Enhanced Isotopic Ratio Outlier Analysis (IROA) Peak Detection and Identification with Ultra-High Resolution GC-Orbitrap/MS: Potential Application for Investigation of Model Organism Metabolomes

 ,
, 
Abstract
1. Introduction
2. Results
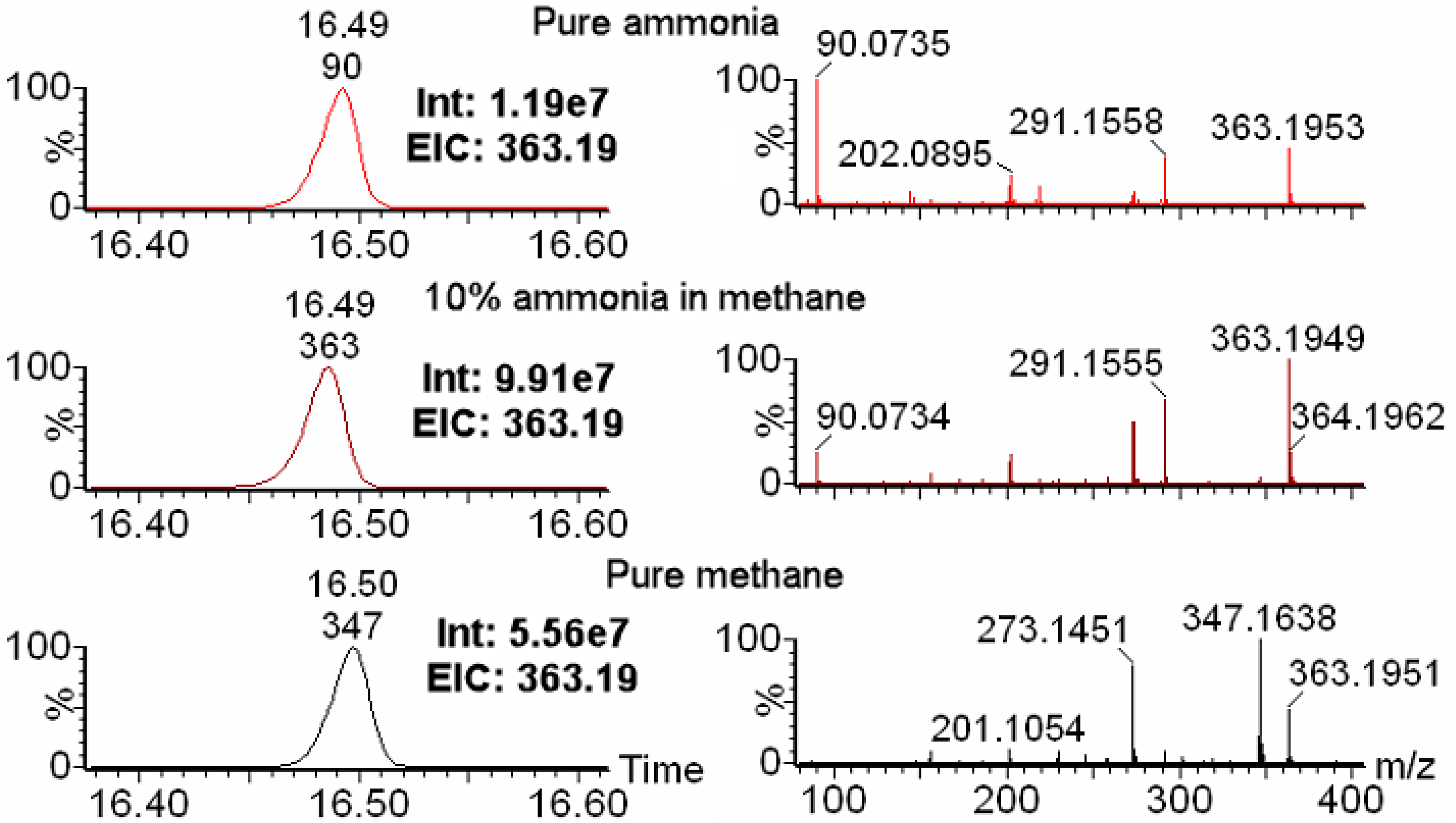
2.1. Choice of Reagent Gas Is Critical for Optimal IROA Pair Detection
2.2. The Wider Dynamic Range of GC-Orbitrap/MS Allows for Increased Detection of IROA Pairs
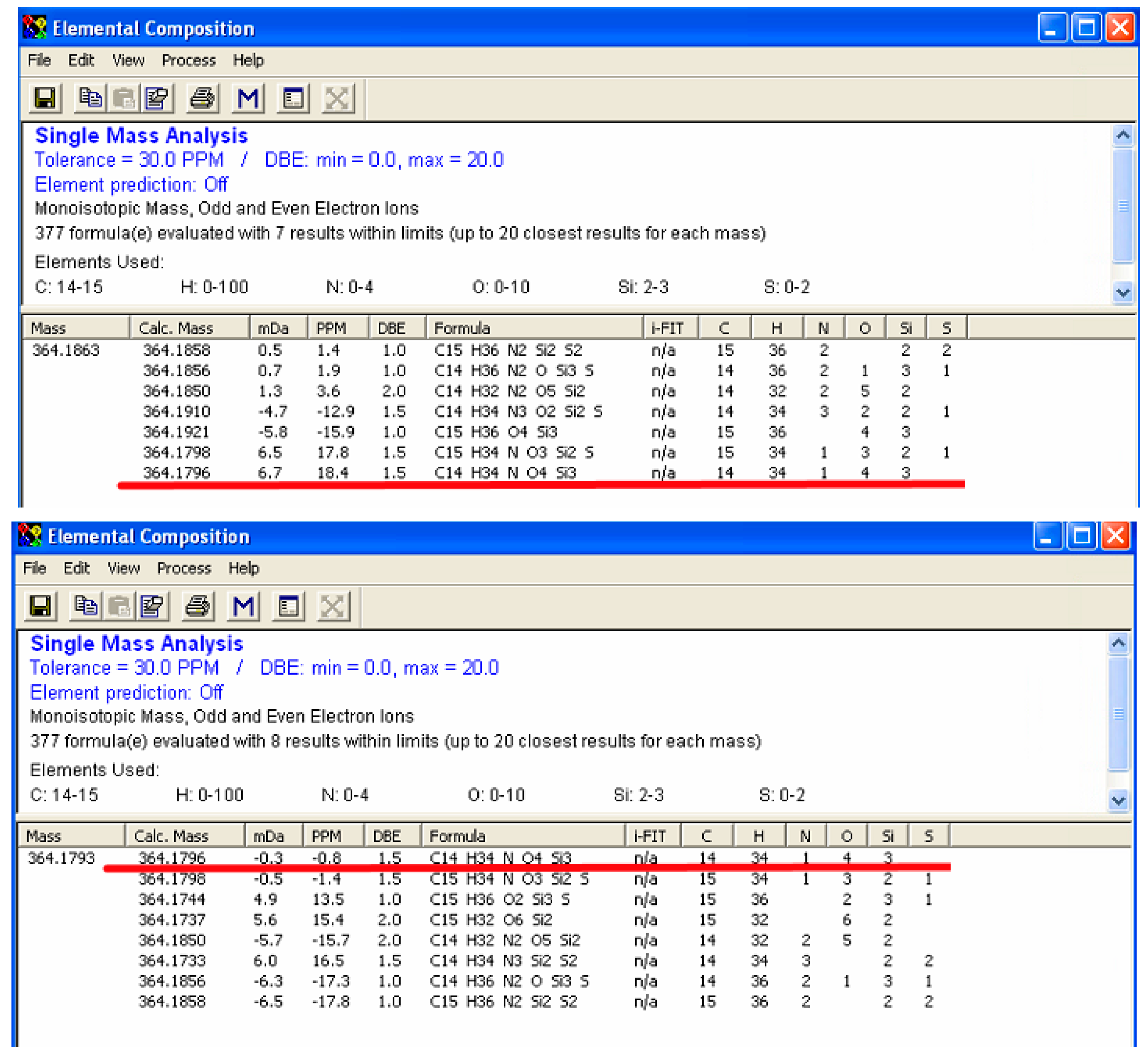
2.3. Enhanced Mass Accuracy Is Associated with Increased Metabolite Identification Capability for the GC-Orbitrap/MS
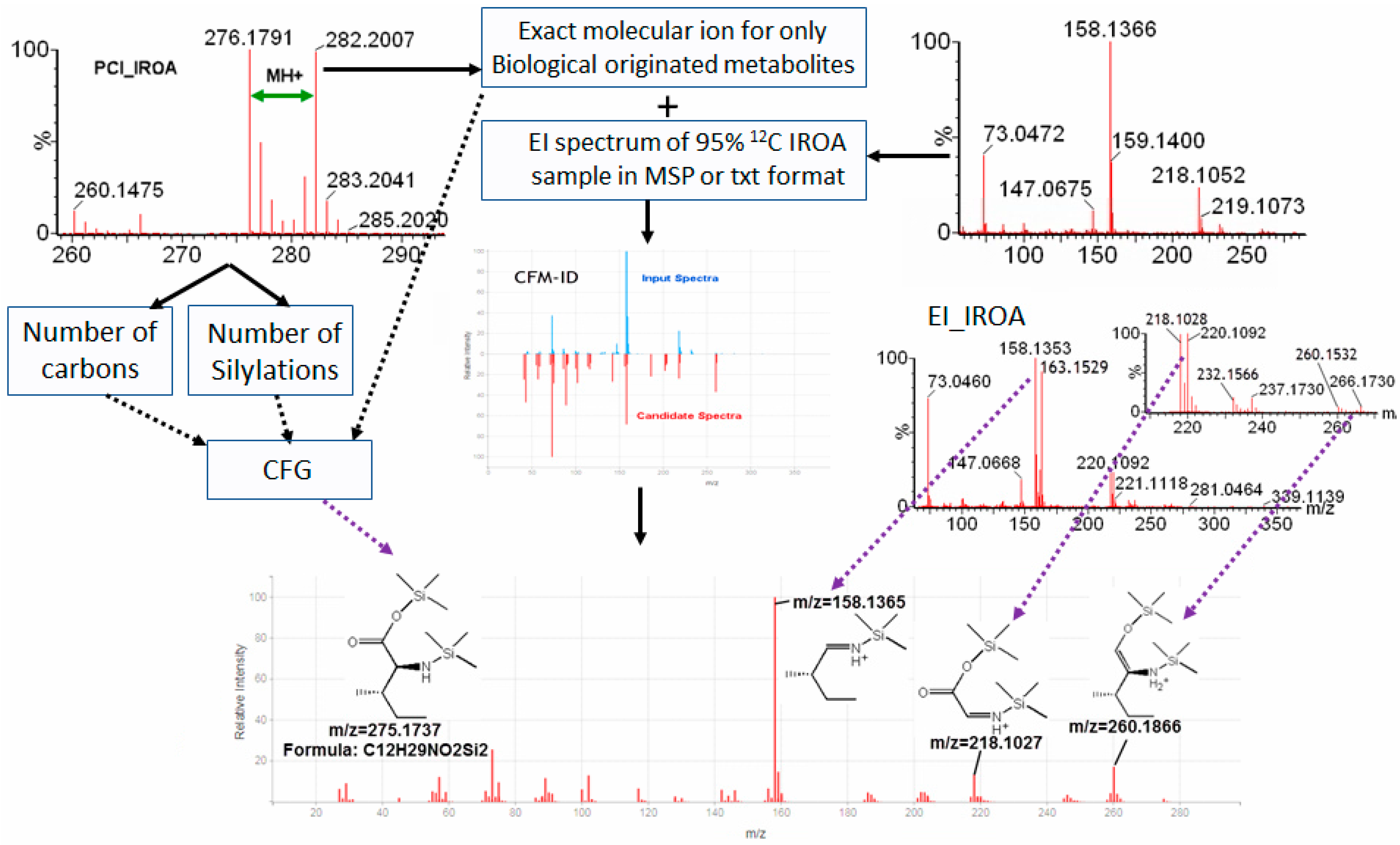
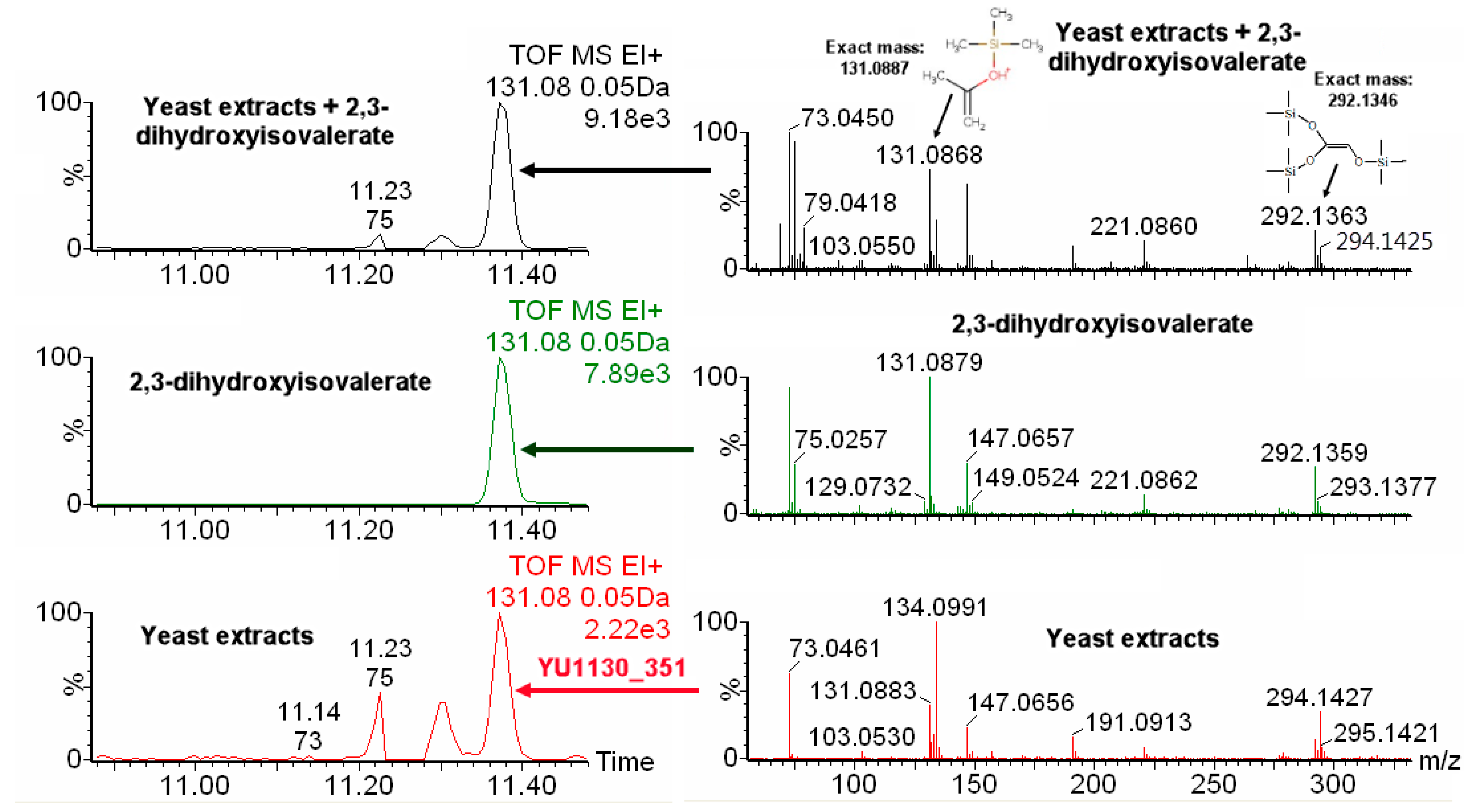
2.4. IROA Enhanced Unknown Metabolites Identification with In Silico Fragmentation
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kurland, I.J.; Accili, D.; Burant, C.; Fischer, S.M.; Kahn, B.B.; Newgard, C.B.; Ramagiri, S.; Ronnett, G.V.; Ryals, J.A.; Sanders, M.; et al. Application of combined omics platforms to accelerate biomedical discovery in diabesity. Ann. N. Y. Acad. Sci. 2013, 1287, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Nie, W.; Yan, L.; Lee, Y.H.; Guha, C.; Kurland, I.J.; Lu, H. Advanced mass spectrometry-based multi-omics technologies for exploring the pathogenesis of hepatocellular carcinoma. Mass Spectrom. Rev. 2014, 35, 331–349. [Google Scholar] [CrossRef] [PubMed]
