The Role of the Ω-Loop in Regulation of the Catalytic Activity of TEM-Type β-Lactamases

 ,
,
Abstract

1. Introduction
2. Structural Relationship between Penicillin-Binding Proteins and β-Lactamases
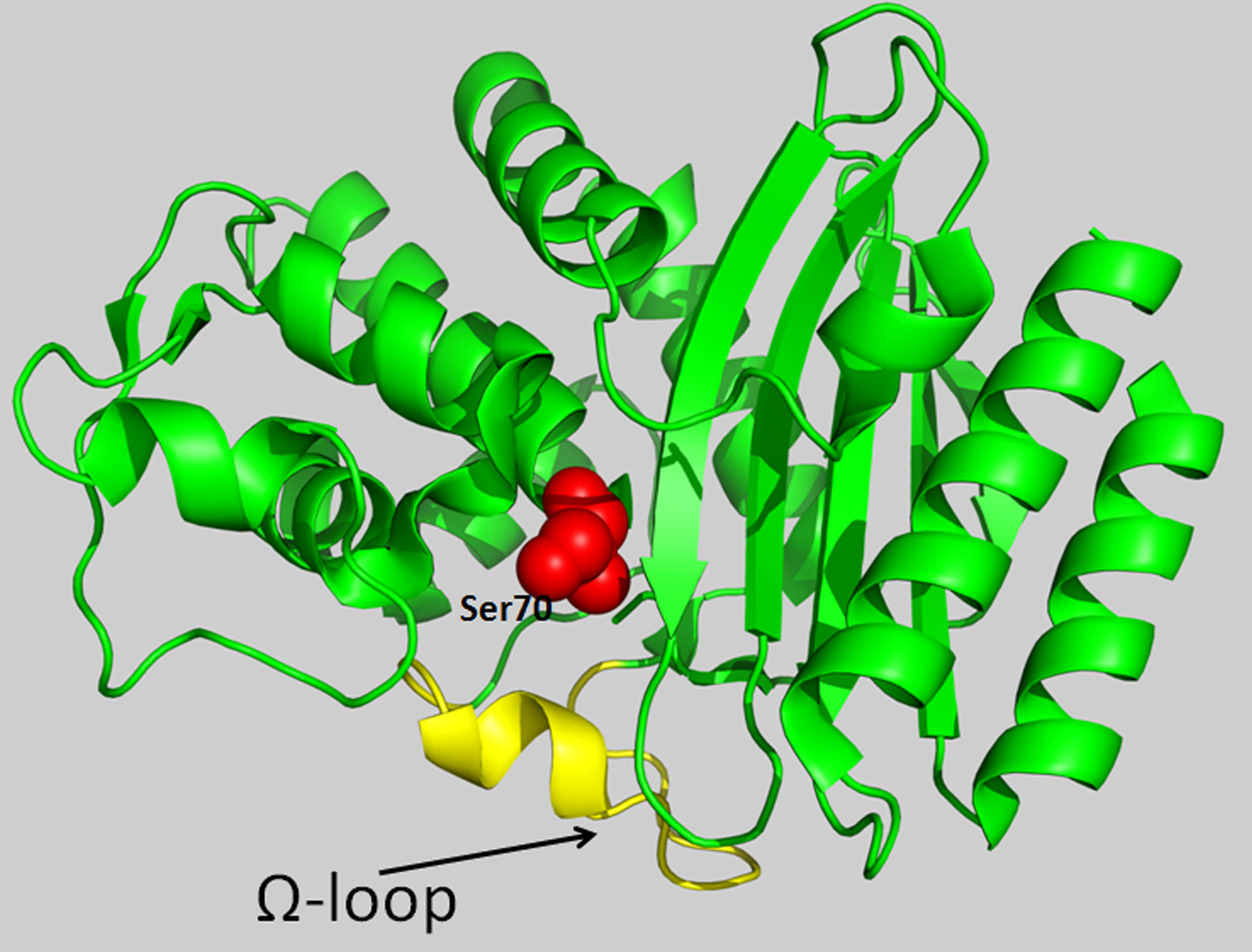
3. Origin and Structure of the Ω-Loop in Class A β-Lactamases
4. The Role of the Ω-Loop in the Catalytic Cycle of TEM Type β-Lactamases
5. The Effect of Mutations in Ω-Loop Residues on Properties of TEM Type β-Lactamases
6. The Molecular Dynamics of the Ω-Loop of Class A β-Lactamases and Its Mobility
7. Current Approaches for Overcoming the Resistance Conferred by β-Lactamases
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Klein, E.Y.; van Boeckel, T.P.; Martinez, E.M.; Pant, S.; Gandra, S.; Levin, S.A.; Goossens, H.; Laxminarayan, R. Global increase and geographic convergence in antibiotic consumption between 2000 and 2015. Proc. Natl. Acad. Sci. USA 2018, 115, E3463–E3470. [Google Scholar] [CrossRef] [PubMed]
- WHO Report on Surveillance of Antibiotic Consumption: 2016–2018 Early Implementation. Available online: https://www.who.int/medicines/areas/rational_use/oms-amr-amc-report-2016-2018/en/ (accessed on 1 October 2019).
