Efficient Extraction from Mice Feces for NMR Metabolomics Measurements with Special Emphasis on SCFAs

 , and
, and
Abstract
:1. Introduction
2. Experimental Design
2.1. Materials
2.2. Equipment
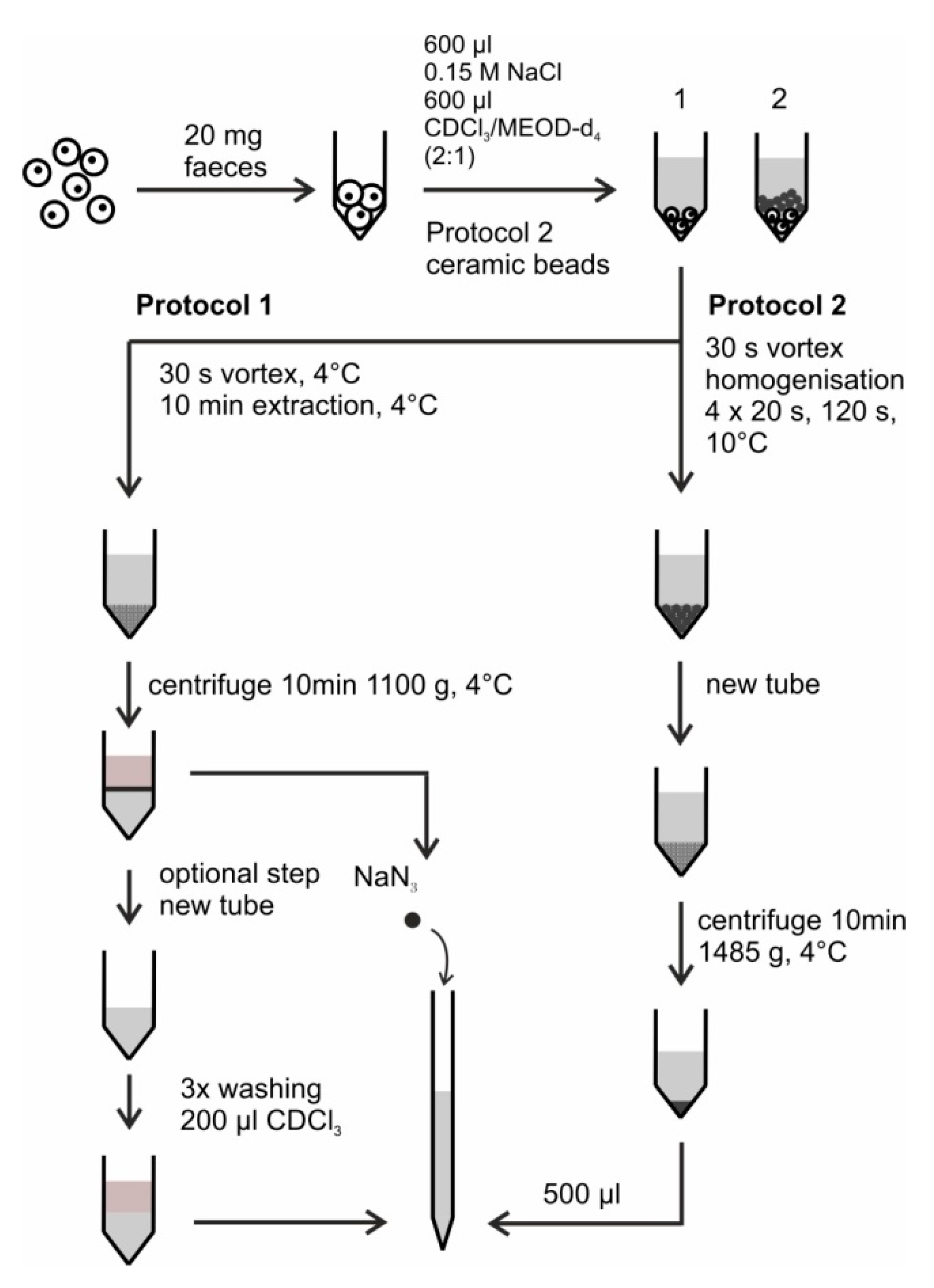
3. Procedure
Time for Completion: Typically the Time for Completion Including Buffer Preparation Is 3:00 Hours.
- (1)
- Weigh 20 mg of the homogenized mouse feces into micro centrifuge tube. Unless otherwise specified and as far as possible, during all the waiting periods keep the samples on dry ice.
- (2)
- Add 600 µL of 0.15 M sodium chloride to the tube. Add additional 600 µL of CDCl3/MeOD-d4 (2:1, v/v). Then vortex the mixture for 30 s.
- (3)
- Place the micro centrifuge tube at 4 °C for the extraction period for 10 min.
- (4)
- Centrifuge the suspension for 10 min at 1100× g and 4 °C.
- (5)
- Transfer the aqueous phase to a new micro centrifuge tube for the washing procedure.
- (6)
- Add 200 μL of CDCl3 to the aqueous phase.
- (7)
- Centrifuge the suspension for 2 min at 1100× g at 4 °C.
- (8)
- Take the chloroform layer away and add fresh amount of 200 µL of CDCl3 to the aqueous phase. Repeat the centrifugation step 7.
- (9)
- Take the chloroform layer again away and add fresh amount of 200 µL of CDCl3 to the aqueous phase.
- (10)
- Perform the third centrifugation for 10 min at 1100× g at 4 °C.
- (11)
- Take the chloroform layer again away. Optional Steps: The described steps from number 5 till 10 are optional. We have realized that these steps help removing the lipid content in our samples originating from mice fed on HFD. However, these steps could only be recommended when no additional small molecular sized metabolites could be also washed out in the process. This should be checked on the samples of interest. Alternatively, the number of washing steps could be reduced to either merely one or two instead of the tested three steps.
- (12)
- Transfer 500 µL of the aqueous phase to an NMR tube, add NaN3 with 1.5 mmol/L concentration and measure the sample by NMR.
- (1)
- Weigh 20 mg of the homogenized mouse feces into the lysing kit tube CK 14 without beads.
- (2)
- Add about 8 ceramic beads (one full spatula) to the solid feces material.
- (3)
- Add 600 µL of 0.15 M sodium chloride to the tube. Additionally add 600 µL of CDCl3/MeOD-d4 (2:1, v/v).
- (4)
- Vortex the suspension for 30 s.
- (5)
- Seal the tube and homogenize it with a Precellys homogenizer. Perform four homogenization cycles at 6000 rpm at 10 °C. Each cycle lasts 20 s and is followed by a waiting time of 120 s at 10 °C.
- (6)
- Transfer the suspension to a new micro centrifuge tube and centrifuge for 10 min at 1485× g and 0 °C.
- (7)
- Transfer 500 µL of the aqueous phase to a NMR tube, mix it with NaN3 with 1.5 mmol/L concentration and measure it by NMR.