- Viant, M.R.; Kurland, I.J.; Jones, M.R.; Dunn, W.M. How close are we to complete annotation of metabolomes? Curr. Opin. Chem. Biol. 2016, 36, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Edison, A.S.; Hall, R.D.; Junot, C.; Karp, P.D.; Kurland, I.J.; Mistrik, R.; Reed, L.K.; Saito, K.; Salek, R.M.; Steinbeck, C.; et al. The time is right to focus on model organism metabolomes. Metabolites 2016, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Blaženović, I.K.T.; Torbašinović, H.; Obrenović, S.; Mehta, S.S.; Tsugawa, H.; Wermuth, T.; Schauer, N.; Jahn, M.; Biedendieck, R.; Jahn, D.; et al. Comprehensive comparison of in silico MS/MS fragmentation tools of the CASMI contest: Database boosting is needed to achieve 93% accuracy. J. Cheminform. 2017, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Clendinen, C.S.; Stupp, G.S.; Ajredini, R.; Lee-McMullen, B.; Beecher, C.; Edison, A.S. An overview of methods using 13C for improved compound identification in metabolomics and natural products. Front. Plant Sci. 2015, 6, 611. [Google Scholar] [CrossRef] [PubMed]
- De Jong, F.A.; Beecher, C. Addressing the current bottlenecks of metabolomics: Isotopic ratio outlier analysis, an isotopic-labeling technique for accurate biochemical profiling. Bioanalysis 2012, 4, 2303–2314. [Google Scholar] [CrossRef] [PubMed]