- Cassini, A.; Högberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A population-level modelling analysis. Lancet Infect. Dis. 2019, 19, 56–66. [Google Scholar] [CrossRef]
- Holmes, A.H.; Moore, L.S.P.; Sundsfjord, A.; Steinbakk, M.; Regmi, S.; Karkey, A.; Guerin, P.J.; Piddock, L.J.V. Understanding the mecha;nisms and drivers of antimicrobial resistance. Lancet 2016, 387, 176–187. [Google Scholar] [CrossRef]
- Bush, K. Past and present perspectives of β-lactamases. Antimicrob. Agents Chemother. 2018, 62, e01076-18. [Google Scholar] [CrossRef]
- Hall, B.G.; Barlow, M. Revised Ambler classification of β-lactamases. J. Antimicrob. Chemother. 2005, 55, 1050–1051. [Google Scholar] [CrossRef]
- Liu, Y.; Li, R.; Xiao, X.; Wang, Z. Molecules that inhibit bacterial resistance enzymes. Molecules 2019, 24, 43. [Google Scholar] [CrossRef]
- Docquier, J.D.; Mangani, S. An update on β-lactamase inhibitor discovery and development. Drug Resist. Updat. 2018, 36, 13–29. [Google Scholar] [CrossRef]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayash, I.Y.; Spencer, J. β-Lactamases and β-lactamase inhibitors in the 21st century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef]
- Van den Akker, F.; Bonomo, R.A. Exploring additional dimensions of complexity in inhibitor design for serine β-lactamases: Mechanistic and intra- and inter-molecular chemistry approaches. Front. Microbiol. 2018, 9, 622. [Google Scholar] [CrossRef]
- Grigorenko, V.G.; Andreeva, I.P.; Rubtsova, M.Y.; Deygen, I.M.; Antipin, R.L.; Majouga, A.G.; Egorov, A.M.; Beshnova, D.A.; Kallio, J.; Hackenberg, C.; et al. Novel non- β-lactam inhibitor of β-lactamase TEM-171 based on acylated phenoxyaniline. Biochimie 2017, 132, 45–53. [Google Scholar] [CrossRef]
- Kreß, N.; Halder, J.M.; Rapp., L.R.; Hauer, B. Unlocked potential of dynamic elements in protein structures: Channels and loops. Curr. Opin. Chem. Biol. 2018, 47, 109–116. [Google Scholar] [CrossRef]
- Papaleo, E.; Saladino, G.; Lambrughi, M.; Lindorff-Larsen, K.; Gervasio, F.L.; Nussinov, R. The role of protein loops and linkers in conformational dynamics and allostery. Chem. Rev. 2016, 116, 6391–6423. [Google Scholar] [CrossRef] [PubMed]
- Mager, P.P.; Walther, H. A Hydrophilic omega-loop (Tyr181 to Tyr188) in the nonsubstrate binding area of HIV-1 reverse transcriptase. Drug Des. Discov. 1996, 14, 225–239. [Google Scholar] [PubMed]
- Krishna, M.M.; Lin, Y.; Rumbley, J.N.; Englander, S.W. Cooperative omega loops in cytochrome c: Role in folding and function. J. Mol. Biol. 2003, 331, 29–36. [Google Scholar] [CrossRef]
- Pimenta, A.C.; Fernandes, R.; Moreira, I.S. Evolution of drug resistance: Insight on TEM β-lactamases structure and activity and β-lactam antibiotics. Mini Rev. Med. Chem. 2014, 14, 111–122. [Google Scholar] [CrossRef]
- Egorov, A.; Ulyashova, M.; Rubtsova, M. Impact of key and secondary drug resistance mutations on structure and activity of β-lactamases. In Antibiotic Drug Resistance, 1st ed.; Martínez, G.L.C., Igrejas, G., Eds.; John Wiley & Sons Inc.: New York, NY, USA, 2019; pp. 121–140. [Google Scholar] [CrossRef]
