levetiracetam | Epilepsy | Anticonvulsant; anti-inflammatory | Yes | [302] |
| Minocycline | IS | Controversial | No | [294,295] |
| Minocycline | PD | Controversial | No | [280,281] |
| Naloxone | PD | Neuroprotectant | No | [283] |
| Nerinetide | IS | Neuroprotectant | No | [298]; NCT04462536 |
| Nilvadipine | AD | Controversial | No | NCT02017340 |
| Nimodipine | AD | Anti-inflammatory | No | [286] |
| Nimodipine | PD | Neuroprotectant | No | [284] |
| Ocrelizumab | MS | Neuroprotectant | Yes | NCT04544436; NCT04387734 |
| Ofatumumab | MS | Anti-inflammatory | No | NCT04486716 |
| Pioglitazone | AD | Controversial | No | [290] |
| Semaglutide | PD | Anti-inflammatory | No | [285];NCT03659682 |
| TLR4 deletion | PD | Neuroprotectant; Anti-inflammatory | No | [278] |
| Tocotrienols | PD | Antioxidant | No | NCT04491383 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vezzani, B.; Carinci, M.; Patergnani, S.; Pasquin, M.P.; Guarino, A.; Aziz, N.; Pinton, P.; Simonato, M.; Giorgi, C. The Dichotomous Role of Inflammation in the CNS: A Mitochondrial Point of View. Biomolecules 2020, 10, 1437. https://doi.org/10.3390/biom10101437
Vezzani B, Carinci M, Patergnani S, Pasquin MP, Guarino A, Aziz N, Pinton P, Simonato M, Giorgi C. The Dichotomous Role of Inflammation in the CNS: A Mitochondrial Point of View. Biomolecules. 2020; 10(10):1437. https://doi.org/10.3390/biom10101437
Chicago/Turabian StyleVezzani, Bianca, Marianna Carinci, Simone Patergnani, Matteo P. Pasquin, Annunziata Guarino, Nimra Aziz, Paolo Pinton, Michele Simonato, and Carlotta Giorgi. 2020. "The Dichotomous Role of Inflammation in the CNS: A Mitochondrial Point of View" Biomolecules 10, no. 10: 1437. https://doi.org/10.3390/biom10101437
APA StyleVezzani, B., Carinci, M., Patergnani, S., Pasquin, M. P., Guarino, A., Aziz, N., Pinton, P., Simonato, M., & Giorgi, C. (2020). The Dichotomous Role of Inflammation in the CNS: A Mitochondrial Point of View. Biomolecules, 10(10), 1437. https://doi.org/10.3390/biom10101437