Analysis of Intracellular Metabolites from Microorganisms: Quenching and Extraction Protocols



Abstract
:1. Introduction
- (a)
- Growing the microorganism under study using an appropriate culture media;
- (b)
- Sample collection at suitable or desired stage of growth and quenching of microbial cells;
- i
- Separation of microbial cells from growth media: The supernatant is used for extracellular metabolite analysis;
- ii
- Microbial cells are used for intracellular metabolite analysis;
- (c)
- Extraction of intracellular metabolites;
- (d)
- Metabolite analysis of intra- and extracellular metabolites using an appropriate instrumental approach.
2. Cell Metabolism and Metabolite Turnover
3. Microbial Cell Envelopes and Leakage of Intracellular Metabolites
3.1. The Microbial Cell Envelope
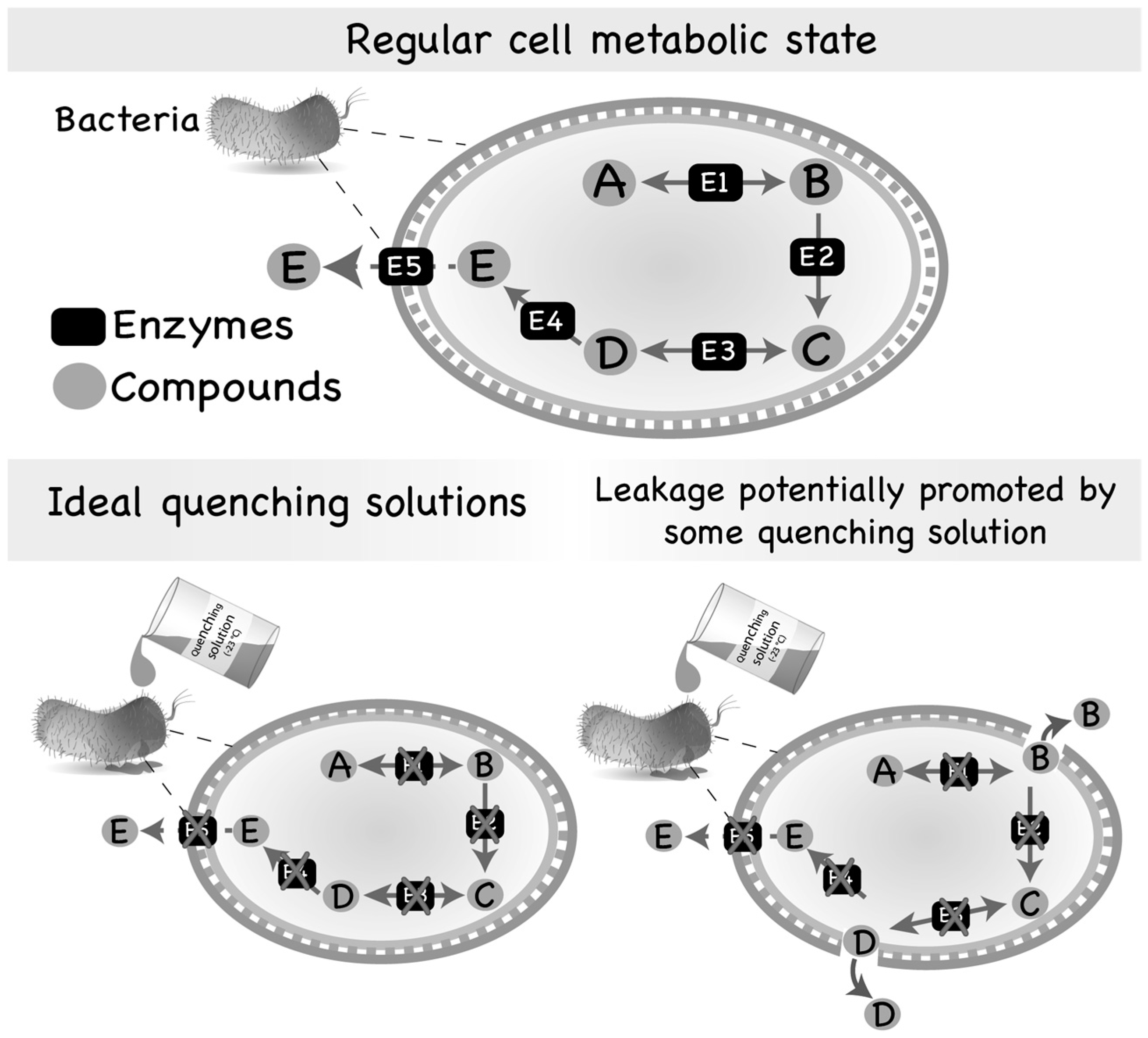
3.2. Leakage of Intracellular Metabolites
4. Overview of Available Quenching Methods for Microbial Cultures
4.1. Bacterial Cells
4.2. Yeast Cells
4.3. Filamentous Fungi and Bacteria
4.4. Protozoa
4.5. Microalgae
5. Devices for Fast Sampling
5.1. Sampling from Culture Flasks
5.2. Sampling from Bioreactors
Stopped-Flow Systems
6. Extraction of Intracellular Metabolites: Disruption Methods for Microbial Cell Envelopes
6.1. The Extraction of Intracellular Metabolites by Chemical Lysis
6.1.1. Boiling Ethanol
6.1.2. Cold Methanol
6.1.3. Buffered Methanol–Chloroform–Water
6.1.4. Hot Water
6.1.5. Acidic Extraction
6.1.6. Alkaline Extraction
6.2. Mechanical Disruption of Cell Walls
6.2.1. Supercritical Fluid Extraction (SFE)
6.2.2. Pressurized Liquid Extraction (PLE)
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fiehn, O. Metabolomics-the link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Bino, R.J.; Hall, R.D.; Fiehn, O.; Kopka, J.; Saito, K.; Draper, J.; Nikolau, B.J.; Mendes, P.; Roessner-Tunali, U.; Beale, M.H.; et al. Potential of metabolomics as a functional genomics tool. Trends Plant Sci. 2004, 9, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Mandal, R.; Stanislaus, A.; Ramirez-Gaona, M. Cancer metabolomics and the human metabolome database. Metabolites 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, P.D.; German, A.J.; Noble, P.J.M. Metabolomics: An emerging post-genomic tool for nutrition. Br. J. Nutr. 2004, 92, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov. 2016, 15, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Aggio, R.B.M.; Obolonkin, V.; Villas-Boas, S.G. Sonic vibration affects the metabolism of yeast cells growing in liquid culture: A metabolomic study. Metabolomics 2012, 8, 670–678. [Google Scholar] [CrossRef]
- Patejko, M.; Jacyna, J.; Markuszewski, M.J. Sample preparation procedures utilized in microbial metabolomics: An overview. J. Chromatogr. B 2017, 1043, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Van der Werf, M.J. Metabolomics: A revolutionary tool to optimize microbial processes. Abstr. Pap. Am. Chem. Soc. 2003, 226, U84. [Google Scholar]
- Villas-Boas, S.G.; Roeseener, U.; Hansen, M.A.E.; Smedsgaard, J.; Nielsen, J. Metabolomics Analysis: An Introduction; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Canelas, A.B.; Ras, C.; ten Pierick, A.; van Dam, J.C.; Heijnen, J.J.; Van Gulik, W.M. Leakage-free rapid quenching technique for yeast metabolomics. Metabolomics 2008, 4, 226–239. [Google Scholar] [CrossRef]