- Vollmer, W.; Blanot, D.; de Pedro, M.A. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008, 32, 149–167. [Google Scholar] [CrossRef]
- Zapun, A.; Contreras-Martel, C.; Vernet, T. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol. Rev. 2008, 32, 361–385. [Google Scholar] [CrossRef]
- Ghuysen, J.M. Serine β-lactamases and penicillin-binding proteins. Annu. Rev. Microbiol. 1991, 45, 37–67. [Google Scholar] [CrossRef]
- Lupoli, T.J.; Tsukamoto, H.; Doud, E.H.; Wang, T.S.A.; Walker, S.; Kahne, D. Transpeptidase-mediated incorporation of D-amino acids into bacterial peptidoglycan. J. Am. Chem. Soc. 2011, 133, 10748–10751. [Google Scholar] [CrossRef]
- Ghuysen, J.M. Penicillin-binding proteins. Wall peptidoglycan assembly and resistance to penicillin: Facts, doubts and hopes. Int. J. Antimicrob. Agents 1997, 8, 45–60. [Google Scholar] [CrossRef]
- Tipper, D.J.; Strominger, J.L. Mechanism of action of penicillins: A proposal based on their structural similarity to acyl-D-alanyl-D-alanine. Proc. Natl. Acad. Sci. USA 1965, 54, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Paterson, G.K.; Harrison, E.M.; Holmes, M.A. The emergence of mecC methicillin-resistant Staphylococcus aureus. Trends Microbiol. 2014, 22, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Fuda, C.; Suvorov, M.; Vakulenko, S.B.; Mobashery, S. The basis for resistance to beta-lactam antibiotics by penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus. J. Biol. Chem. 2004, 279, 40802–40806. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Bradford, P.A. β-Lactams and β-lactamase inhibitors: An overview. Cold Spring Harb. Perspect. Med. 2016, 6, a025247. [Google Scholar] [CrossRef] [PubMed]
- Massova, I.; Mobashery, S. Kinship and diversification of bacterial penicillin-binding proteins and β-lactamases. Antimicrob. Agents Chemother. 1998, 42, 1–17. [Google Scholar] [CrossRef]
- Kar, D.; Pandey, S.D.; Mallick, S.; Dutta, M.; Ghosh, A.S. Substitution of alanine at position 184 with glutamic acid in Escherichia coli PBP5 Ω-like loop introduces a moderate cephalosporinase activity. Protein J. 2018, 37, 122–131. [Google Scholar] [CrossRef]
- Palzkill, T. Structural and mechanistic basis for extended-spectrum drug-resistance mutations in altering the specificity of TEM, CTX-M, and KPC β-lactamases. Front. Mol. Biosci. 2018, 5, 1–19. [Google Scholar] [CrossRef]
- Stec, B.; Holtz, K.M.; Wojciechowski, C.L.; Kantrowitz, E.R. Structure of the wild-type TEM-1 β-lactamase at 1.55 Å and the mutant enzyme Ser70Ala at 2.1 Å suggest the mode of noncovalent catalysis for the mutant enzyme. Acta Crystallogr. Sect. D Biol. Crystallogr. 2005, 61, 1072–1079. [Google Scholar] [CrossRef]
- Majiduddin, F.K.; Materon, I.C.; Palzkill, T.G. Molecular analysis of beta-lactamase structure and function. Int. J. Med. Microbiol. 2002, 292, 127–137. [Google Scholar] [CrossRef]
