Kinetic, Thermodynamic, and Crystallographic Studies of 2-Triazolylthioacetamides as Verona Integron-Encoded Metallo-β-Lactamase 2 (VIM-2) Inhibitor

 ,
,
Abstract
1. Introduction
2. Materials and Methods
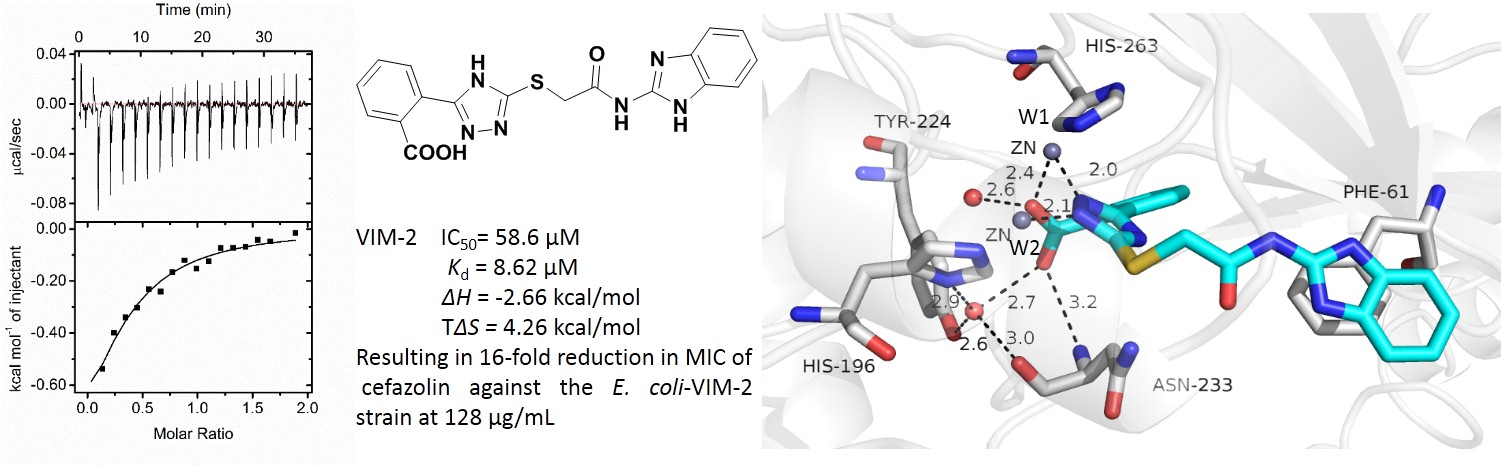
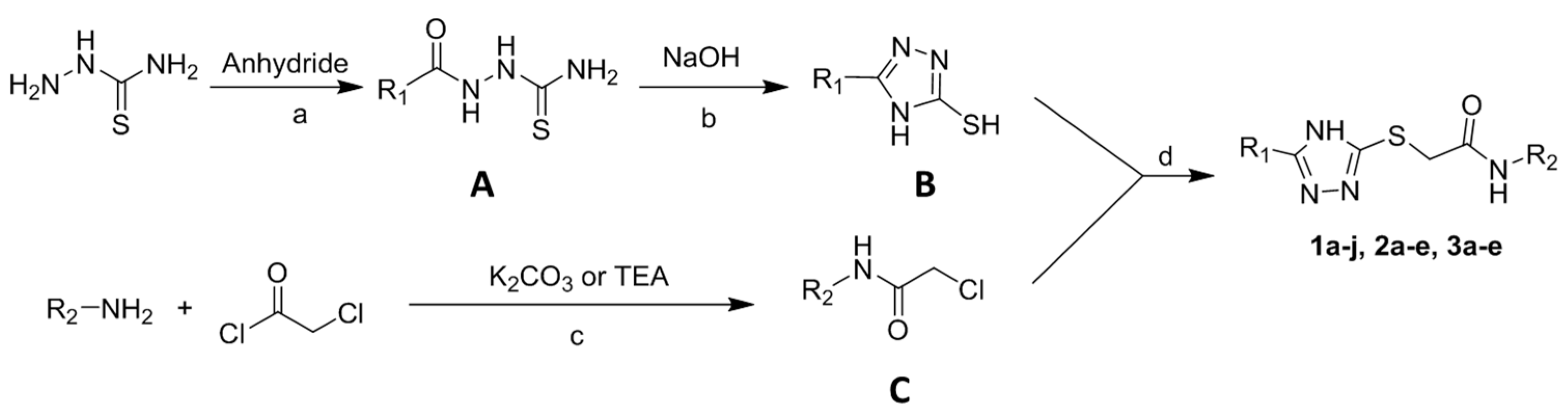
2.1. Synthesis of 2-Triazolylthioacetamides
2.2. Over-Expression and Purification of VIM-2
2.3. Inhibition Studies
2.4. Antibacterial Activity Assays
2.5. Evaluation of Binding Affinity
2.6. Crystallization, X-ray Data Collection, and Structure Determination
3. Results and Discussion
3.1. Synthesis of 2-Triazolylthioacetamides
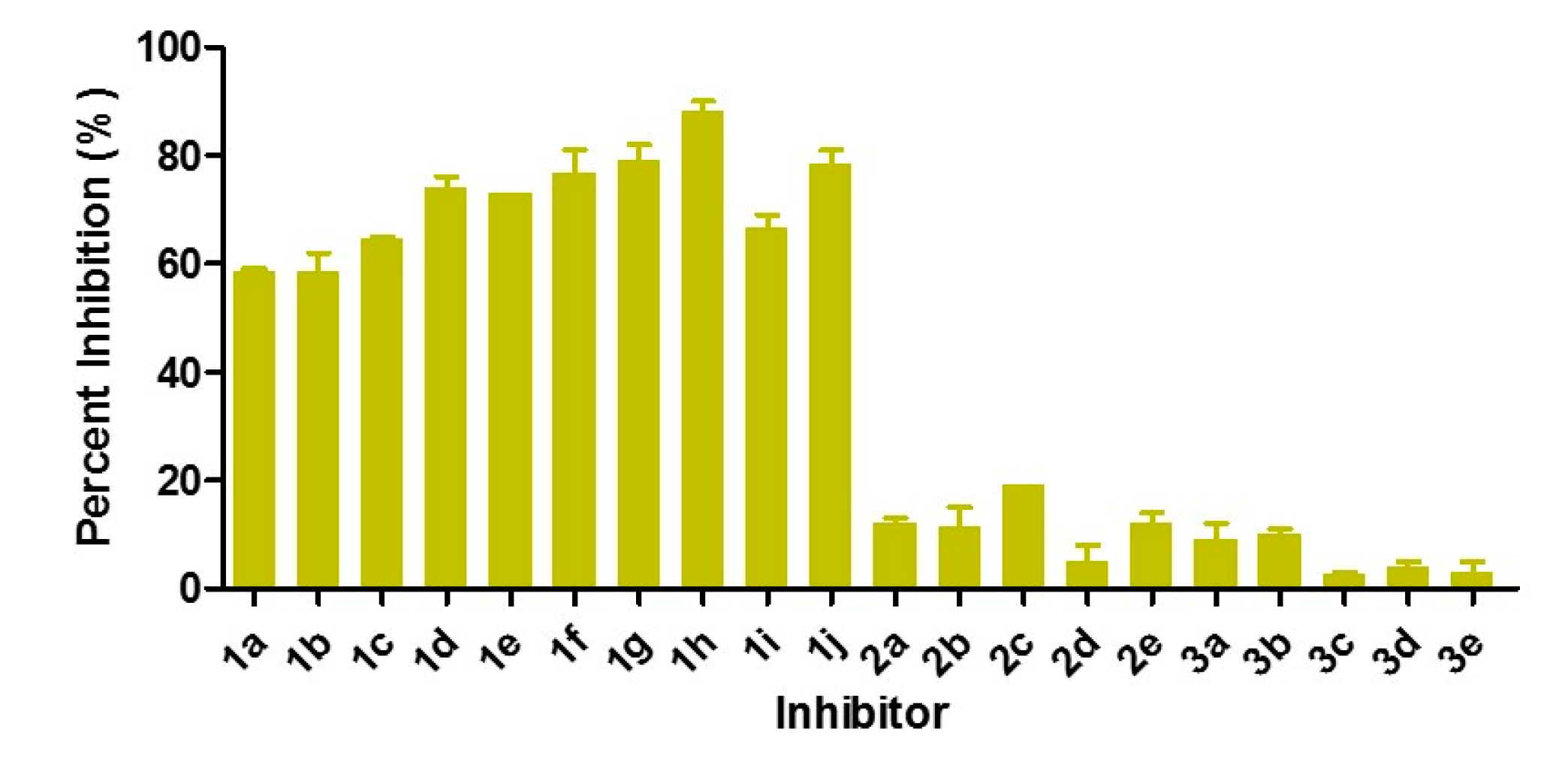
3.2. Inhibition of VIM-2 by 2-Triazolylthioactamides
3.3. Antibacterial Activity Assays
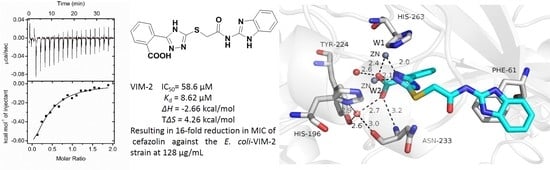
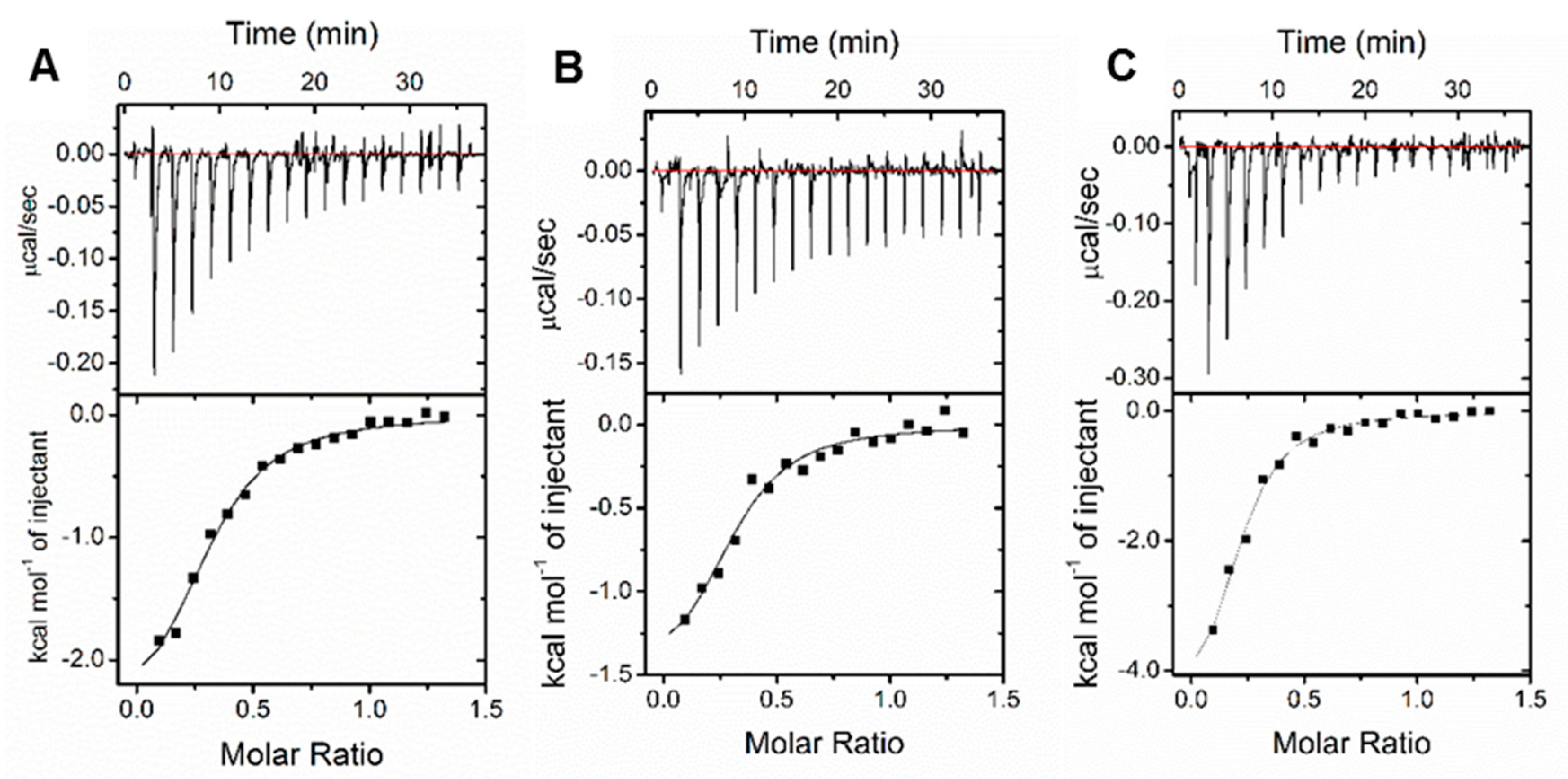
3.4. Binding Affinity Evaluation
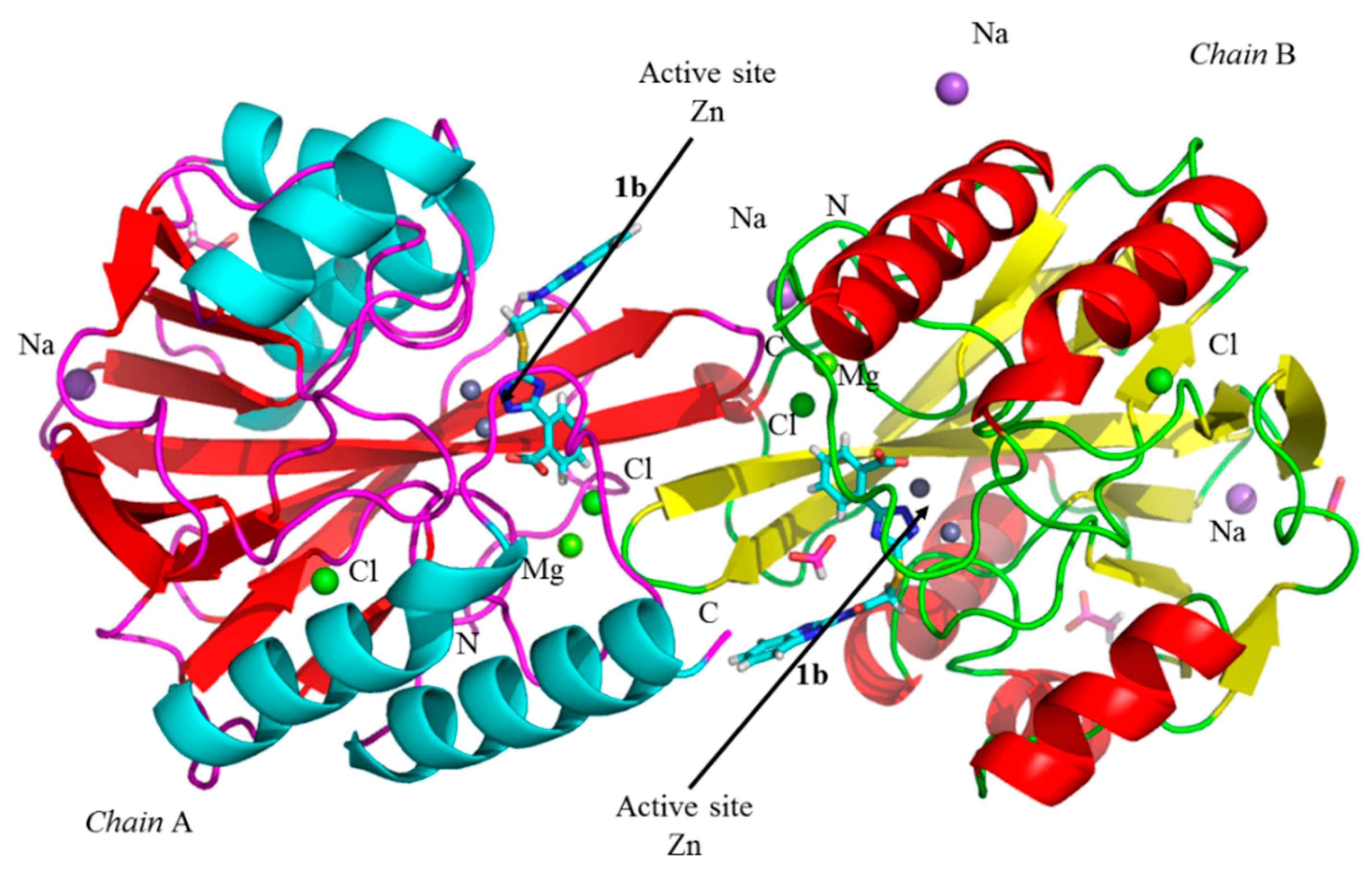
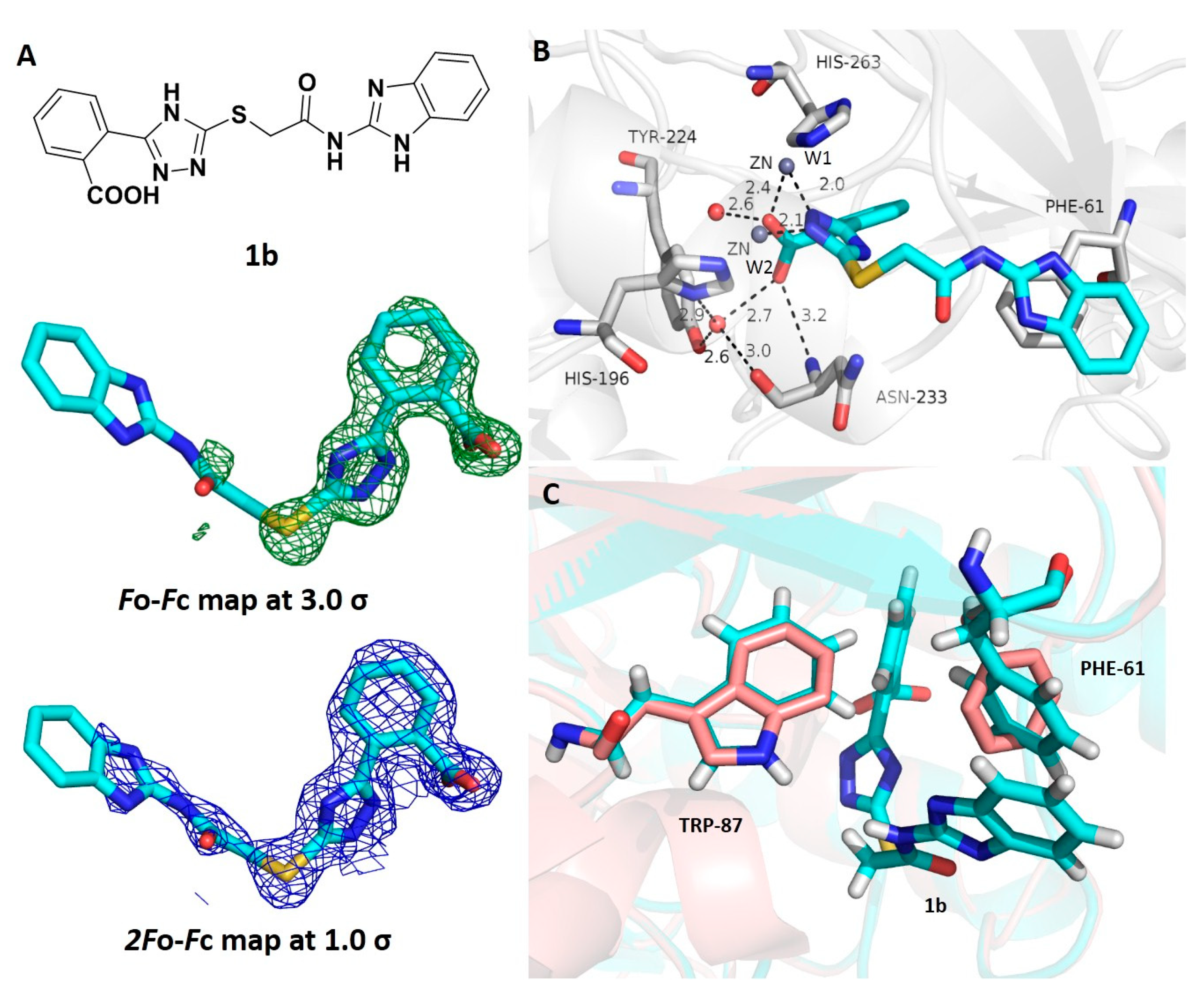
3.5. Crystallographic Analyses
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fisher, J.F.; Meroueh, S.O.; Mobashery, S. Bacterial resistance to β-lactam antibiotics: Compelling opportunism, compelling opportunity. Chem. Rev. 2005, 105, 395–424. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.S.; Apisarnthanarak, A.; Hsu, L.Y. Mechanisms of β-lactam antimicrobial resistance and epidemiology of major community- and healthcare-associated multidrug-resistant bacteria. Adv. Drug. Delivery. Rev. 2014, 78, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Jacoby, G.A. Updated functional classification of β-lactamases. Antimicrob Agents Chemother 2010, 54, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Bebrone, D.C.; Lassaux, P.; Vercheval, L.; Sohier, J.; Jehaes, A.; Sauvage, E.; Galleni, M. Current challenges in antimicrobial chemotherapy: Focus on β-lactamase inhibition. Drugs 2010, 70, 651–679. [Google Scholar] [CrossRef]