- Edison, A.S.; Clendinen, C.S.; Ajredini, R.; Beecher, C.; Ponce, F.V.; Stupp, G.S. Metabolomics and natural-products strategies to study chemical ecology in nematodes. Integr. Comp. Biol. 2015, 55, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Stupp, G.S.; Clendinen, C.S.; Ajredini, R.; Szewc, M.A.; Garrett, T.; Menger, R.F.; Yost, R.A.; Beecher, C.; Edison, A.S. Isotopic ratio outlier analysis global metabolomics of Caenorhabditis elegans. Anal. Chem. 2013, 85, 11858–11865. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Moir, R.; Willis, I.; Beecher, C.; Tsai, Y.H.; Garrett, T.J.; Yost, R.A.; Kurland, I.J. Isotopic Ratio Outlier Analysis of the S. cerevisiae Metabolome Using Accurate Mass Gas Chromatography/Time-of-Flight Mass Spectrometry: A New Method for Discovery. Anal. Chem. 2016, 88, 2747–2754. [Google Scholar] [CrossRef] [PubMed]
- Giavalisco, P.; Kohl, K.; Hummel, J.; Seiwert, B.; Willmitzer, L. 13C isotope-labeled metabolomes allowing for improved compound annotation and relative quantification in liquid chromatography-mass spectrometry-based metabolomic research. Anal. Chem. 2009, 81, 6546–6551. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Mashego, M.R.; van Dam, J.C.; Proell, A.M.; Vinke, J.L.; Ras, C.; van Winden, W.A.; van Gulik, W.M.; Heijnen, J.J. Quantitative analysis of the microbial metabolome by isotope dilution mass spectrometry using uniformly 13C-labeled cell extracts as internal standards. Anal. Biochem. 2005, 336, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.D.; Yuan, J.; Kimball, E.H.; Rabinowitz, J.D. Absolute quantitation of intracellular metabolite concentrations by an isotope ratio-based approach. Nat. Protoc. 2008, 3, 1299–1311. [Google Scholar] [CrossRef] [PubMed]