- Ness, S.; Martin, R.; Kindler, A.M.; Paetzel, M.; Gold, M.; Jensen, S.E.; Jones, J.B.; Strynadka, N.C. Structure-based design guides the improved efficacy of deacylation transition state analogue inhibitors of TEM-1 β-lactamase. Biochemistry 2000, 39, 5312–5321. [Google Scholar] [CrossRef]
- Zhang, W.; Shi, Q.; Meroueh, S.O.; Vakulenko, S.B.; Mobashery, S. Catalytic mechanism of penicillin-binding protein 5 of Escherichia coli. Biochemistry 2007, 46, 10113–10121. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.D.; Kumarasiri, M.; Zhang, W.; Hesek, D.; Lee, M.; Toth, M.; Vakulenko, S.; Fisher, J.F.; Mobashery, S.; Chena, Y. Structural analysis of the role of Pseudomonas aeruginosa penicillin-binding protein 5 in β-lactam resistance. Antimicrob. Agents Chemother. 2013, 57, 3137–3146. [Google Scholar] [CrossRef] [PubMed]
- Lobkovsky, E.; Moews, P.C.; Liu, H.; Zhao, H.; Frère, J.M.; Knox, J.R. Evolution of an enzyme activity: Crystallographic structure at 2-Å resolution of cephalosporinase from the ampC gene of Enterobacter cloacae P99 and comparison with a class A penicillinase. Proc. Natl. Acad. Sci. USA 1993, 90, 11257–11261. [Google Scholar] [CrossRef] [PubMed]
- Vanhove, M.; Raquet, X.; Palzkill, T.; Pain, R.H.; Frère, J.M. The rate-limiting step in the folding of the cis-Pro167Thr mutant of TEM-1 beta-lactamase is the trans to cis isomerization of a non-proline peptide bond. Proteins 1996, 25, 104–111. [Google Scholar] [CrossRef]
- Vanhove, M.; Lejeune, A.; Pain, R.H. Beta-lactamases as models for protein-folding studies. Cell. Mol. Life Sci. 1998, 54, 372–377. [Google Scholar] [CrossRef]
- Bös, F.; Pleiss, J. Multiple molecular dynamics simulations of TEM beta-lactamase: Dynamics and water binding of the omega-loop. Biophys. J. 2009, 97, 2550–2558. [Google Scholar] [CrossRef]
- Wang, X.; Minasov, G.; Shoichet, B.K. Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J. Mol. Biol. 2002, 320, 85–95. [Google Scholar] [CrossRef]
- Kuzin, A.P.; Nukaga, M.; Nukaga, Y.; Hujer, A.; Bonomo, R.A.; Knox, J.R. Inhibition of the SHV-1 beta-lactamase by sulfones: Crystallographic observation of two reaction intermediates with tazobactam. Biochemistry 2001, 40, 1861–1866. [Google Scholar] [CrossRef]
- Ghiglione, B.; Rodriguez, M.M.; Herman, R.; Curto, L.M.; Dropa, M.; Bouillenne, F.; Kerff, F.; Galleni, M.; Charlier, P.; Gutkind, G.; et al. Structural and kinetic insights into the “cceftazidimase” behavior of the extended-spectrum beta-lactamase CTX-M-96. Biochemistry 2015, 54, 5072–5082. [Google Scholar] [CrossRef]
- Morinaka, A.; Tsutsumi, Y.; Yamada, M.; Suzuki, K.; Watanabe, T.; Abe, T.; Furuuchi, T.; Inamura, S.; Sakamaki, Y.; Mitsuhashi, N.; et al. OP0595, a new diazabicyclooctane: Mode of action as a serine beta-lactamase inhibitor, antibiotic and beta-lactam ‘enhancer’. J. Antimicrob. Chemother. 2015, 70, 2779–2786. [Google Scholar] [CrossRef]