- Ehmann, D.E.; Jahić, H.; Ross, P.L.; Gu, R.F.; Hu, J.; Durandréville, T.F.; Lahiri, S.; Thresher, J.; Livchak, S.; Gao, N. Kinetics of avibactam inhibition against class A, C, and D β-lactamases. J. Biol. Chem. 2013, 288, 27960–27971. [Google Scholar] [CrossRef]
- Hinchliffe, P.; González, M.M.; Mojica, M.F.; González, J.M.; Castillo, V.; Saiz, C.; Kosmopoulou, M.; Tooke, C.L.; Llarrull, L.I.; Mahler, G. Cross-class metallo-β-lactamase inhibition by bisthiazolidines reveals multiple binding modes. Proc. Natl. Acad. Sci. USA 2016, 113, E3745. [Google Scholar] [CrossRef]
- Brem, J.; Berkel, S.S.V.; Zollman, D.; Lee, S.Y.; Gileadi, O.; Mchugh, P.J.; Walsh, T.R.; Mcdonough, M.A.; Schofield, C.J. Structural basis of metallo-β-lactamase inhibition by captopril stereoisomers. Antimicrob. Agents Chemother. 2015, 60, 142–150. [Google Scholar] [CrossRef]
- Skagseth, S.; Akhter, S.; Paulsen, M.H.; Muhammad, Z.; Lauksund, S.; Samuelsen, Ø.; Leiros, H.K.S.; Bayer, A. Metallo-β-lactamase inhibitors by bioisosteric replacement: Preparation, activity and binding. Eur. J. Med. Chem. 2017, 135, 159–173. [Google Scholar] [CrossRef]
- Arjomandi, O.K.; Hussein, W.M.; Vella, P.; Yusof, Y.; Sidjabat, H.E.; Schenk, G.; Mcgeary, R.P. Design, synthesis, and invitro and biological evaluation of potent amino acid-derived thiol inhibitors of the metallo-β-lactamase IMP-1. Eur. J. Med. Chem. 2016, 114, 318–327. [Google Scholar] [CrossRef]