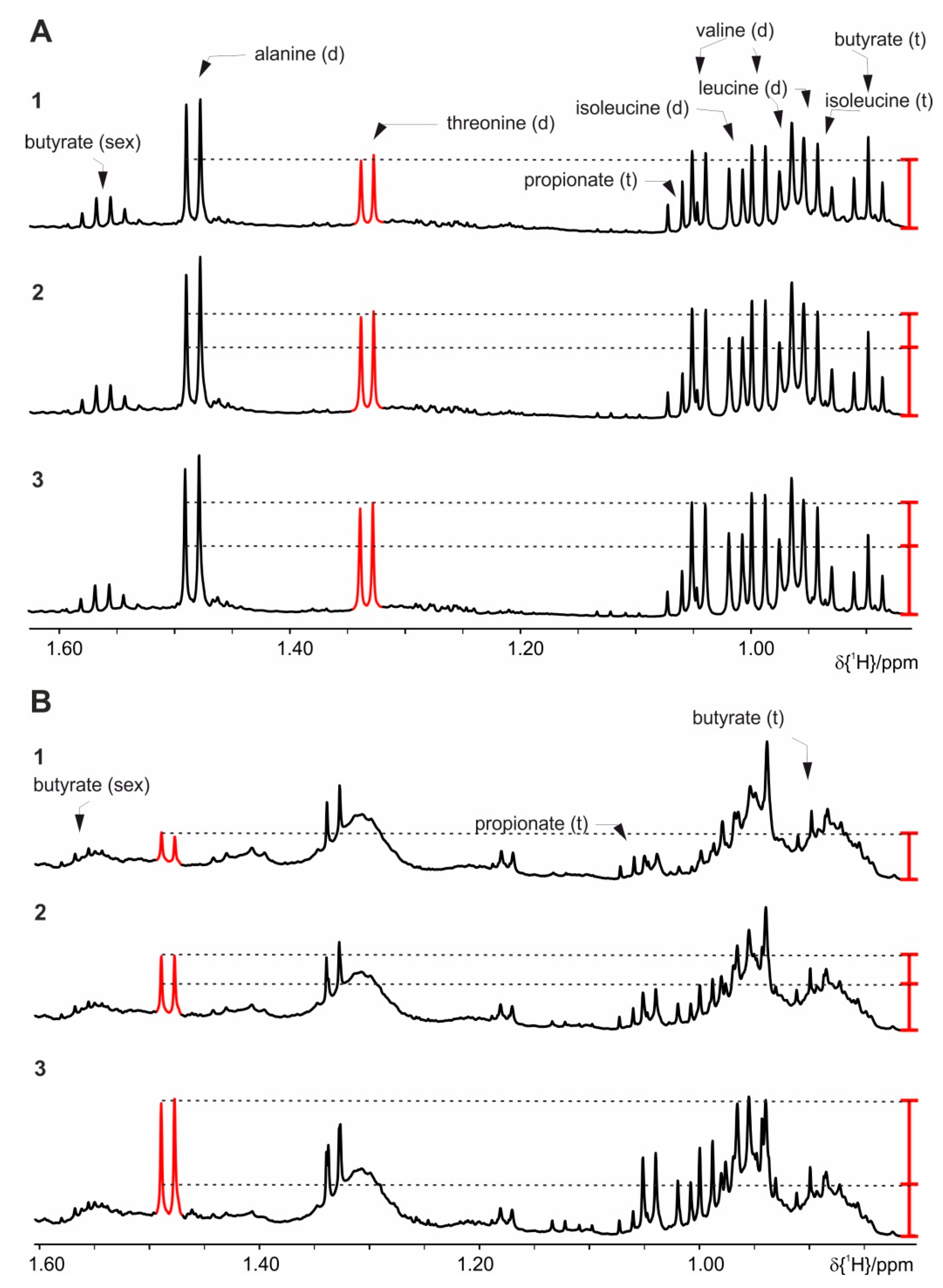
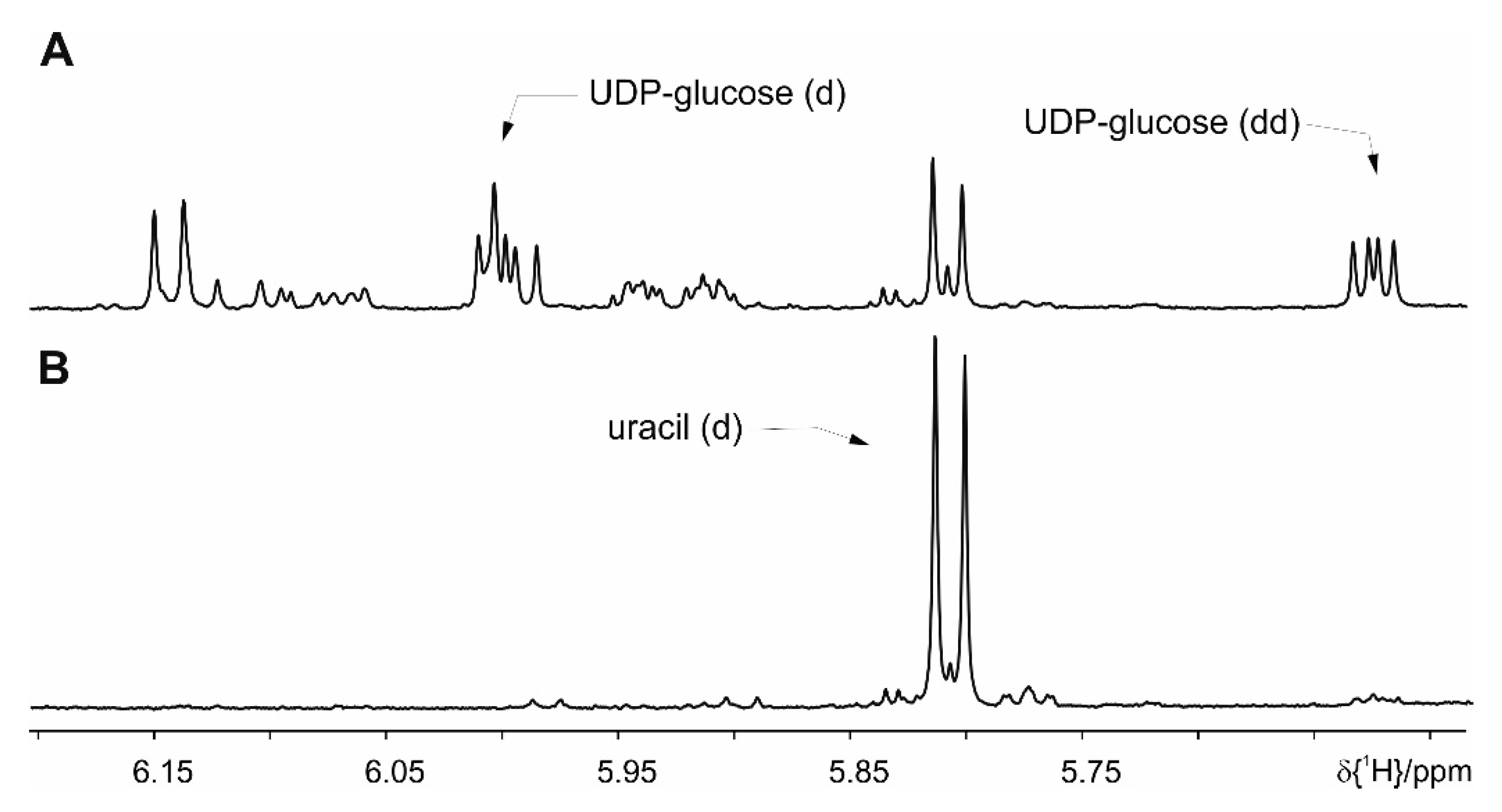
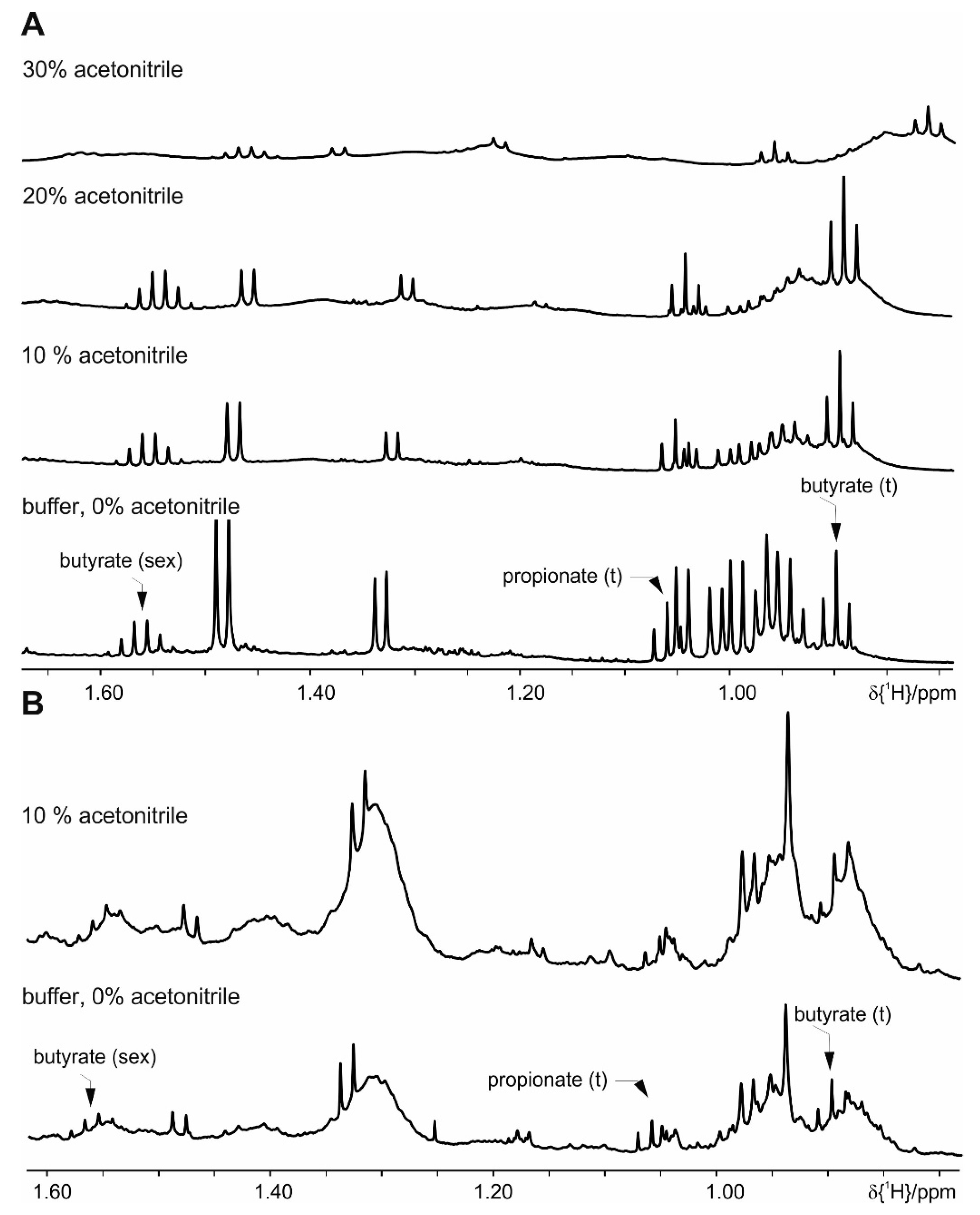
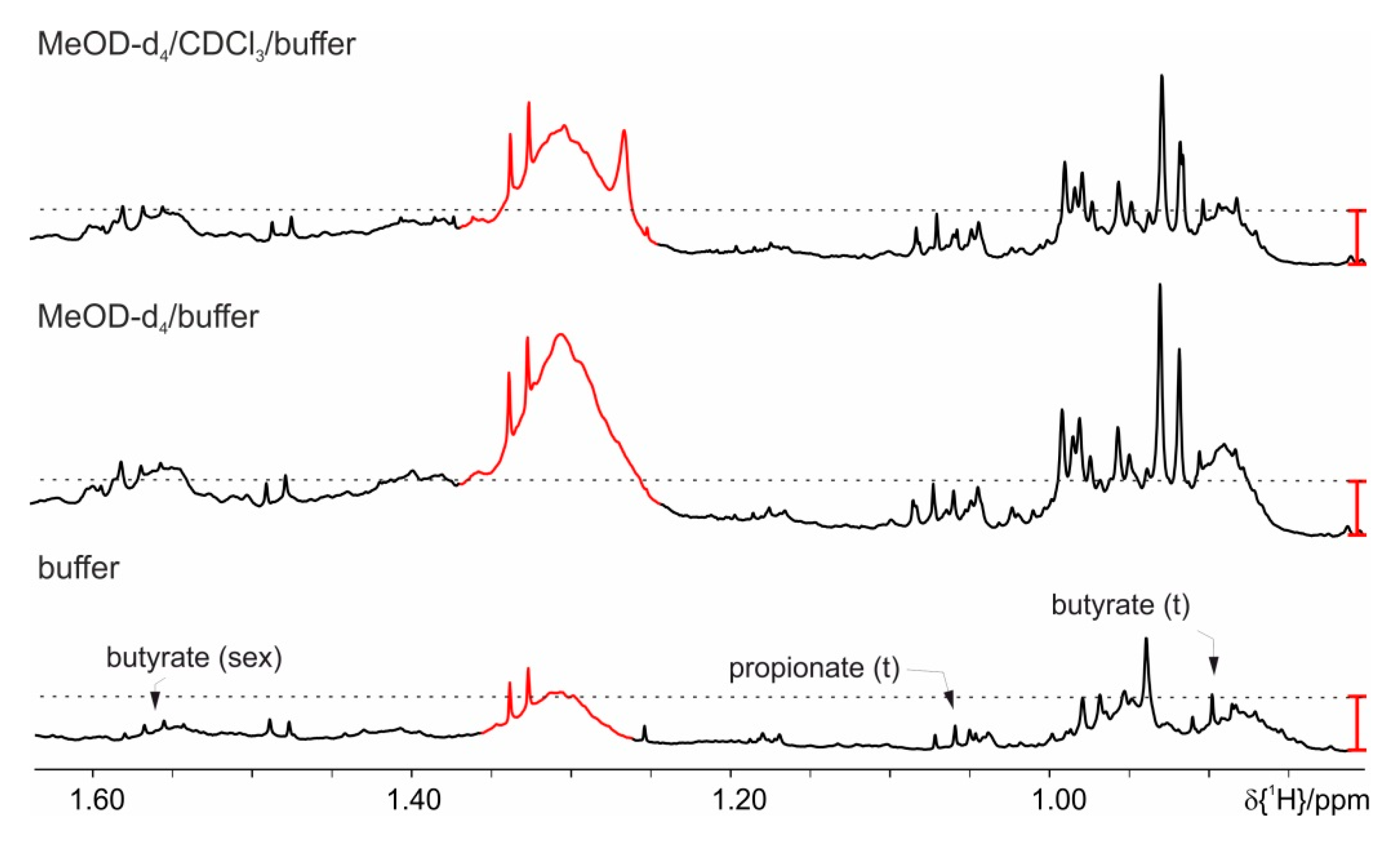
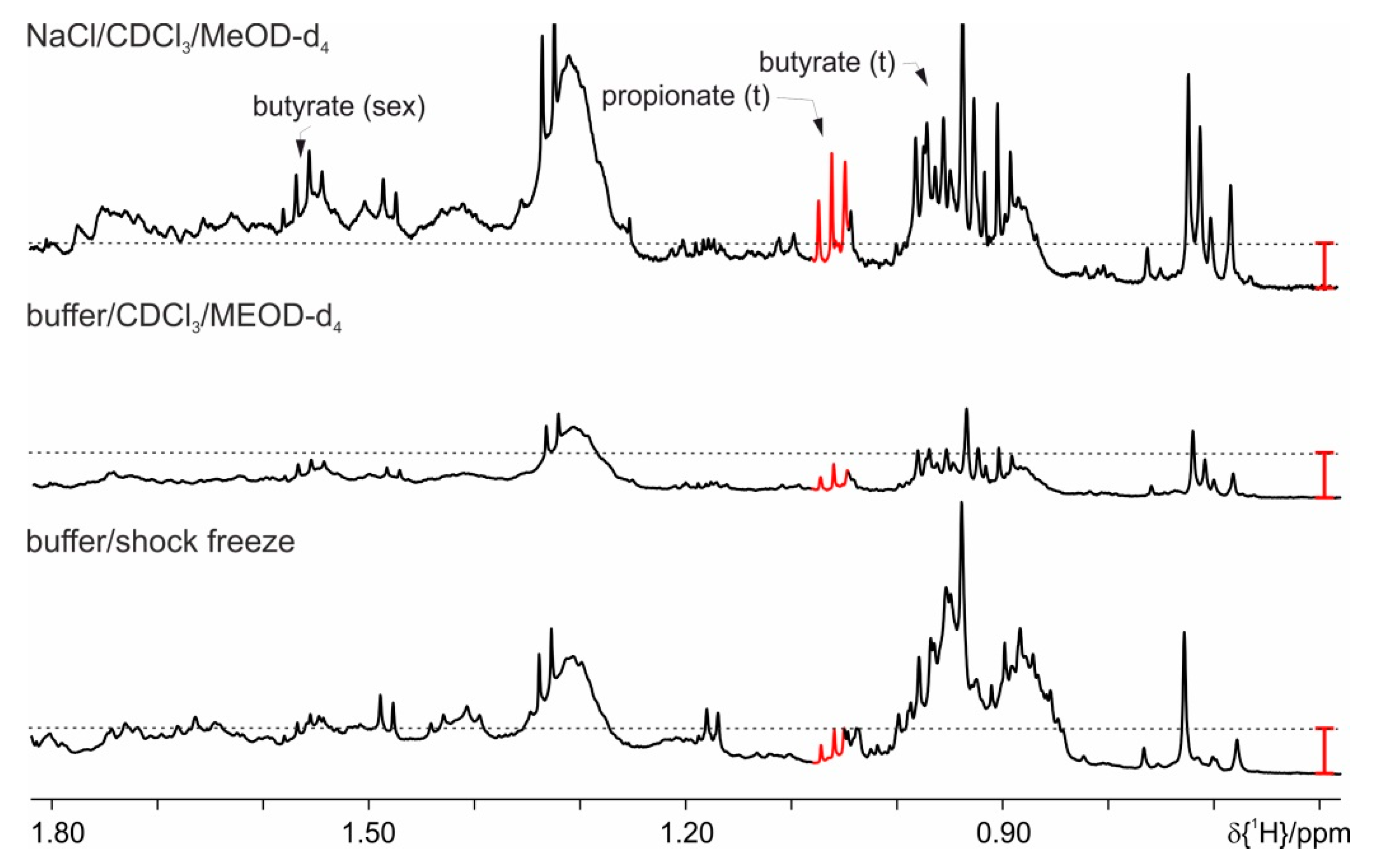
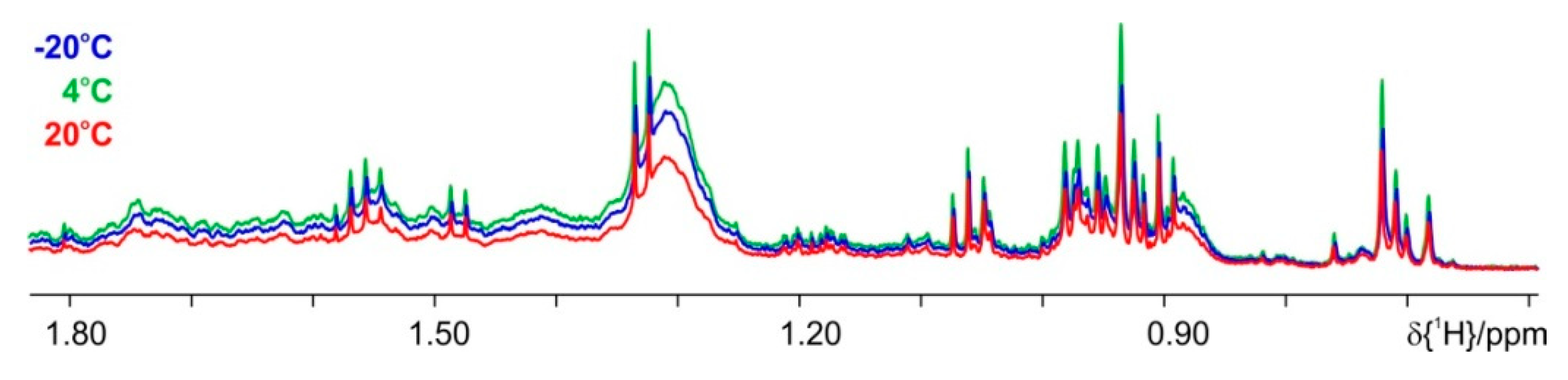
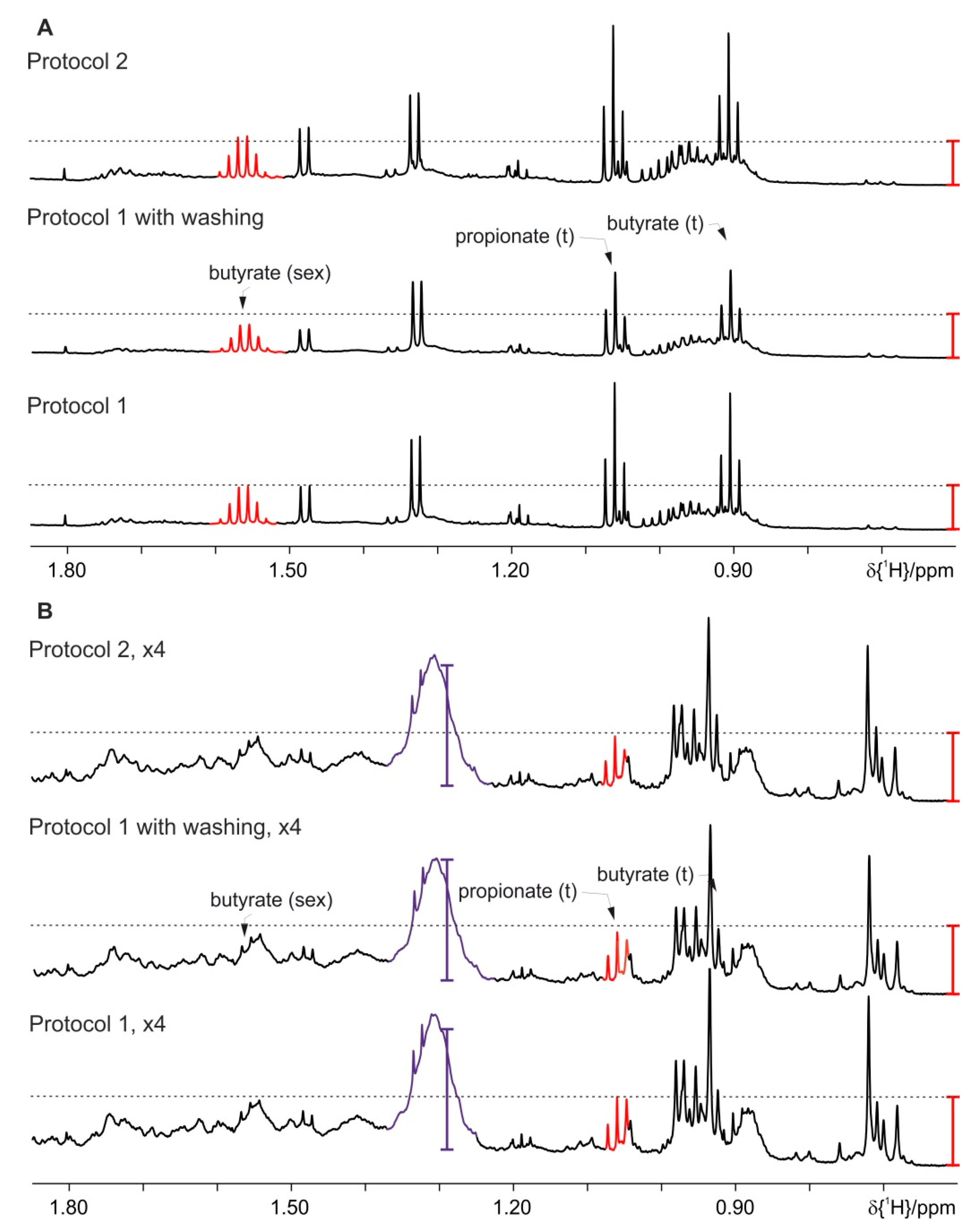
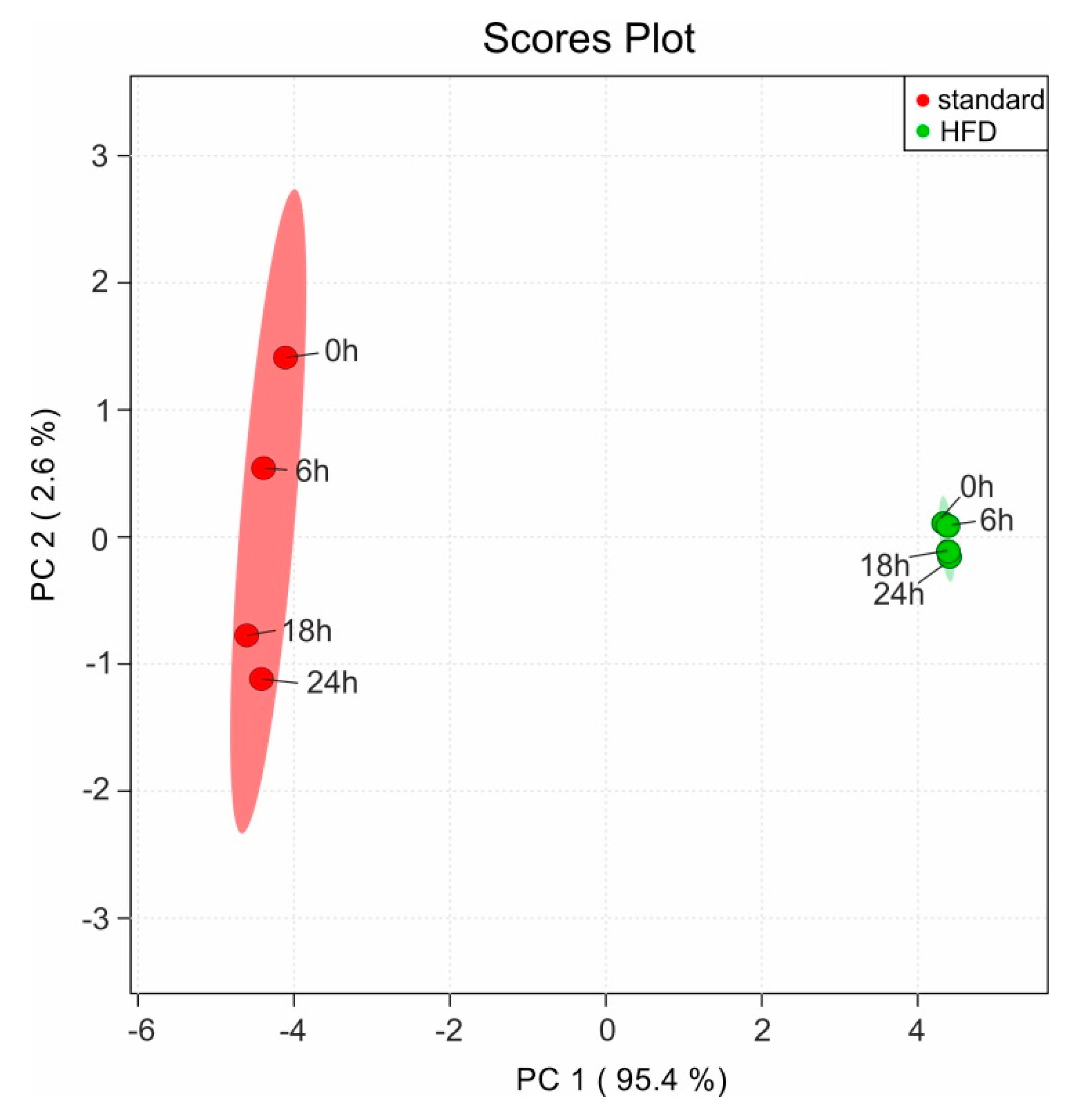
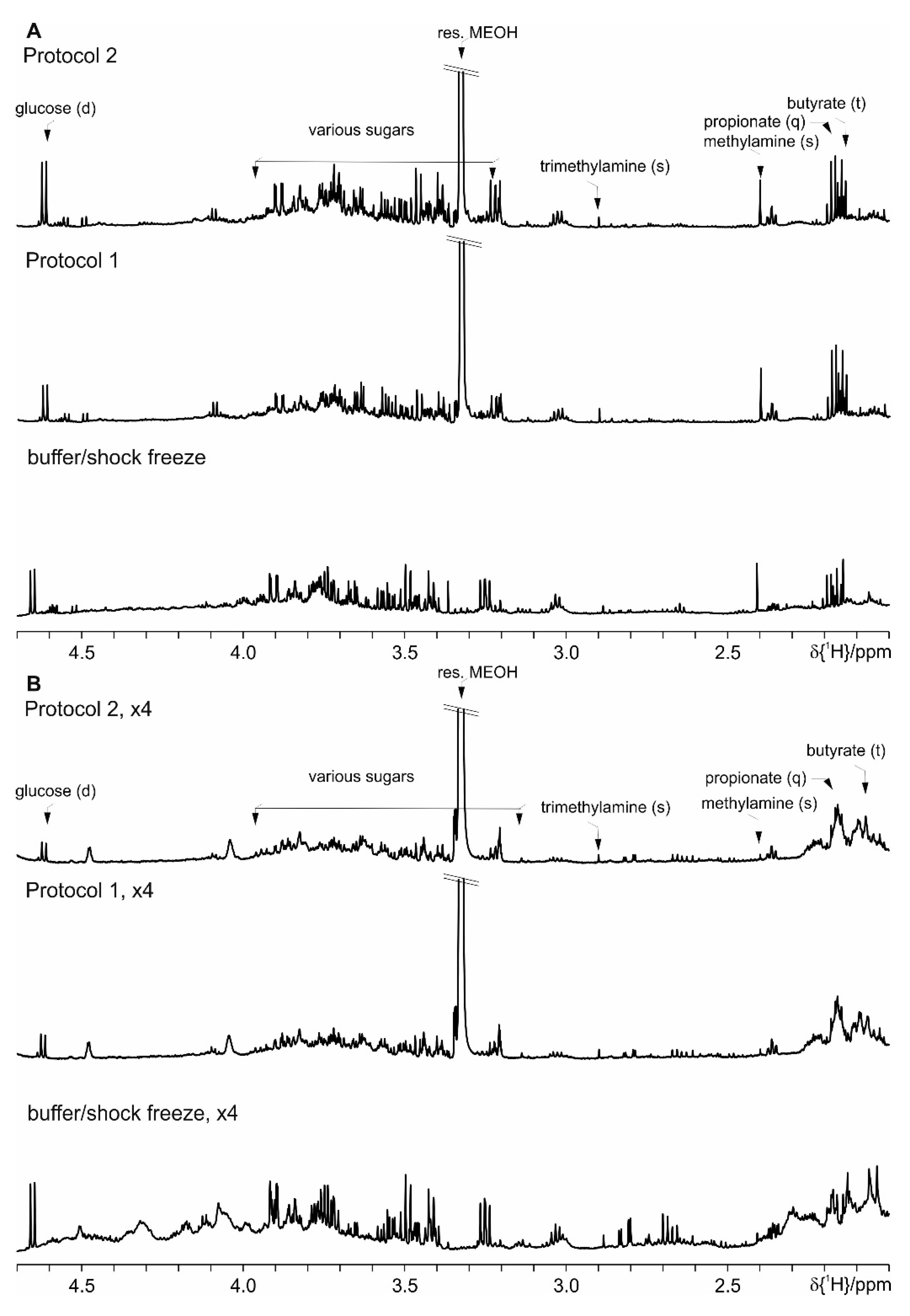
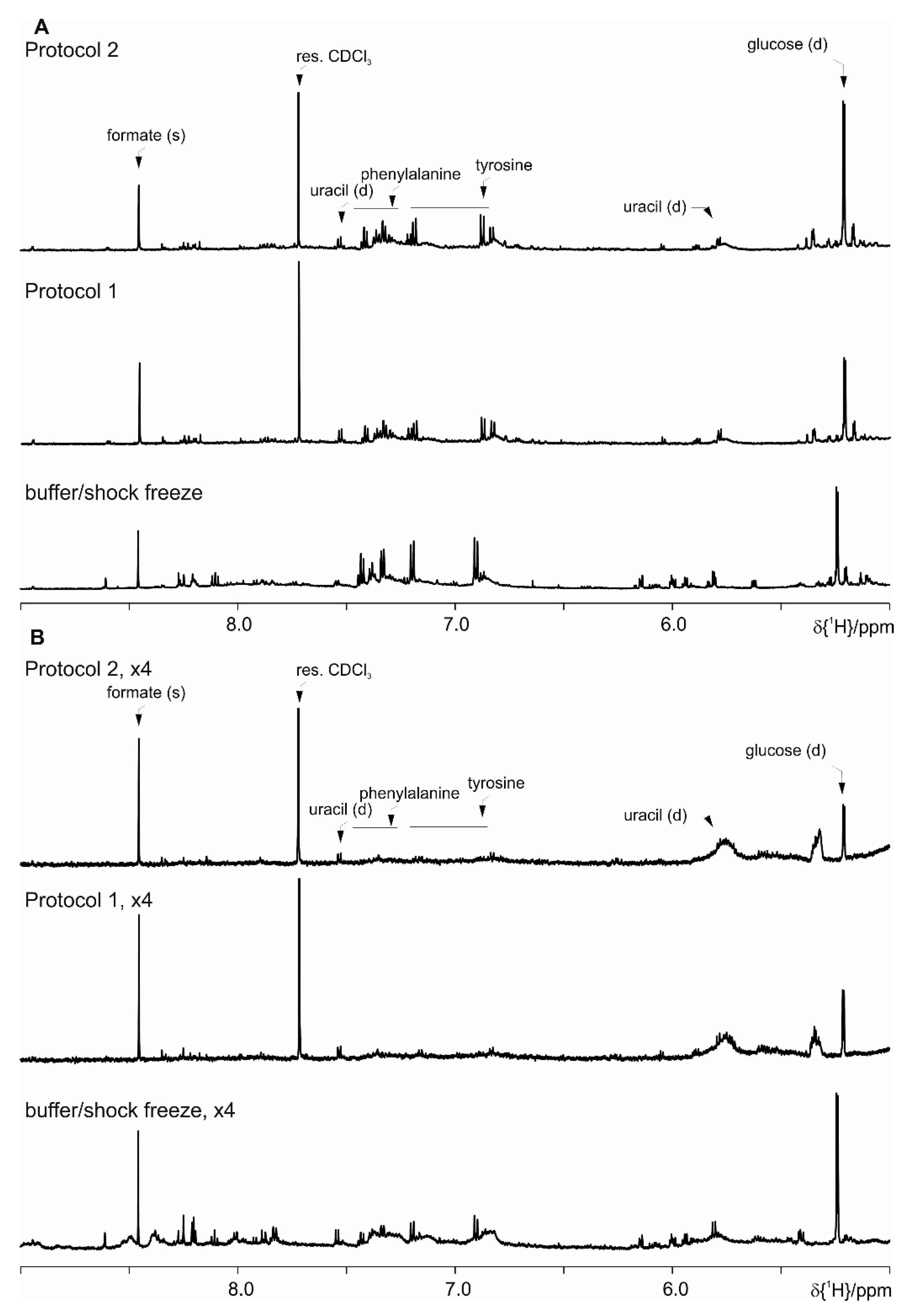
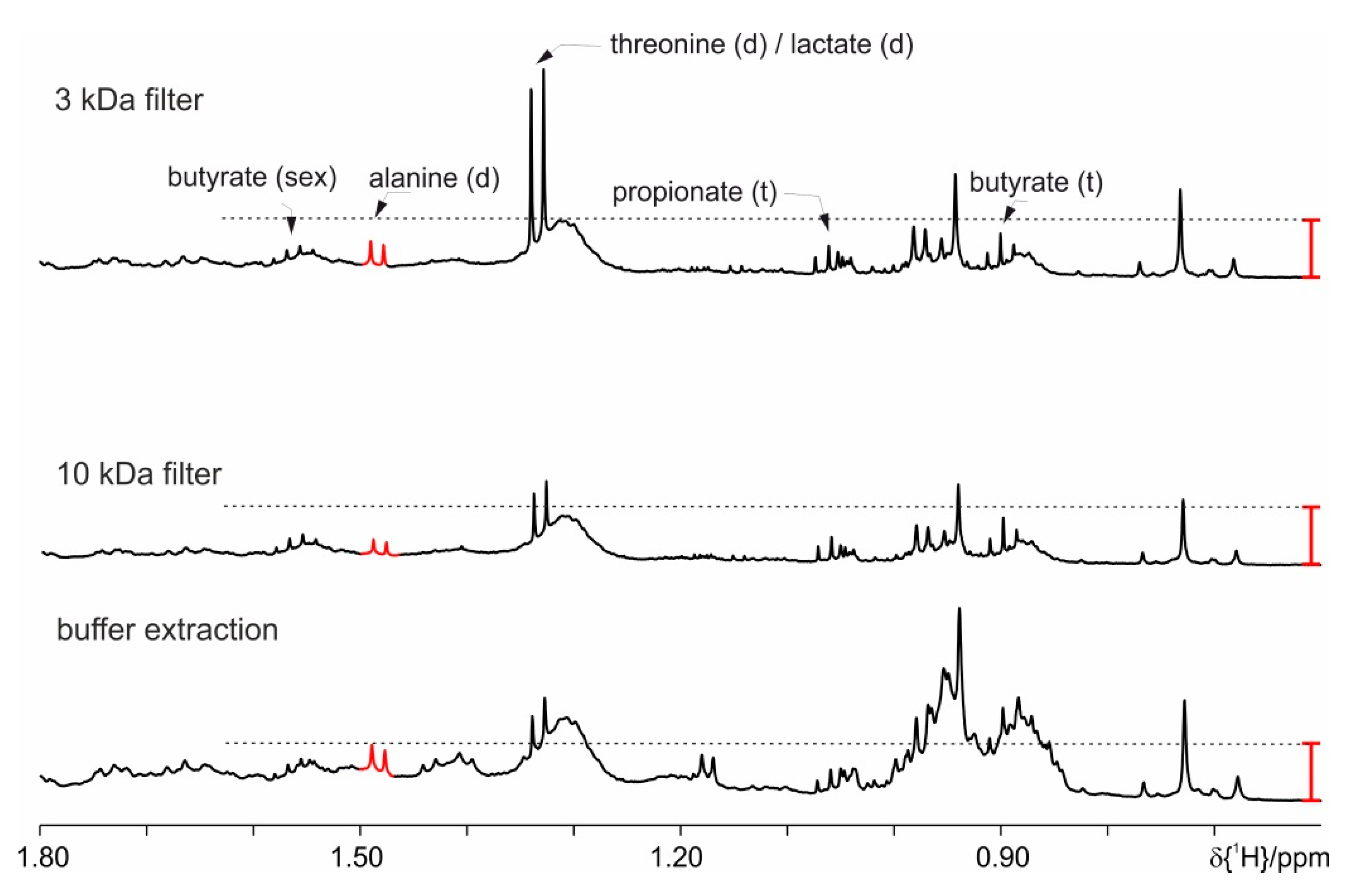
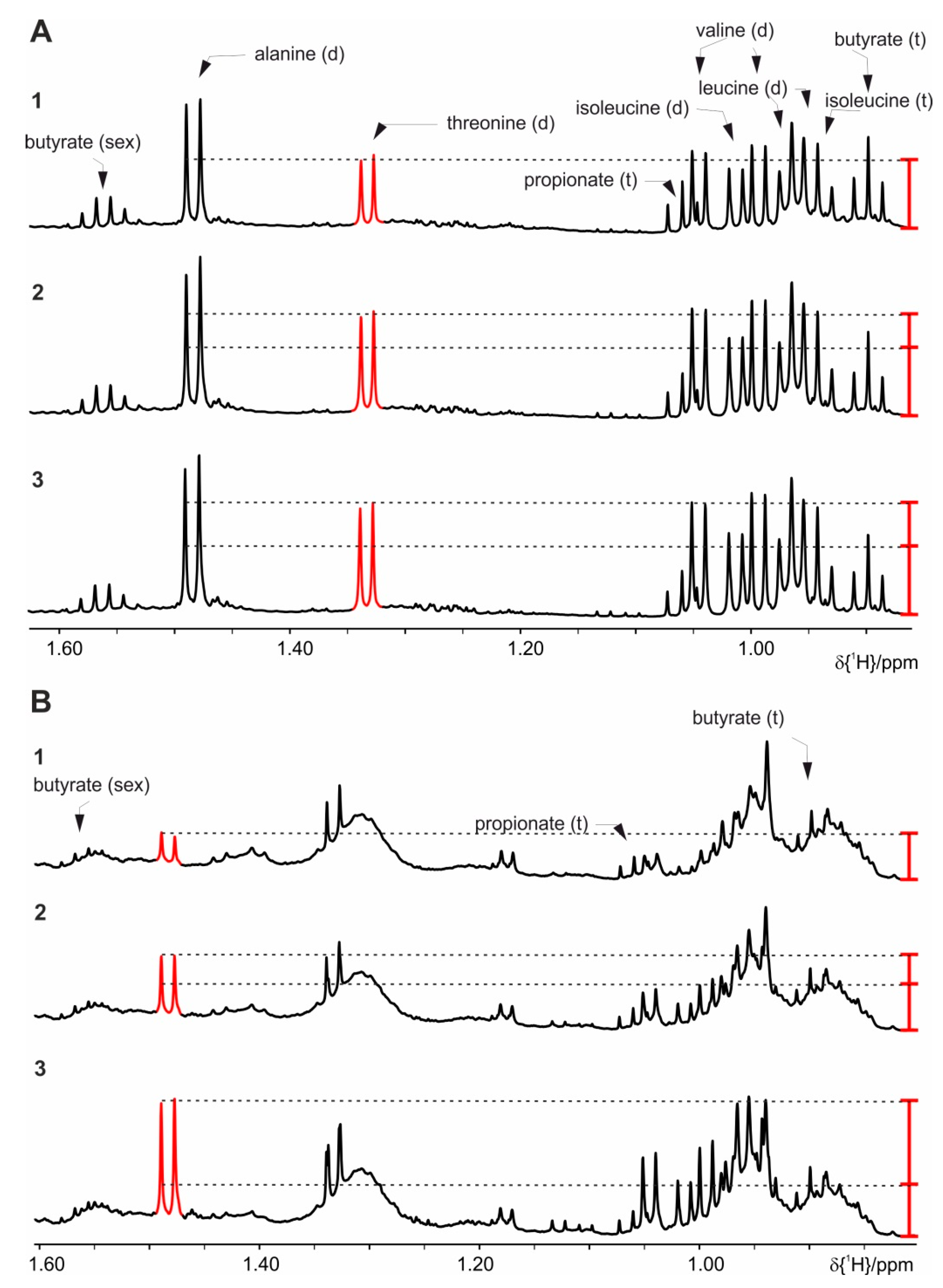
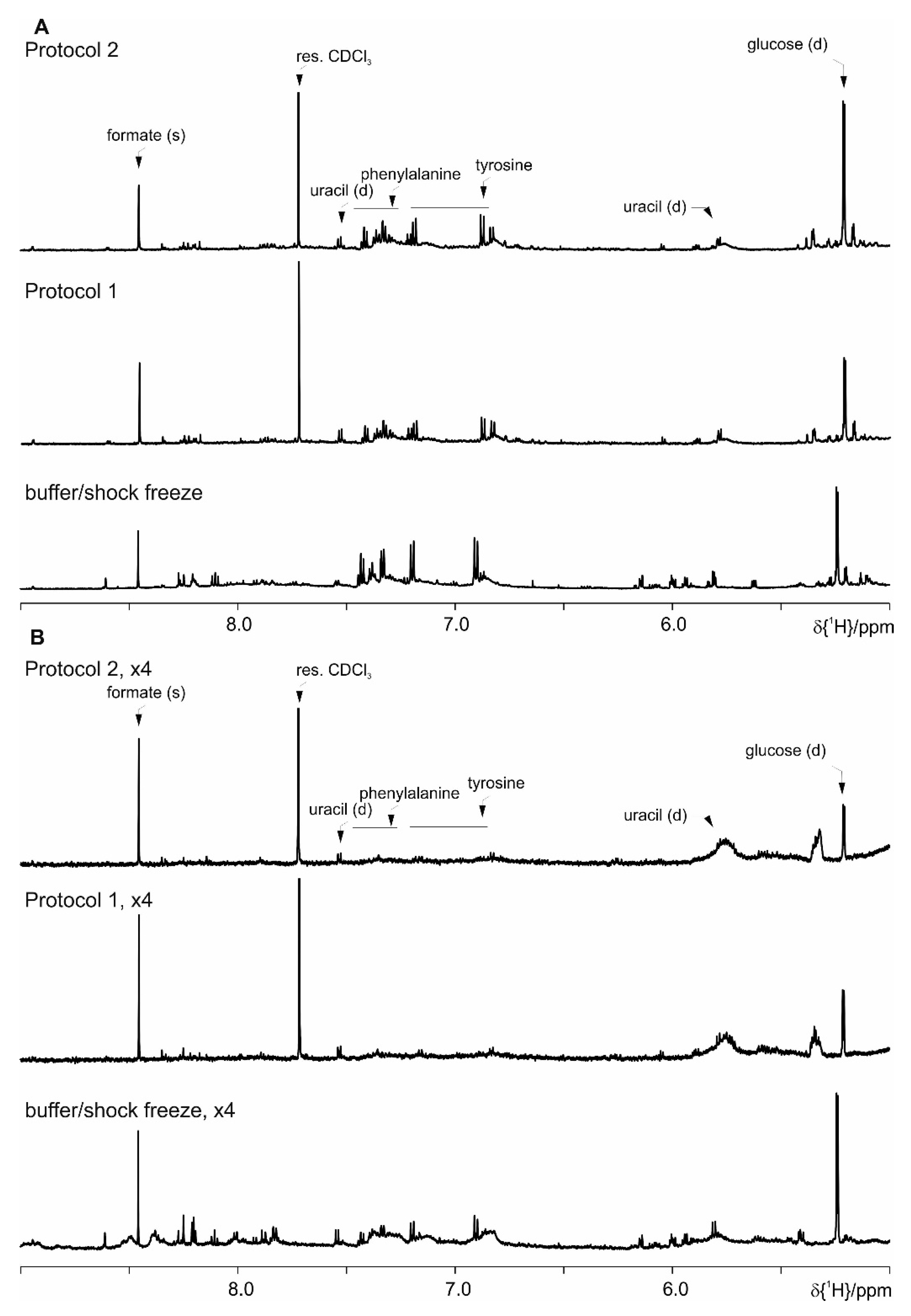
4. Results
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Scully, T. Public health: Society at large. Nature 2014, 508, S50–S51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köster, I.; Huppertz, E.; Hauner, H.; Schubert, I. Direct costs of diabetes mellitus in Germany—CoDiM 2000–2007. Exp. Clin. Endocrinol. Diabetes 2011, 119, 377–385. [Google Scholar] [CrossRef]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef] [PubMed]
- Shortt, C.; Hasselwander, O.; Meynier, A.; Nauta, A.; Fernández, E.N.; Putz, P.; Rowland, I.; Swann, J.; Türk, J.; Vermeiren, J.; et al. Systematic review of the effects of the intestinal microbiota on selected nutrients and non-nutrients. Eur. J. Nutr. 2018, 57, 25–49. [Google Scholar] [CrossRef]
- Graf, D.; Di Cagno, R.; Fåk, F.; Flint, H.J.; Nyman, M.; Saarela, M.; Watzl, B. Contribution of diet to the composition of the human gut microbiota. Microb. Ecol. Health Dis. 2015, 26, 26164. [Google