- Da Luz, J.A.; Hans, E.; Zeng, A.P. Automated fast filtration and on-filter quenching improve the intracellular metabolite analysis of microorganisms. Eng. Life Sci. 2014, 14, 135–142. [Google Scholar] [CrossRef]
- Canelas, A.B.; ten Pierick, A.; Ras, C.; Seifar, R.M.; van Dam, J.C.; van Gulik, W.M.; Heijnen, J.J. Quantitative evaluation of intracellular metabolite extraction techniques for yeast metabolomics. Anal. Chem. 2009, 81, 7379–7389. [Google Scholar] [CrossRef] [PubMed]
- Bolten, C.J.; Kiefer, P.; Letisse, F.; Portais, J.-C.; Wittmann, C. Sampling for metabolome analysis of microorganisms. Anal. Chem. 2007, 79, 3843–3849. [Google Scholar] [CrossRef] [PubMed]
- Van Gulik, W.M.; Canelas, A.B.; Taymaz-Nikerel, H.; Douma, R.D.; de Jonge, L.P.; Heijnen, J.J. Fast sampling of the cellular metabolome. Methods Mol. Biol. 2012, 881, 279–306. [Google Scholar]
- Baidoo, E.E.K.; Benke, P.I.; Keasling, J.D. Mass spectrometry-based microbial metabolomics. Methods Mol. Biol. 2012, 881, 215–278. [Google Scholar]
- Buchholz, A.; Hurlebaus, J.; Wandrey, C.; Takors, R. Metabolomics: Quantification of intracellular metabolite dynamics. Biomol. Eng. 2002, 19, 5–15. [Google Scholar] [CrossRef]
- Hajjaj, H.; Blanc, P.J.; Goma, G.; Francois, J. Sampling techniques and comparative extraction procedures for quantitative determination of intra- and extracellular metabolites in filamentous fungi. Fems Microbiol. Lett. 1998, 164, 195–200. [Google Scholar] [CrossRef]
- Villas-Boas, S.G.; Hojer-Pedersen, J.; Akesson, M.; Smedsgaard, J.; Nielsen, J. Global metabolite analysis of yeast: Evaluation of sample preparation methods. Yeast 2005, 22, 1155–1169. [Google Scholar] [CrossRef] [PubMed]
- Behrends, V.; Williams, H.D.; Bundy, J.G. Metabolic footprinting: Extracellular metabolomic analysis. Methods Mol. Biol. 2014, 1149, 281–292. [Google Scholar]
- Mo, M.L.; Palsson, B.O.; Herrgard, M.J. Connecting extracellular metabolomic measurements to intracellular flux states in yeast. BMC Syst. Biol. 2009, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Villas-Boas, S.G.; Bruheim, P. Cold glycerol-saline: The promising quenching solution for accurate intracellular metabolite analysis of microbial cells. Anal. Biochem. 2007, 370, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.C.; Mukhopadhyay, R.; Wen, B.N.; Gitai, Z.; Wingreen, N.S. Cell shape and cell-wall organization in gram-negative bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 19282–19287. [Google Scholar] [CrossRef] [PubMed]
- Bartholomew, J.W.; Mittwer, T. The gram stain. Bacteriol. Rev. 1952, 16, 1–29. [Google Scholar] [PubMed]
- Northcote, D.H.; Goulding, K.J.; Horne, R.W. Chemical composition and structure of the cell wall of hydrodictyon-africanum yaman. Biochem. J. 1960, 77, 503. [Google Scholar] [CrossRef] [PubMed]
- Lipke, P.N.; Ovalle, R. Cell wall architecture in yeast: New structure and new challenges. J. Bacteriol. 1998, 180, 3735–3740. [Google Scholar] [PubMed]
- Bowman, S.M.; Free, S.J. The structure and synthesis of the fungal cell wall. Bioessays 2006, 28, 799–808. [Google Scholar] [CrossRef] [PubMed]
- VanWinkleSwift, K.P.; Rickoll, W.L. The zygospore wall of chlamydomonas monoica (chlorophyceae): Morphogenesis and evidence for the presence of sporopollenin. J. Phycol. 1997, 33, 655–665. [Google Scholar] [CrossRef]
- Damiani, M.C.; Leonardi, P.I.; Pieroni, O.I.; Caceres, E.J. Ultrastructure of the cyst wall of haematococcus pluvialis (chlorophyceae): Wall development and behaviour during cyst germination. Phycologia 2006, 45, 616–623. [Google Scholar] [CrossRef]