- Nitanai, Y.; Shimamura, T.; Uchiyama, T.; Ishii, Y.; Takehira, M.; Yutani, K.; Matsuzawa, H.; Miyano, M. The catalytic efficiency (kcat/Km) of the class A beta-lactamase Toho-1 correlates with the thermal stability of its catalytic intermediate analog. Biochim. Biophys. Acta 2010, 1804, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Sanschagrin, F.; Passmore, L.; De Castro, L.; Levesque, R.C.; Strynadka, N.C. Insights into the molecular basis for the carbenicillinase activity of PSE-4 beta-lactamase from crystallographic and kinetic studies. Biochemistry 2001, 40, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Petrella, S.; Ziental-Gelus, N.; Mayer, C.; Renard, M.; Jarlier, V.; Sougakoff, W. Genetic and structural insights into the dissemination potential of the extremely broad-spectrum class A beta-lactamase KPC-2 identified in an Escherichia coli strain and an Enterobacter cloacae strain isolated from the same patient in France. Antimicrob. Agents Chemother. 2008, 52, 3725–3736. [Google Scholar] [CrossRef]
- Calvopina, K.; Hinchliffe, P.; Brem, J.; Heesom, K.J.; Johnson, S.; Cain, R.; Lohans, C.T.; Fishwick, C.W.G.; Schofield, C.J.; Spencer, J.; et al. Structural/mechanistic insights into the efficacy of nonclassical beta-lactamase inhibitors against extensively drug resistant Stenotrophomonas maltophilia clinical isolates. Mol. Microbiol. 2017, 106, 492–504. [Google Scholar] [CrossRef]
- Fonseca, F.; Chudyk, E.I.; van der Kamp, M.W.; Correia, A.; Mulholland, A.J.; Spencer, J. The basis for carbapenem hydrolysis by class A beta-lactamases: A combined investigation using crystallography and simulations. J. Am. Chem. Soc. 2012, 134, 18275–18285. [Google Scholar] [CrossRef]
- Pan, X.; He, Y.; Lei, J.; Huang, X.; Zhao, Y. Crystallographic snapshots of class A β-lactamase catalysis reveal structural changes that facilitate β-lactam hydrolysis. J. Biol. Chem. 2017, 292, 4022–4033. [Google Scholar] [CrossRef]
- Medeiros, A.A. Beta-lactamases: Quality and resistance. Clin. Microbiol. Infect. 1997, 3 (Suppl. 4), S2–S9. [Google Scholar] [CrossRef]
- Roccatano, D.; Sbardella, G.; Aschi, M.; Amicosante, G.; Bossa, C.; Di Nola, A.; Mazza, F. Dynamical aspects of TEM-1 beta-lactamase probed by molecular dynamics. J. Comput. Aided Mol. Des. 2005, 19, 329–340. [Google Scholar] [CrossRef]
- Escobar, W.A.; Tan, A.K.; Lewis, E.R.; Fink, A.L. Site-directed mutagenesis of glutamate- 166 in β-lactamase leads to a branched path mechanism. Biochemistry 1994, 33, 7619–7626. [Google Scholar] [CrossRef]
- Dellus-Gur, E.; Elias, M.; Caselli, E.; Prati, F.; Salverda, M.L.M.; De Visser, J.A.G.M.; Fraser, J.S.; Tawfik, D.S. Negative epistasis and evolvability in TEM-1 β-lactamase—The thin line between an enzyme’s conformational freedom and disorder. J. Mol. Biol. 2015, 427, 2396–2409. [Google Scholar] [CrossRef]
- Raquet, X.; Lamotte-Brasseur, J.; Fonze, E.; Goussard, S.; Courvalin, P.; Frère, J.M. TEM beta-lactamase mutants hydrolysing third-generation cephalosporins—A kinetic and molecular modeling analysis. J. Mol. Microbiol. 1994, 244, 625–639. [Google Scholar] [CrossRef]