Scholar] [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Backhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Aliaga, I. Model organisms in molecular nutrition research. Mol. Nutr. Food Res. 2012, 56, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Salter, A.M. Animal Models in Nutrition Research. In Nutrition Research Methodologies; Lovegrove, J., Sharma, S., Hodson, L., Eds.; John Wiley & Sons: Chichester, UK, 2015; Chapter 18; pp. 265–277. [Google Scholar]
- Stephen, A.M.; Cummings, J.H. The microbial contribution to human faecal mass. J. Med. Microbiol. 1980, 13, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Gratton, J.; Phetcharaburanin, J.; Mullish, B.H.; Williams, H.R.T.; Thursz, M.; Nicholson, J.K.; Holmes, E.; Marchesi, J.R.; Li, J.V. Optimized Sample Handling Strategy for Metabolic Profiling of Human Feces. Anal. Chem. 2016, 88, 4661–4668. [Google Scholar] [CrossRef]
- Tooth, D.; Garsed, K.; Singh, G.; Marciani, L.; Lam, C.; Fordham, I.; Fields, A.; Banwait, R.; Lingaya, M.; Layfield, R.; et al. Characterisation of faecal protease activity in irritable bowel syndrome with diarrhoea: Origin and effect of gut transit. Gut 2014, 63, 753–760. [Google Scholar] [CrossRef]
- Carroll, I.M.; Ringel-Kulka, T.; Ferrier, L.; Wu, M.C.; Siddle, J.P.; Bueno, L.; Ringel, Y. Fecal protease activity is associated with compositional alterations in the intestinal microbiota. PLoS ONE 2013, 8, e78017. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; An, Y.; Yao, J.; Wang, Y.; Tang, H. An optimised sample preparation method for NMR-based faecal metabonomic analysis. Analyst 2010, 135, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, S.; Yde, C.C.; Schmedes, M.S.; Jensen, H.M.; Meier, S.; Bertram, H.C. Strategy for Nuclear-Magnetic-Resonance-Based Metabolomics of Human Feces. Anal. Chem. 2015, 87, 5930–5937. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, J.; Li, J.V.; Zhou, N.-Y.; Tang, H.; Wang, Y. Gut microbiota composition modifies fecal metabolic profiles in mice. J. Proteome Res. 2013, 12, 2987–2999. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, S.; Westerhuis, J.A.; Ouwehand, A.C.; Saarinen, M.T.; Forssten, S.D.; Jensen, H.M.; Young, J.F.; Bertram, H.C.; Yde, C.C. Gut microbial activity as influenced by fiber digestion: Dynamic metabolomics in an in vitro colon simulator. Metabolomics 2016, 12, 1362. [Google Scholar] [CrossRef]
- Deda, O.; Gika, H.G.; Wilson, I.D.; Theodoridis, G.A. An overview of fecal sample preparation for global metabolic profiling. J. Pharm. Biomed. Anal. 2015, 113, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, G.; Noor, S.O.; Ridgway, K.; Scovell, L.; Jamieson, C.; Johnson, I.T.; Colquhoun, I.J.; Kemsley, E.K.; Narbad, A. Metabolomics of fecal extracts detects altered metabolic activity of gut microbiota in ulcerative colitis and irritable bowel syndrome. J. Proteome Res. 2011, 10, 4208–4218. [Google Scholar] [CrossRef]