- Imam, S.H.; Buchanan, M.J.; Shin, H.C.; Snell, W.J. The chlamydomonas cell-wall-characterization of the wall framework. J. Cell Biol. 1985, 101, 1599–1607. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cai, J.; Jiang, Y.G.; Jiang, X.G.; Zhang, D.Y. Preparation of biosilica structures from frustules of diatoms and their applications: Current state and perspectives. Appl. Microbiol. Biotechnol. 2013, 97, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Svensen, O.; Frette, O.; Erga, S.R. Scattering properties of microalgae: The effect of cell size and cell wall. Appl. Opt. 2007, 46, 5762–5769. [Google Scholar] [CrossRef] [PubMed]
- Lemgruber, L.; Lupetti, P.; De Souza, W.; Vommaro, R.C.; da Rocha-Azevedo, B. The fine structure of the acanthamoeba polyphaga cyst wall. FEMS Microbiol. Lett. 2010, 305, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Naderer, T.; McConville, M.J. The leishmania-macrophage interaction: A metabolic perspective. Cell. Microbiol. 2008, 10, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; Maitra, P. Role of ATP in control of energy metabolism in growing bacteria. J. Gen. Microbiol. 1968, 53, S7–S8. [Google Scholar] [PubMed]
- Weibel, K.E.; Mor, J.R.; Fiechter, A. Rapid sampling of yeast-cells and automated assays of adenylate, citrate, pyruvate and glucose-6-phosphate pools. Anal. Biochem. 1974, 58, 208–216. [Google Scholar] [CrossRef]
- Wollenberger, A.; Ristau, O.; Schoffa, G. Eine einfache technik der extrem schnellen abkuhlung grosserer gewebestucke. Pflug. Arch. Eur. J. Physiol. 1960, 270, 399–412. [Google Scholar] [CrossRef]
- Visser, D.; van Zuylen, G.A.; van Dam, J.C.; Oudshoorn, A.; Eman, M.R.; Ras, C.; van Gulik, W.M.; Frank, J.; van Dedem, G.W.K.; Heijnen, J.J. Rapid sampling for analysis of in vivo kinetics using the bioscope: A system for continuous-pulse experiments. Biotechnol. Bioeng. 2002, 79, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Faijes, M.; Mars, A.E.; Smid, E.J. Comparison of quenching and extraction methodologies for metabolome analysis of lactobacillus plantarum. Microb. Cell Fact. 2007, 6. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, L.P.; Douma, R.D.; Heijnen, J.J.; van Gulik, W.M. Optimization of cold methanol quenching for quantitative metabolomics of penicillium chrysogenum. Metabolomics 2012, 8, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, B.; Francois, J.; Renaud, M. A rapid and reliable method for metabolite extraction in yeast using boiling buffered ethanol. Yeast 1997, 13, 1347–1355. [Google Scholar] [CrossRef]
- Wittmann, C.; Kromer, J.O.; Kiefer, P.; Binz, T.; Heinzle, E. Impact of the cold shock phenomenon on quantification of intracellular metabolites in bacteria. Anal. Biochem. 2004, 327, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Franzen, J.S.; Binkley, S.B. Comparison of acid-soluble nucleotides in Escherichia-coli at different growth rates. J. Biol. Chem. 1961, 236, 515–519. [Google Scholar] [PubMed]
- Cole, H.A.; Wimpenny, J.W.; Hughes, D.E. ATP pool in Escherichia coli. I. Measurement of pool using a modified luciferase assay. Biochim. Biophys. Acta 1967, 143, 445–453. [Google Scholar] [CrossRef]
- Strange, R.E.; Wade, H.E.; Dark, F.A. Effect of starvation on adenosine triphosphate concentration in aerobacter aerogenes. Nature 1963, 199, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Williamson, D.H.; Lund, P.; Krebs, H.A. Redox state of free nicotinamide-adenine dinucleotide in cytoplasm and mitochondria of rat liver. Biochem. J. 1967, 103, 514. [Google Scholar] [CrossRef] [PubMed]