- King, A.M.; Reidyu, S.A.; Wang, W.; King, D.T.; Pascale, G.D.; Strynadka, N.C.; Walsh, T.R.; Coombes, B.K.; Wright, G.D. Aspergillomarasmine a overcomes metallo-β-lactamase antibiotic resistance. Nature 2014, 510, 503–506. [Google Scholar] [CrossRef]
- Christopeit, T.; Carlsen, T.J.; Helland, R.; Leiros, H.S. Discovery of novel inhibitor scaffolds against the metallo-β-lactamase VIM-2 by SPR based fragment screening. J. Med. Chem. 2015, 58, 8671–8682. [Google Scholar] [CrossRef] [PubMed]
- Spicer, T.; Minond, D.; Enogieru, I.; Saldanha, S.A.; Mercer, B.A.; Allais, C.; Liu, Q.; Roush, W.R. Probe Reports from the NIH Molecular Libraries Program; NCBI: Bethesda, MD, USA, 2012. [Google Scholar]
- Brem, J.; Berkel, S.S.V.; Aik, W.S.; Rydzik, A.M.; Avison, M.B.; Pettinati, I.; Umland, K.D.; Kawamura, A.; Spencer, J.; Claridge, T.D.; et al. Rhodanine hydrolysis leads to potent thioenolate mediated metallo-β-lactamase inhibition. Nat. Chem. 2014, 6, 1084–1090. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Chen, C.; Wang, W.M.; Xu, L.W.; Yang, K.W.; Oelschlaeger, P.; He, Y. Rhodanine as a potent scaffold for the development of broad-spectrum metallo-β-lactamase inhibitors. ACS Med. Chem. Lett. 2018, 9, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Brem, J.; Cain, R.; Cahill, S.; Mcdonough, M.A.; Clifton, I.J.; Jiménez-Castellanos, J.C.; Avison, M.B.; Spencer, J.; Fishwick, C.W.; Schofield, C.J. Structural basis of metallo-β-lactamase, serine-β-lactamase and penicillin-binding protein inhibition by cyclic boronates. Nat. Commun. 2016, 7, 12406–12413. [Google Scholar] [CrossRef]