- Vakulenko, S.B.; Taibi-Tronche, P.; Tóth, M.; Massova, I.; Lerner, S.A.; Mobashery, S. Effects on substrate profile by mutational substitutions at positions 164 and 179 of the class A TEM(pUC19) beta-lactamase from Escherichia coli. J. Biol. Chem. 1999, 274, 23052–23060. [Google Scholar] [CrossRef] [PubMed]
- Hart, K.M.; Ho, C.M.; Dutta, S.; Gross, M.L.; Bowman, G.R. Modelling proteins’ hidden conformations to predict antibiotic resistance. Nat. Commun. 2016, 7, 12965. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, V.B.; Allen, J.; Camps, M.; Karchin, R. Network models of TEM β-lactamase mutations coevolving under antibiotic selection show modular structure and anticipate evolutionary trajectories. PLoS Comput. Biol. 2011, 7, e1002184. [Google Scholar] [CrossRef]
- Chaibi, E.B.; Sirot, D.; Paul, G.; Labia, R. Inhibitor-resistant TEM β-lactamases: Phenotypic, genetic and biochemical characteristics. J. Antimicrob. Chemother. 1999, 43, 447–458. [Google Scholar] [CrossRef]
- Stojanoski, V.; Chow, D.C.; Hu, L.; Sankaran, B.; Gilbert, H.F.; Prasad, B.V.; Palzkill, T. A triple mutant in the X-loop of TEM-1 β-lactamase changes the substrate profile via a large conformational change and an altered general base for catalysis. J. Biol. Chem. 2015, 290, 10382–10394. [Google Scholar] [CrossRef]
- Palzkill, T.; Le, Q.Q.; Venkatachalam, K.V.; LaRocco, M.; Ocera, H. Evolution of antibiotic resistance: Several different amino acid substitutions in an active site loop alter the substrate profile of β-lactamase. Mol. Microbiol. 1994, 12, 217–229. [Google Scholar] [CrossRef]
- Petrosino, J.F.; Palzkill, T. Systematic mutagenesis of the active site omega-loop of TEM-1 beta-lactamase. J. Bacteriol. 1996, 178, 1821–1828. [Google Scholar] [CrossRef]
- Fisette, O.; Gagné, S.; Lagüe, P. Molecular dynamics of class A β-lactamases—effects of substrate binding. Biophys. J. 2012, 103, 1790–1801. [Google Scholar] [CrossRef]
- Shcherbinin, D.S.; Rubtsova, M.Y.; Grigorenko, V.G.; Uporov, I.V.; Veselovsky, A.V.; Egorov, A.M. Investigation of the role of mutations M182T and Q39K in structure of β-lactamase TEM-72 by molecular dynamics method. Biochem. (Moscow) Suppl. Ser. B Biomed. Chem. 2017, 11, 120–127. [Google Scholar] [CrossRef]
- Shcherbinin, D.; Veselovsky, A.; Rubtsova, M.; Grigorenko, V.; Egorov, A. The impact of long-distance mutations on the Ω-loop conformation in TEM type β-lactamases. J. Biomol. Struct. Dyn. 2019, 1–8. [Google Scholar] [CrossRef]
- Morin, S.; Gagné, S.M. NMR Dynamics of PSE-4 β-lactamase: An interplay of ps-ns order and ms-ms motions in the active site. Biophys. J. 2009, 96, 4681–4691. [Google Scholar] [CrossRef]
- Meroueh, S.O.; Roblin, P.; Golemi, D.; Maveyraud, L.; Vakulenko, S.B.; Zhang, Y.; Samama, J.P.; Mobashery, S. Molecular dynamics at the root of expansion of function in the M69L inhibitor-resistant TEM beta-lactamase from Escherichia coli. J. Am. Chem. Soc. 2002, 14, 9422–9423. [Google Scholar] [CrossRef]