- Weindl, D.; Wegner, A.; Jager, C.; Hiller, K. Isotopologue ratio normalization for non-targeted metabolomics. J. Chromatogr. A 2015, 1389, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Blank, L.M.; Desphande, R.R.; Schmid, A.; Hayen, H. Analysis of carbon and nitrogen co-metabolism in yeast by ultrahigh-resolution mass spectrometry applying 13C- and 15N-labeled substrates simultaneously. Anal. Bioanal. Chem. 2012, 403, 2291–2305. [Google Scholar] [CrossRef] [PubMed]
- Brauer, M.J.; Saldanha, A.J.; Dolinski, K.; Botstein, D. Homeostatic adjustment and metabolic remodeling in glucose-limited yeast cultures. Mol. Biol. Cell 2005, 16, 2503–2517. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.; Schmidt, S.; Muller-Hannemann, M.; Neumann, S. In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC Bioinform. 2010, 11, 148. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.; Pon, A.; Wilson, M.; Greiner, R.; Wishart, D. CFM-ID: A web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res. 2014, 42, W94–W99. [Google Scholar] [CrossRef] [PubMed]
- Ruttkies, C.; Schymanski, E.L.; Wolf, S.; Hollender, J.; Neumann, S. MetFrag relaunched: Incorporating strategies beyond in silico fragmentation. J. Cheminform. 