- Cahill, S.T.; Cain, R.; Wang, D.Y.; Lohans, C.T.; Wareham, D.W.; Oswin, H.P.; Mohammed, J.; Spencer, J.; Fishwick, C.W.G.; McDonough, M.A.; et al. Cyclic boronates inhibit all classes of β-lactamases. Antimicrob. Agents Chemother 2017, e02260-16. [Google Scholar] [CrossRef]
- Chiou, J.; Wan, S.; Chan, K.F.; So, P.K.; He, D.; Chan, E.W.; Chan, T.H.; Wong, K.Y.; Tao, J.; Chen, S. Ebselen as a potent covalent inhibitor of new delhi metallo-β-lactamase (NDM-1). Chem. Commun. 2015, 51, 9543–9546. [Google Scholar] [CrossRef]
- Chen, C.; Xiang, Y.; Yang, K.W.; Zhang, Y.; Wang, W.M.; Su, J.P.; Ge, Y.; Liu, Y. A protein structure-guided covalent scaffold selectively targets the B1 and B2 subclass metallo-β-lactamases. Chem. Commun. 2018, 54, 4802–4805. [Google Scholar] [CrossRef]
- Bush, K. The ABCD’s of β-lactamase nomenclature. J. Infect Chemother. 2013, 19, 549–559. [Google Scholar] [CrossRef]
- Poirel, L.; Naas, T.; Nicolas, D.; Collet, L.; Bellais, S.; Cavallo, J.D.; Nordmann, P. Characterization of VIM-2, a carbapenem-hydrolyzing metallo-β-lactamase and its plasmid- and integron-borne gene from a pseudomonas aeruginosa clinical isolate in france. Antimicrob Agents Chemother. 2000, 44, 891–897. [Google Scholar] [CrossRef]
- Aitha, M.; Marts, A.R.; Bergstrom, A.; Møller, A.J.; Moritz, L.; Turner, L.; Nix, J.C.; Bonomo, R.A.; Page, R.C.; Tierney, D.L.; et al. Biochemical, mechanistic, and spectroscopic characterization of metallo-β-lactamase VIM-2. Biochemistry 2014, 53, 7321–7331. [Google Scholar] [CrossRef]