- Meneksedag, D.; Dogan, A.; Kanlikilicer, P.; Ozkirimli, E. Communication between the active site and the allosteric site in class A beta-lactamases. Comput. Biol. Chem. 2013, 43, 1–10. [Google Scholar] [CrossRef]
- Drawz, S.M.; Bonomo, R.A. Three decades of β-lactamase inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [Google Scholar] [CrossRef]
- King, D.T.; Sobhanifar, S.; Strynadka, N.C.J. One ring to rule them all: Current trends in combating bacterial resistance to the β-lactams. Protein Sci. 2016, 25, 787–803. [Google Scholar] [CrossRef]
- Wright, H.; Bonomo, R.A.; Paterson, D.L. New agents for the treatment of infections with Gram-negative bacteria: Restoring the miracle or false dawn? Clin. Microbiol. Infect. 2017, 23, 704–712. [Google Scholar] [CrossRef]
- Tuon, F.F.; Rocha, J.L.; Formigoni-Pinto, M.R. Pharmacological aspects and spectrum of action of ceftazidime-avibactam: A systematic review. Infection 2018, 46, 165–181. [Google Scholar] [CrossRef]
- Blizzard, T.A.; Chen, H.; Kim, S.; Wu, J.; Bodner, R.; Gude, C.; Imbriglio, J.; Young, K.; Park, Y.W.; Ogawa, A.; et al. Discovery of MK-7655, a beta-lactamase inhibitor for combination with Primaxin(R). Bioorganic Med. Chem. Lett. 2014, 24, 780–785. [Google Scholar] [CrossRef]
- Papp-Wallace, K.M.; Nguyen, N.Q.; Jacobs, M.R.; Bethel, C.R.; Barnes, M.D.; Kumar, V.; Bajaksouzian, S.; Rudin, S.D.; Rather, P.N.; Bhavsar, S.; et al. Strategic approaches to overcome resistance against Gram-negative pathogens using beta-lactamase inhibitors and beta-lactam enhancers: Activity of three novel diazabicyclooctanes WCK 5153, zidebactam (WCK 5107), and WCK 4234. J. Med. Chem. 2018, 61, 4067–4086. [Google Scholar] [CrossRef]
- Moya, B.; Barcelo, I.M.; Bhagwat, S.; Patel, M.; Bou, G.; Papp-Wallace, K.M.; Bonomo, R.A.; Oliver, A. WCK 5107 (zidebactam) and WCK 5153 are novel inhibitors of PBP2 showing potent “beta-lactam enhancer” activity against Pseudomonas aeruginosa, including multidrug-resistant metallo beta-lactamase-producing high-risk clones. Antimicrob. Agents Chemother. 2017, 61, e02529-16. [Google Scholar] [CrossRef] [PubMed]
- Giddins, M.J.; Macesic, N.; Annavajhala, M.K.; Stump, S.; Khan, S.; McConville, T.H.; Mehta, M.; Gomez-Simmonds, A.; Uhlemann, A.C. Successive emergence of ceftazidime-avibactam resistance through distinct genomic adaptations in blaKPC-2-harboring Klebsiella pneumoniae sequence type 307 isolates. Antimicrob. Agents Chemother. 2018, 62, e02101-17. [Google Scholar] [CrossRef] [PubMed]
- Both, A.; Buttner, H.; Huang, J.; Perbandt, M.; Belmar Campos, C.; Christner, M.; Maurer, F.P.; Kluge, S.; König, C.; Aepfelbacher, M.; et al. Emergence of ceftazidime/avibactam non-susceptibility in an MDR Klebsiella pneumonia isolate. J. Antimicrob. Chemother. 2017, 72, 2483–2488. [Google Scholar] [CrossRef] [PubMed]
- Hecker, S.J.; Reddy, K.R.; Totrov, M.; Hirst, G.C.; Lomovskaya, O.; Griffith, D.C.; King, P.; Tsivkovski, R.; Sun, D.; Sabet, M.; et al. Discovery of a cyclic boronic acid beta-lactamase inhibitor (RPX7009) with utility vs class A serine carbapenemases. J. Med. Chem. 2015, 58, 3682–3692. [Google Scholar] [CrossRef] [PubMed]