- Deda, O.; Chatziioannou, A.C.; Fasoula, S.; Palachanis, D.; Raikos, Ν.; Theodoridis, G.A.; Gika, H.G. Sample preparation optimization in fecal metabolic profiling. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1047, 115–123. [Google Scholar] [CrossRef]
- Karu, N.; Deng, L.; Slae, M.; Guo, A.C.; Sajed, T.; Huynh, H.; Wine, E.; Wishart, D.S. A review on human fecal metabolomics: Methods, applications and the human fecal metabolome database. Anal. Chim. Acta 2018, 1030, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Jambocus, N.G.S.; Saari, N.; Ismail, A.; Khatib, A.; Mahomoodally, M.F.; Hamid, A.A. An Investigation into the Antiobesity Effects of Morinda citrifolia L. Leaf Extract in High Fat Diet Induced Obese Rats Using a (1)H NMR Metabolomics Approach. J. Diabetes Res. 2016, 2016, 2391592. [Google Scholar] [CrossRef]
- Lin, X.; Zhao, L.; Tang, S.; Zhou, Q.; Lin, Q.; Li, X.; Zheng, H.; Gao, H. Metabolic effects of basic fibroblast growth factor in streptozotocin-induced diabetic rats: A 1H NMR-based metabolomics investigation. Sci. Rep. 2016, 6, 36474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, D.M.; Deltimple, N.; van Velzen, E.; van Dorsten, F.A.; Bingham, M.; Vaughan, E.E.; van Duynhoven, J. (1)H NMR metabolite profiling of feces as a tool to assess the impact of nutrition on the human microbiome. NMR Biomed. 2008, 21, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Kromke, M.; Palomino-Schätzlein, M.; Mayer, H.; Pfeffer, S.; Pineda-Lucena, A.; Luy, B.; Hausberg, M.; Muhle-Goll, C. Profiling human blood serum metabolites by nuclear magnetic resonance spectroscopy: A comprehensive tool