- Garciasaez, I.; Docquier, J.D.; Rossolini, G.M.; Dideberg, O. The three-dimensional structure of VIM-2, a Zn-β-lactamase from pseudomonas aeruginosa in its reduced and oxidised form. J. Mol. Biol. 2008, 375, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Zhang, Y.L.; Kang, J.S.; Oelschlaeger, P.; Xiao, L.; Nie, S.S.; Yang, K.W. Triazolylthioacetamide: A valid scaffold for the development of new delhi metallo-β-lactmase-1 (NDM-1) inhibitors. ACS Med. Chem. Lett. 2016, 7, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Yang, K.W.; Zhou, Y.J.; Lacuran, A.E.; Oelschlaeger, P.; Crowder, M.W. Diaryl-substituted azolylthioacetamides: Inhibitor discovery of new delhi metallo-β-lactamase-1 (NDM-1). Chem. Med. Chem. 2014, 9, 2445–2448. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.K.; Kang, J.S.; Oelschlaeger, P.; Yang, K.W. Azolylthioacetamide: A highly promising scaffold for the development of metallo-β-lactamase inhibitors. ACS Med. Chem. Lett. 2015, 6, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Chang, Y.N.; Ge, Y.; Kang, J.S.; Zhang, Y.L.; Liu, X.L.; Oelschlaeger, P.; Yang, K.W. Azolylthioacetamides as a potent scaffold for the development of metallo-β-lactamase inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 5225–5229. [Google Scholar] [CrossRef] [PubMed]
- Christopeit, T.; Yang, K.W.; Yang, S.K.; Leiros, H.K.S. The structure of the metallo-β-lactamase VIM-2 in complex with a triazolylthioacetamide inhibitor. Acta Crystallogr. 2016, 72, 813–819. [Google Scholar] [CrossRef]
- Wikler, M.A. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard; Clinical & Laboratory Standards Institute: Wayne, PA, USA, 2012. [Google Scholar]