- Rojas, L.J.; Taracila, M.A.; Papp-Wallace, K.M.; Bethel, C.R.; Caselli, E.; Romagnoli, C.; Winkler, M.L.; Spellberg, B.; Prati, F.; Bonomo, R.A. Boronic acid transition state inhibitors active against KPC and other class A beta-lactamases: Structure-activity relationships as a guide to inhibitor design. Antimicrob. Agents Chemother. 2016, 60, 1751–1759. [Google Scholar] [CrossRef]
- Wu, G.; Cheon, E. Meropenem-vaborbactam for the treatment of complicated urinary tract infections including acute pyelonephritis. Expert Opin. Pharmacother. 2018, 19, 1495–1502. [Google Scholar] [CrossRef]
- Krajnc, A.; Lang, P.A.; Panduwawala, T.D.; Brem, J.; Schofield, S.J. Will morphing boron-based inhibitors beat the β-lactamases? Curr. Opin. Chem. Biol. 2019, 50, 101–110. [Google Scholar] [CrossRef]
- Antipin, R.L.; Beshnova, D.A.; Petrov, R.A.; Shiryaeva, A.S.; Andreeva, I.P.; Grigorenko, V.G.; Rubtsova, M.Y.; Majouga, A.G.; Lamzin, V.S.; Egorov, A.M. Synthesis, SAR and molecular docking study of novel non-β-lactam inhibitors of TEM type β-lactamase. Bioorg. Med. Chem. Lett. 2017, 27, 1588–1592. [Google Scholar] [CrossRef]
- Hujer, A.M.; Bethel, C.R.; Bonomo, R.A. Antibody mapping of the linear epitopes of CMY-2 and SHV-1 β-lactamases. Antimicrob. Agents Chemother. 2004, 48, 3980–3988. [Google Scholar] [CrossRef][Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| β-Lactamase | PDB ID | Amino Acid Sequence of the Ω-Loop (Residues from 164 to 179, Glu166 is Highlighted in Bold) |
|---|---|---|
| TEM-1 | 1ERO [32] | RWEPELNEAIPNDERD |
| TEM-64 | 1JWZ [39] | SWEPELNEAIPNDERD |
| SHV-1 | 1VM1 [40] | RWETELNEALPGDARD |
| CTX-M-12 | 3ZNY [41] | RTEPTLNTAIPGDPRD |
| CTX-M-44 | 4X69 [42] | RTEPTLNTAIPGDPRD |
| Toho-1 | 2ZQA [43] | RTAPTLNTAIPGDPRD |
| Pse-4 | 1G6A [44] | RIEPDLNEGKLGDLRD |
| Kpc-2 | 3DW0 [45] | RWELELNSAIPGDARD |
| L2 | 5NE2 [46] | RLEPELNSFAKGDPRD |
| Sfc-1 | 4EQI [47] | RWELELNSAIPGDDRD |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Egorov, A.; Rubtsova, M.; Grigorenko, V.; Uporov, I.; Veselovsky, A. The Role of the Ω-Loop in Regulation of the Catalytic Activity of TEM-Type β-Lactamases. Biomolecules 2019, 9, 854. https://doi.org/10.3390/biom9120854
Egorov A, Rubtsova M, Grigorenko V, Uporov I, Veselovsky A. The Role of the Ω-Loop in Regulation of the Catalytic Activity of TEM-Type β-Lactamases. Biomolecules. 2019; 9(12):854. https://doi.org/10.3390/biom9120854
Chicago/Turabian StyleEgorov, Alexey, Maya Rubtsova, Vitaly Grigorenko, Igor Uporov, and Alexander Veselovsky. 2019. "The Role of the Ω-Loop in Regulation of the Catalytic Activity of TEM-Type β-Lactamases" Biomolecules 9, no. 12: 854. https://doi.org/10.3390/biom9120854
APA StyleEgorov, A., Rubtsova, M., Grigorenko, V., Uporov, I., & Veselovsky, A. (2019). The Role of the Ω-Loop in Regulation of the Catalytic Activity of TEM-Type β-Lactamases. Biomolecules, 9(12), 854. https://doi.org/10.3390/biom9120854