for the evaluation of hemodialysis efficiency. Transl. Res. 2016, 171, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Weger, B.D.; Weger, M.; Görling, B.; Schink, A.; Gobet, C.; Keime, C.; Poschet, G.; Jost, B.; Krone, N.; Hell, R.; et al. Extensive Regulation of Diurnal Transcription and Metabolism by Glucocorticoids. PLoS Genet. 2016, 12, e1006512. [Google Scholar] [CrossRef] [PubMed]
- Rist, M.J.; Roth, A.; Frommherz, L.; Weinert, C.H.; Krüger, R.; Merz, B.; Bunzel, D.; Mack, C.; Egert, B.; Bub, A.; et al. Metabolite patterns predicting sex and age in participants of the Karlsruhe Metabolomics and Nutrition (KarMeN) study. PLoS ONE 2017, 12, e0183228. [Google Scholar] [CrossRef] [PubMed]
- Kraus, D.; Yang, Q.; Kahn, B.B. Lipid Extraction from Mouse Feces. Bio. Protoc. 2015, 1–5. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Xia, J. MetaboAnalystR: An R package for flexible and reproducible analysis of metabolomics data. Bioinformatics 2018, 34, 4313–4314. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S. Using MetaboAnalyst 3.0 for Comprehensive Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2016, 55, 14.10.1–14.10.91. [Google Scholar] [CrossRef]
- Xia, J.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. MetaboAnalyst 3.0--making metabolomics more meaningful. Nucleic Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef]
- Van den Berg, R.A.; Hoefsloot, H.C.J.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genomics 2006, 7, 142. [Google Scholar] [CrossRef] [PubMed]