- Faupel, R.P.; Seitz, H.J.; Tarnowski, W.; Thiemann, V.; Weiss, C. Problem of tissue sampling from experimental-animals with respect to freezing technique, anoxia, stress and narcosis-new method for sampling rat-liver tissue and physiological values of glycolytic intermediates and related compounds. Arch. Biochem. Biophys. 1972, 148, 509–522. [Google Scholar] [CrossRef]
- Saez, M.J.; Lagunas, R. Determination of intermediary metabolites in yeast-critical-examination of effect of sampling conditions and recommendations for obtaining true levels. Mol. Cell. Biochem. 1976, 13, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Ruijter, G.J.G.; Visser, J. Determination of intermediary metabolites in Aspergillus niger. J. Microbiol. Methods 1996, 25, 295–302. [Google Scholar] [CrossRef]
- Dekoning, W.; Vandam, K. A method for the determination of changes of glycolytic metabolites in yeast on a subsecond time scale using extraction at neutral ph. Anal. Biochem. 1992, 204, 118–123. [Google Scholar] [CrossRef]
- Moritz, B.; Striegel, K.; de Graaf, A.A.; Sahm, H. Kinetic properties of the glucose-6-phosphate and 6-phosphogluconate dehydrogenases from corynebacterium glutamicum and their application for predicting pentose phosphate pathway flux in vivo. Eur. J. Biochem. 2000, 267, 3442–3452. [Google Scholar] [CrossRef] [PubMed]
- Maharjan, R.P.; Ferenci, T. Global metabolite analysis: The influence of extraction methodology on metabolome profiles of Escherichia coli. Anal. Biochem. 2003, 313, 145–154. [Google Scholar] [CrossRef]
- Tredwell, G.D.; Edwards-Jones, B.; Leak, D.J.; Bundy, J.G. The development of metabolomic sampling procedures for Pichia pastoris, and baseline metabolome data. PLoS ONE 2011, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Jensen, N.B.S.; Jokumsen, K.V.; Villadsen, J. Determination of the phosphorylated sugars of the Embden-Meyerhoff-Parnas pathway in Lactococcus lactis using a fast sampling technique and solid phase extraction. Biotechnol. Bioeng. 1999, 63, 356–362. [Google Scholar] [CrossRef]
- Chassagnole, C.; Noisommit-Rizzi, N.; Schmid, J.W.; Mauch, K.; Reuss, M. Dynamic modeling of the central carbon metabolism of Escherichia coli. Biotechnol. Bioeng. 2002, 79, 53–73. [Google Scholar] [CrossRef] [PubMed]
- Japelt, K.B.; Christensen, J.H.; Villas-Boas, S.G. Metabolic fingerprinting of Lactobacillus paracasei: The optimal quenching strategy. Microb. Cell Fact. 2015, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Bolling, C.; Fiehn, O. Metabolite profiling of Chlamydomonas reinhardtii under nutrient deprivation. Plant Physiol. 2005, 139, 1995–2005. [Google Scholar] [CrossRef] [PubMed]
- De Souza, D.P.; Saunders, E.C.; McConville, M.J.; Likic, V.A. Progressive peak clustering in GC-MS metabolomic experiments applied to leishmania parasites. Bioinformatics 2006, 22, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Smart, K.F.; Aggio, R.B.M.; Van Houtte, J.R.; Villas-Boas, S.G. Analytical platform for metabolome analysis of microbial cells using methyl chloroformate derivatization followed by gas chromatography-mass spectrometry. Nat. Protoc. 2010, 5, 1709–1729. [Google Scholar] [CrossRef] [PubMed]
- Leder, I.G. Interrelated effects of cold shock and osmotic pressure on the permeability of the Escherichia coli membrane to permease accumulated substrates. J. Bacteriol. 1972, 111, 406–415. [Google Scholar]