- Wang, H.L.; Liu, S.; Yu, Z.J.; Wu, C.; Cheng, L.; Wang, Y.; Chen, K.; Zhou, S.; Chen, Q.; Yu, Y. Interactions between sirtuins and fluorogenic small-molecule substrates offer insights into inhibitor design. RSC Adv. 2017, 7, 36214–36222. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.G.; Bunkoczi, V.B.; Chen, I.W.; Davis, N.; Echols, J.J.; Headd, L.W.; Hung, G.J.; Kapral, R.W.; Grosse-Kunstleve, A.J.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. Design of specific inhibitors of phospholipase a2: Crystal structure of a complex formed between phospholipase a2 from daboia russelli pulchella and a designed pentapeptide Leu-Ala-Ile-Tyr-Ser at 2.0 resolution. Protein Eng. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Garau, G.; Garcı’a-Sa’ez, I.; Bebrone, C.; Anne, C.; Mercuri, P.; Galleni, M.; Fre‘re, J.M.; Dideberg, O. Molecular dynamic simulations of the metallo-β-lactamase from Bacteroides fragilis in the presence and absence of a tight-binding inhibitor. Antimicrob. Agents Chemother. 2004, 48, 2347–2349. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.G.; May-Dracka, T.L.; Gagnon, M.M.; Tommasi, R. Trends and exceptions of physical properties on antibacterial activity for gram-positive and gram-negative pathogens. J. Med. Chem. 2014, 57, 10144–10161. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K. Interpreting steep dose-response curves in early inhibitor discovery. J. Med. Chem. 2006, 49, 7274–7277. [Google Scholar] [CrossRef] [PubMed]
- Callies, O.; Hernandez Daranas, A. Application of isothermal titration calorimetry as a tool to study natural product interactions. Nat. Prod. Rep. 2016, 33, 881–904. [Google Scholar] [CrossRef] [PubMed]