2016, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Kind, T.; Nakabayashi, R.; Yukihira, D.; Tanaka, W.; Cajka, T.; Saito, K.; Fiehn, O.; Arita, M. Hydrogen rearrangement rules: Computational MS/MS fragmentation and structure elucidation using MS-FINDER software. Anal. Chem. 2016, 88, 7946–7958. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Weber, R.J.; Allwood, J.W.; Mistrik, R.; Zhu, Z.; Ji, Z.; Chen, S.; Dunn, W.B.; He, S.; Viant, M.R. HAMMER: Automated operation of mass frontier to construct in silico mass spectral fragmentation libraries. Bioinformatics 2014, 30, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; Kind, T.; Fiehn, O. Using accurate mass gas chromatography-mass spectrometry with the MINE database for Epimetabolite Annotation. Anal. Chem. 2017, 89, 10171–10180. [Google Scholar] [CrossRef] [PubMed]
- Rudewicz, P.; Munson, B. Effect of ammonia partial pressure on the sensitivities for oxygenated compounds in ammonia chemical ionization mass spectrometry. Anal. Chem. 1986, 58, 2903–2907. [Google Scholar] [CrossRef]
- Ligon, W.V.; Grade, H. Chemical ionization mass spectrometery utilizing and isotopically labeled reagent gas. J. Am. Soc. Mass Spectrom. 1994, 5, 596–598. [Google Scholar] [CrossRef]
- Sparkman, O.D.; Penton, Z.; Kitson, F.G. Gas Chromatography and Mass Spectrometry: A Practical Guide; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Linster, C.L.; Van Schaftingen, E.; Hanson, A.D. Metabolite damage and its repair or pre-emption. Nat. Chem. Biol. 2013, 9, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Showalter, M.R.; Cajka, T.; Fiehn, O. Epimetabolites: Discovering metabolism beyond building and burning. Curr. Opin. Chem. Biol. 2017, 36, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Henkin, T.M. Riboswitch RNAs: Using RNA to sense cellular metabolism. Genes Dev. 2008, 22, 3383–3390. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Fuchs, R.T.; Grundy, F.J.; Henkin, T.M. Riboswitch RNAs: Regulation of gene expression by direct monitoring of a physiological signal. RNA Biol. 2010, 7, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Jeffryes, J.G.; Colastani, R.L.; Elbadawi-Sidhu, M.; Kind, T.; Niehaus, T.D.; Broadbelt, L.J.; Hanson, A.D.; Fiehn, O.; Tyo, K.E.; Henry, C.S. MINEs: Open access databases of computationally predicted enzyme promiscuity products for untargeted metabolomics. J. Cheminform. 2015, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Crutchfield, C.A.; Lu, W.; Melamud, E.; Rabinowitz, J.D. Mass spectrometry-based metabolomics of yeast. Methods Enzymol. 2010, 470, 393–426. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GC-TOF/MS 5% Ammonia in Methane | GC-Orbitrap/MS Methane | GC-Orbitrap/MS 10% Ammonia in Methane | |
|---|---|---|---|
| Extracted material used | 200 μL | 1 mL | 1 mL |
| Saturation in splitless injection | Yes | No | No |
| IROA peak pairs | 126 | 116 | 244 |
| Annotated | 82 | 46 | 101 |