- Britten, R.J.; McClure, F.T. Amino acid pool in Escherichia-coli. Bacteriol. Rev. 1962, 26, 292. [Google Scholar] [PubMed]
- Smeaton, J.R.; Elliott, W.H. Selective release of ribonuclease-inhibitor from Bacillus subtilis cells by cold shock treatment. Biochem. Biophys. Res. Commun. 1967, 26, 75–81. [Google Scholar] [CrossRef]
- Carnicer, M.; Canelas, A.B.; ten Pierick, A.; Zeng, Z.; van Dam, J.; Albiol, J.; Ferrer, P.; Heijnen, J.J.; van Gulik, W. Development of quantitative metabolomics for Pichia pastoris. Metabolomics 2012, 8, 284–298. [Google Scholar] [CrossRef] [PubMed]
- Link, H.; Fuhrer, T.; Gerosa, L.; Zamboni, N.; Sauer, U. Real-time metabolome profiling of the metabolic switch between starvation and growth. Nat. Methods 2015, 12, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Theobald, U.; Mailinger, W.; Reuss, M.; Rizzi, M. In vivo analysis of glucose-induced fast changes in yeast adenine-nucleotide pool applying a rapid sampling technique. Anal. Biochem. 1993, 214, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Larsson, G.; Tornkvist, M. Rapid sampling, cell inactivation and evaluation of low extracellular glucose concentrations during fed-batch cultivation. J. Biotechnol. 1996, 49, 69–82. [Google Scholar] [CrossRef]
- Schaefer, U.; Boos, W.; Takors, R.; Weuster-Botz, D. Automated sampling device for monitoring intracellular metabolite dynamics. Anal. Biochem. 1999, 270, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, M.; Fuhrman, L.; Ingram, A.; Nickerson, K.W.; Conway, T. Single-run separation and detection of multiple metabolic intermediates by anion-exchange high-performance liquid-chromatography and application to cell pool extracts prepared from Escherichia-coli. Anal. Biochem. 1995, 232, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Schaub, J.; Schiesling, C.; Reuss, M.; Dauner, M. Integrated sampling procedure for metabolome analysis. Biotechnol. Prog. 2006, 22, 1434–1442. [Google Scholar] [CrossRef] [PubMed]
- Lange, H.C.; Eman, M.; van Zuijlen, G.; Visser, D.; van Dam, J.C.; Frank, J.; de Mattos, M.J.T.; Heijnen, J.J. Improved rapid sampling for in vivo kinetics of intracellular metabolites in Saccharomyces cerevisiae. Biotechnol. Bioeng. 2001, 75, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Van Gulik, W.M. Fast sampling for quantitative microbial metabolomics. Curr. Opin. Biotechnol. 2010, 21, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Buziol, S.; Bashir, I.; Baumeister, A.; Claassen, W.; Noisommit-Rizzi, N.; Mailinger, W.; Reuss, M. New bioreactor-coupled rapid stopped-flow sampling technique for measurements of metabolite dynamics on a subsecond time scale. Biotechnol. Bioeng. 2002, 80, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Mashego, M.R.; van Gulik, W.M.; Vinke, J.L.; Visser, D.; Heijnen, J.J. In vivo kinetics with rapid perturbation experiments in Saccharomyces cerevisiae using a second-generation bioscope. Metab. Eng. 2006, 8, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Duportet, X.; Aggio, R.B.M.; Carneiro, S.; Villas-Boas, S.G. The biological interpretation of metabolomic data can be misled by the extraction method used. Metabolomics 2012, 8, 410–421. [Google Scholar] [CrossRef]