- Li, G.B.; Abboud, M.I.; Brem, J.; Someya, H.; Lohans, C.T.; Yang, S.Y.; Spencer, J.; Wareham, D.W.; McDonough, M.A.; Schofield, C.J. NMR-filtered virtual screening leads to non-metal chelating metallo-beta-lactamase inhibitors. Chem. Sci. 2017, 8, 928–937. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compds | IC50 (μM) | clogP | Compds | IC50 (μM) | clogP |
|---|---|---|---|---|---|
| 1a | 40.8 ± 0.3 | 2.28 | 1f | 45.8 ± 0.2 | 1.88 |
| 1b | 58.6 ± 0.1 | 2.18 | 1g | 31.4± 0.4 | 3.03 |
| 1c | 49.1 ± 0.1 | 2.74 | 1h | 20.6 ± 0.1 | 3.05 |
| 1d | 52.2 ± 0.5 | 3.05 | 1i | 45.2 ± 0.1 | 2.14 |
| 1e | 38.2 ± 0.3 | 2.23 | 1j | 57.6 ± 0.4 | 2.19 |
| (A) | |||||||
| Compds | MIC (μg/mL) | Compds | MIC (μg/mL) | ||||
| Control | 128 | 1f | 32 | ||||
| 1a | 32 | 1g | 16 | ||||
| 1b | 32 | 1h | 16 | ||||
| 1c | 32 | 1i | 32 | ||||
| 1d | 32 | 1j | 16 | ||||
| 1e | 16 | ||||||
| (B) | |||||||
| Compds\conc | 0 | 16 | 32 | 64 | 128 | ||
| 1b | 128 | 32 | 32 | 16 | 8 | ||
| 1c | 128 | 32 | 32 | 16 | 8 | ||
| 1g | 128 | 16 | 16 | 8 | 4 | ||
| 1h | 128 | 16 | 8 | 8 | 4 | ||
| Compounds | Ka (M−1) | Kd (μM) | ΔH (kcal/mol) | TΔS (kcal/mol) | ΔG (kcal/mol) |
|---|---|---|---|---|---|
| 1b | 1.16 × 105 | 8.62 | −2.66 | 4.26 | −6.92 |
| 1c | 1.26 × 105 | 7.94 | −1.61 | 5.34 | −6.95 |
| 1h | 1.56 × 105 | 6.41 | −5.10 | 1.98 | −7.08 |
| Structure (PDB ID) | 6A61 |
|---|---|
| Data Collection | SSRF BL18u |
| Space group | C 1 2 1 |
| a, b, c (Å) | 103.759, 79.421, 67,981 |
| α, β, γ (°) | 90, 130.938, 90 |
| a Mol/ASU | 2 |
| Resolution range (Å) | 39.19–1.78 (1.839–1.78) |
| Number of unique reflections | 39948 |
| Completeness (%) | 99.13 |
| I/σ(I) | 6.3 (3.8) |