- Smilde, A.K.; van der Werf, M.J.; Bijlsma, S.; van der Werff-van der Vat, B.J.C.; Jellema, R.H. Fusion of mass spectrometry-based metabolomics data. Anal. Chem. 2005, 77, 6729–6736. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protocol | Freeze Cycle 1 | Ultra-Sonication | Details | Reference |
|---|---|---|---|---|
| Basic | 30 s vortex | Wu [13] | ||
| ultrasonic | 3 × 30 s | kept at 4 °C, vortex in between | Lamichhane [14] | |
| Shock freeze 1 | 3 × 30 s | vortex before freezing | Lamichhane [14] | |
| Combination of ultra-sonication and shock freeze 1 | 3 × 30 s | 3 × 30 s | kept to 4 °C vortex in between the runs | Lamichhane [14] |
| Double extraction, 1st fraction | 3 × 30 s | kept at 4 °C, vortex in between | Wu [13] | |
| Double extraction, 2nd fraction | 3 × 30 s | kept at 4 °C, vortex in between | Wu [13] | |
| Double extraction, fractions combined | 3 × 30 s | kept at 4 °C, vortex in between, lyophilised at −80 °C overnight | Wu [13] | |
| Beads | ceramic beads 15 min by 99 rpm juddered | |||
| Acetonitrile (10%, 20%, or 30%) | 3 × 30 s | tempered to 4 °C vortex in between | ||
| EDTA | 2 mM EDTA, 30 s vortex | |||
| 8.75 M TCA/buffer (1:34 v:v) | 10 min incubation in fridge (6 °C) | |||
| Heating (40 °C, 60 °C or 90 °C) | 10 min | |||
| Filter (10 kDa or 3 kDa) | Supernatant 15 or 30 min at 15,000× g centrifuged |
| Extraction | Solvent 1 | Centrifugation | Details | Reference |
|---|---|---|---|---|