- Park, C.; Yun, S.; Lee, S.Y.; Park, K.; Lee, J. Metabolic profiling of Klebsiella oxytoca: Evaluation of methods for extraction of intracellular metabolites using UPLC/Q-TOF-MS. Appl. Biochem. Biotechnol. 2012, 167, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, D.Y.; Wohlgemuth, G.; Park, H.S.; Fiehn, O.; Kim, K.H. Evaluation and optimization of metabolome sample preparation methods for Saccharomyces cerevisiae. Anal. Chem. 2013, 85, 2169–2176. [Google Scholar] [CrossRef] [PubMed]
- Groussac, E.; Ortiz, M.; Francois, J. Improved protocols for quantitative determination of metabolites from biological samples using high performance ionic-exchange chromatography with conductimetric and pulsed amperometric detection. Enzym. Microb. Technol. 2000, 26, 715–723. [Google Scholar] [CrossRef]
- Hans, M.A.; Heinzle, E.; Wittmann, C. Quantification of intracellular amino acids in batch cultures of Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 2001, 56, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Jernejc, K. Comparison of different methods for metabolite extraction from Aspergillus niger mycelium. Acta Chim. Slov. 2004, 51, 567–578. [Google Scholar]
- El Rammouz, R.; Letisse, F.; Durand, S.; Portais, J.-C.; Moussa, Z.W.; Fernandez, X. Analysis of skeletal muscle metabolome: Evaluation of extraction methods for targeted metabolite quantification using liquid chromatography tandem mass spectrometry. Anal. Biochem. 2010, 398, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Bolten, C.J.; Wittmann, C. Appropriate sampling for intracellular amino acid analysis in five phylogenetically different yeasts. Biotechnol. Lett. 2008, 30, 1993–2000. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Long, D.; Ji, J.; Yang, W.; Zeng, Z.; Guo, S.; Ji, Z.; Qi, G.; Chen, S. Sample preparation for the metabolomics investigation of poly-gamma-glutamate-producing bacillus licheniformis by GC-MS. J. Microbiol. Methods 2013, 94, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Villas-Boas, S.G.; Moxley, J.F.; Akesson, M.; Stephanopoulos, G.; Nielsen, J. High-throughput metabolic state analysis: The missing link in integrated functional genomics of yeasts. Biochem. J. 2005, 388, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Hiller, J.; Franco-Lara, E.; Weuster-Botz, D. Metabolic profiling of Escherichia coli cultivations: Evaluation of extraction and metabolite analysis procedures. Biotechnol. Lett. 2007, 29, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Dai, J.; Liu, W.-W.; Ling, X.; Zhu, P.-F.; Zhao, Z.-J. Comparison of extraction methods for E. coli metabolome analysis using liquid chromatography tandem mass spectrometry. Chin. J. Anal. Chem. 2011, 39, 534–539. [Google Scholar]
- Abdullah, M.I.; Young, J.C.; Games, D.E. Supercritical-fluid extraction of carboxylic and fatty-acids from Agaricus spp. mushrooms. J. Agric. Food Chem. 1994, 42, 718–722. [Google Scholar] [CrossRef]
- Cocks, S.; Wrigley, S.K.; Chicarellirobinson, M.I.; Smith, R.M. High-performance liquid-chromatography comparison of supercritical-fluid extraction and solvent-extraction of microbial fermentation products. J. Chromatogr. A 1995, 697, 115–122. [Google Scholar] [CrossRef]
- Gharaibeh, A.A.; Voorhees, K.J. Characterization of lipid fatty acids in whole-cell microorganisms using in situ supercritical fluid derivatization/extraction and gas chromatography mass spectrometry. Anal. Chem. 1996, 68, 2805–2810. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.B.; Lee, S.Y.; Lee, E.K.; Haam, S.J.; Kim, W.S. Separation of astaxanthin from red yeast Phaffia rhodozyma by supercritical carbon dioxide extraction. Biochem. Eng. J. 2002, 11, 181–187. [Google Scholar] [CrossRef]