| Wilson B Factor (Å2) | 16.10 |
| Overall B factor (Å2) | 24.15 |
| Protein B factor (Å2) | 22.80 |
| Ligand B factor (Å2) | 50.62 |
| Water B factor (Å2) | 31.79 |
| b RMSD from Ideal Bond Length (Å) | 0.006 |
| b RMSD from Ideal Angles (°) | 0.83 |
| Final Rwork (%) | 17.65% |
| Final Rfree (%) | 20.63% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, Y.; Zhang, Y.-J.; Ge, Y.; Zhou, Y.; Chen, C.; Wahlgren, W.Y.; Tan, X.; Chen, X.; Yang, K.-W. Kinetic, Thermodynamic, and Crystallographic Studies of 2-Triazolylthioacetamides as Verona Integron-Encoded Metallo-β-Lactamase 2 (VIM-2) Inhibitor. Biomolecules 2020, 10, 72. https://doi.org/10.3390/biom10010072
Xiang Y, Zhang Y-J, Ge Y, Zhou Y, Chen C, Wahlgren WY, Tan X, Chen X, Yang K-W. Kinetic, Thermodynamic, and Crystallographic Studies of 2-Triazolylthioacetamides as Verona Integron-Encoded Metallo-β-Lactamase 2 (VIM-2) Inhibitor. Biomolecules. 2020; 10(1):72. https://doi.org/10.3390/biom10010072
Chicago/Turabian StyleXiang, Yang, Yue-Juan Zhang, Ying Ge, Yajun Zhou, Cheng Chen, Weixiao Yuan Wahlgren, Xiangshi Tan, Xi Chen, and Ke-Wu Yang. 2020. "Kinetic, Thermodynamic, and Crystallographic Studies of 2-Triazolylthioacetamides as Verona Integron-Encoded Metallo-β-Lactamase 2 (VIM-2) Inhibitor" Biomolecules 10, no. 1: 72. https://doi.org/10.3390/biom10010072
APA StyleXiang, Y., Zhang, Y.-J., Ge, Y., Zhou, Y., Chen, C., Wahlgren, W. Y., Tan, X., Chen, X., & Yang, K.-W. (2020). Kinetic, Thermodynamic, and Crystallographic Studies of 2-Triazolylthioacetamides as Verona Integron-Encoded Metallo-β-Lactamase 2 (VIM-2) Inhibitor. Biomolecules, 10(1), 72. https://doi.org/10.3390/biom10010072