| Average mass difference for 55 metabolites (ppm) | 32.2 ± 38.8 | - | 1.48 ± 1.25 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, Y.; Moir, R.D.; Willis, I.M.; Seethapathy, S.; Biniakewitz, R.C.; Kurland, I.J. Enhanced Isotopic Ratio Outlier Analysis (IROA) Peak Detection and Identification with Ultra-High Resolution GC-Orbitrap/MS: Potential Application for Investigation of Model Organism Metabolomes. Metabolites 2018, 8, 9. https://doi.org/10.3390/metabo8010009
Qiu Y, Moir RD, Willis IM, Seethapathy S, Biniakewitz RC, Kurland IJ. Enhanced Isotopic Ratio Outlier Analysis (IROA) Peak Detection and Identification with Ultra-High Resolution GC-Orbitrap/MS: Potential Application for Investigation of Model Organism Metabolomes. Metabolites. 2018; 8(1):9. https://doi.org/10.3390/metabo8010009
Chicago/Turabian StyleQiu, Yunping, Robyn D. Moir, Ian M. Willis, Suresh Seethapathy, Robert C. Biniakewitz, and Irwin J. Kurland. 2018. "Enhanced Isotopic Ratio Outlier Analysis (IROA) Peak Detection and Identification with Ultra-High Resolution GC-Orbitrap/MS: Potential Application for Investigation of Model Organism Metabolomes" Metabolites 8, no. 1: 9. https://doi.org/10.3390/metabo8010009
APA StyleQiu, Y., Moir, R. D., Willis, I. M., Seethapathy, S., Biniakewitz, R. C., & Kurland, I. J. (2018). Enhanced Isotopic Ratio Outlier Analysis (IROA) Peak Detection and Identification with Ultra-High Resolution GC-Orbitrap/MS: Potential Application for Investigation of Model Organism Metabolomes. Metabolites, 8(1), 9. https://doi.org/10.3390/metabo8010009