- Smith, R.M. Before the injection-modern methods of sample preparation for separation techniques. J. Chromatogr. A 2003, 1000, 3–27. [Google Scholar] [CrossRef]
- Khosravi-Darani, K.; Vasheghani-Farahani, E. Application of supercritical fluid extraction in biotechnology. Crit. Rev. Biotechnol. 2005, 25, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Ariza, J.L.; de la Torre, M.A.C.; Giraldez, I.; Morales, E. Speciation analysis of selenium compounds in yeasts using pressurised liquid extraction and liquid chromatography-microwave-assisted digestion-hydride generation-atomic fluorescence spectrometry. Anal. Chim. Acta 2004, 524, 305–314. [Google Scholar] [CrossRef]
- Rodriguez-Meizoso, I.; Jaime, L.; Santoyo, S.; Cifuentes, A.; Reina, G.G.-B.; Senorans, F.J.; Ibanez, E. Pressurized fluid extraction of bioactive compounds from phormidium species. J. Agric. Food Chem. 2008, 56, 3517–3523. [Google Scholar] [CrossRef] [PubMed]
- Marcinowska, R.; Trygg, J.; Wolf-Watz, H.; Mortiz, T.; Surowiec, I. Optimization of a sample preparation method for the metabolomic analysis of clinically relevant bacteria. J. Microbiol. Methods 2011, 87, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Entian, K.D.; Zimmermann, F.K.; Scheel, I. A partial defect in carbon catabolite repression in mutants of Saccharomyces cerevisiae with reduced hexose phosphyorylation. Mol. Gen. Genet. MGG 1977, 156, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Gale, E.F. The assimilation of amino-acids by bacteria; the passage of certain amino-acids across the cell wall and their concentration in the internal environment of Streptococcus faecalis. J. Gen. Microbiol. 1947, 1, 53–76. [Google Scholar] [PubMed]
- Bagnara, A.S.; Finch, L.R. Quantitative extraction and estimation of intracellular nucleoside triphosphates of Escherichia coli. Anal. Biochem. 1972, 45, 24–34. [Google Scholar] [PubMed]


{kind=link}
| Year | Quenching Method | Organism | Reference |
|---|---|---|---|
| 1963 | Perchloric acid solution | Bacteria (Aerobacter aerogenes) | [44] |
| 1976 | Fast filtration followed by liquid nitrogen immersion of biomass | Yeast | [47] |
| 1992 | Cold-methanol (60% v/v) solution | Yeast (Saccharomyces cerevisiae) | [49] |
| 1996 | Buffered methanol (60% v/v) solution at −45 °C | Filamentous fungi (Aspergillus niger) | [48] |
| 1998 | Dropping mycelium cultures in liquid nitrogen or spraying the culture on a cold methanol (60% v/v) solution followed by rapid centrifugation | Filamentous fungi (Monascus ruber) | [17] |
| 2004 | Quick filtration | Bacterium (Corynebacterium glutamicum) | [41] |
| 2005 | 32.5% methanol solution in water supplemented with CaCl2, MgCl2 and KCl | Microalgae (Chlamydomonas reinhardtii) | [56] |
| 2006 | Immersion of culture flasks to ethanol–dry ice bath | Protozoa (Leishmania donovani) | [57] |
| 2007 | 60% v/v cold-methanol solution with different additives | Bacteria (Lactobacillus plantarum) | [38] |
| 2007 | Fast filtration | Bacteria (Bacillus subtilis, Corynebacterium glutamicum, Escherichia coli, Gluconobacter oxydans, Pseudomonas putida, and Zymononas mobilis) | [13] |
| 2007 | Cold glycerol–saline solution | Bacteria and yeast (Pseudomonas fluorescens, Streptomyces coelicolor and Saccharomyces cerevisiae) | [21] |
| 2008 | Pure methanol at −40 °C | Yeast (Saccharomyces cerevisiae) | [10] |