| MeOD-d4/CDCl3/D2O (or buffer 2) | 300/300/350 (1:1:1.166) | 30 min, 1400× g, 4 °C, 10 min 15,000× g, 4 °C, each separate phase | Phase separation overnight | Lamichhane [14] |
| CD2Cl2/buffer | 600/600 (1:1) | 30 min 15,000× g, 4 °C, aqueous phase 10 min 15,000× g, 4 °C | - | - |
| 0.15 M NaCl/CDCl3/MeOD-d4 | 600/400/200 (3:2:1) | RT, 10 min, 1100× g | - | Kraus [27] 3 |
| 0.15 M NaCl/CDCl3/MeOD-d4 | 600/400/200 (3:2:1) | 30 s vortex, 4 °C, 10 min, 1100× g | Optionally three CDCl3 washing steps | Protocol 1, this work |
| 0.15 M NaCl/CDCl3/MeOD-d4 | 600/400/200 (3:2:1) | 30 s vortex, homogenizer four times 20 s, 10 °C, 6000 rpm, centrifuged 0 °C 1485× g for 10 min | - | Protocol 2, this work |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hauser, A.; Eisenmann, P.; Muhle-Goll, C.; Luy, B.; Dötsch, A.; Graf, D.; Tzvetkova, P. Efficient Extraction from Mice Feces for NMR Metabolomics Measurements with Special Emphasis on SCFAs. Metabolites 2019, 9, 55. https://doi.org/10.3390/metabo9030055
Hauser A, Eisenmann P, Muhle-Goll C, Luy B, Dötsch A, Graf D, Tzvetkova P. Efficient Extraction from Mice Feces for NMR Metabolomics Measurements with Special Emphasis on SCFAs. Metabolites. 2019; 9(3):55. https://doi.org/10.3390/metabo9030055
Chicago/Turabian StyleHauser, Adrian, Philipp Eisenmann, Claudia Muhle-Goll, Burkhard Luy, Andreas Dötsch, Daniela Graf, and Pavleta Tzvetkova. 2019. "Efficient Extraction from Mice Feces for NMR Metabolomics Measurements with Special Emphasis on SCFAs" Metabolites 9, no. 3: 55. https://doi.org/10.3390/metabo9030055