| 2010 | Cold glycerol solution and fast filtration | Bacteria, yeast and filamentous fungi | [58] |
| 2011 | Comparison of four different quenching method based on aqueous cold-methanol solution | Yeast (Pichia pastoris) | [52] |
| 2012 | 40% v/v of methanol solution at −20 °C | Mould (Penicillium chrysogenum) | [39] |
| 2014 | Automated fast filtration and on-filter quenching | Bacteria (Escherichia coli) | [11] |
| Extraction Method | Extracted Metabolites | Microorganisms | References |
|---|---|---|---|
| CHEMICAL EXTRACTIONS | |||
| Boiling ethanol | Polar (thermostable) | Sacharomyces cerevisiae, Aspergillus sp., Penicillium chrysogenum, Monascus ruber, Klebsiella oxytoca, Escherichia coli, Enterococcus faecalis and Lactobacillus plantarum | [12,17,18,37,38,40,51,73,74,75,76,77,78,79] |
| Cold methanol | Polar and mid polar | Sacharomyces cerevisiae, Aspergillus sp., Zymomonas mobilis, Penicillium sp., Klebsiella oxytoca, Bacillus subtilis, Pseudomonas putida, Gluconobacter oxydans, Corynebacterium glutamicum, Escherichia coli, Enterococcus faecalis and Lactobacillus plantarum | [13,18,39,51,73,75,80,81] |
| Buffered methanol–water–chloroform | Polar and non-polar | Sacharomyces cerevisiae, Escherichia coli, Bacillus licheniformis and Klebsiella oxytoca | [12,18,51,74,81,82] |
| Hot water | Polar (thermostable) | Sacharomyces cerevisiae, Escherichia coli and Klebsiella oxytoca | [12,74,83] |
| Acidic extraction | Polar and acid stable | Monascus ruber, Sacharomyces cerevisiae, Aspergillus niger, Klebsiella oxytoca, Bacillus licheniformis and Escherichia coli | [17,18,38,74,78,81,84] |
| Alkaline extraction | Polar and alkali stable | Monascus ruber, Sacharomyces cerevisiae, Aspergillus niger, Klebsiella oxytoca and Escherichia coli | [17,18,51,74,76,78] |
| MECHANICAL EXTRACTIONS | |||
| Superficial fluid extraction | Non-polar to mid polar | Agaricus sp., Gram-positive and Gram-negative bacteria | [85,86,87,88,89,90] |
| Pressurised liquid extraction | Secondary metabolites | Yeasts and microalgae | [89,91,92] |
| Microwave | Thermostable metabolites | Yeasts | [91] |
| COMBINATION OF CHEMICAL AND MECHANICAL EXTRACTION | |||
| Pure cold methanol coupled to sonication | Polar, mid polar and stable | Sacharomyces cerevisiae, Aspergillus sp., Escherichia coli and Enterococcus faecalis | [73] |
| Methanol and bead mill | Polar and mid polar | Clinically relevant bacteria | [93] |
| Cold methanol–water solution coupled to freeze–thaw cycles | Polar and mid polar | S. cerevisiae, Aspergillus sp., Escherichia coli and Enterococcus faecalis | [73] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinu, F.R.; Villas-Boas, S.G.; Aggio, R. Analysis of Intracellular Metabolites from Microorganisms: Quenching and Extraction Protocols. Metabolites 2017, 7, 53. https://doi.org/10.3390/metabo7040053
Pinu FR, Villas-Boas SG, Aggio R. Analysis of Intracellular Metabolites from Microorganisms: Quenching and Extraction Protocols. Metabolites. 2017; 7(4):53. https://doi.org/10.3390/metabo7040053
Chicago/Turabian StylePinu, Farhana R., Silas G. Villas-Boas, and Raphael Aggio. 2017. "Analysis of Intracellular Metabolites from Microorganisms: Quenching and Extraction Protocols" Metabolites 7, no. 4: 53. https://doi.org/10.3390/metabo7040053
APA StylePinu, F. R., Villas-Boas, S. G., & Aggio, R. (2017). Analysis of Intracellular Metabolites from Microorganisms: Quenching and Extraction Protocols. Metabolites, 7(4), 53. https://doi.org/10.3390/metabo7040053