
NMR Chemical Shift Ranges of Urine Metabolites in Various Organic Solvents


Abstract
:1. Introduction
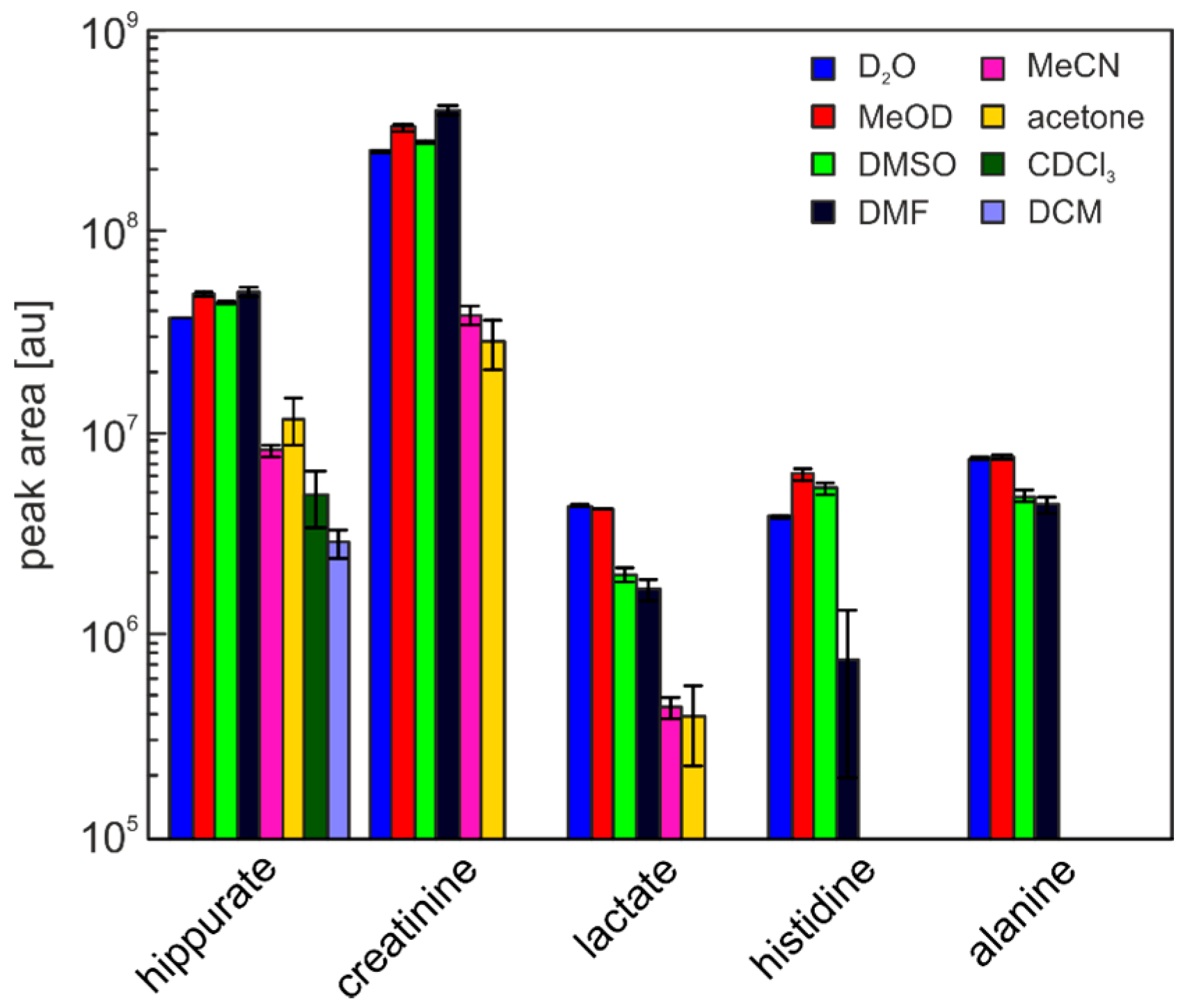
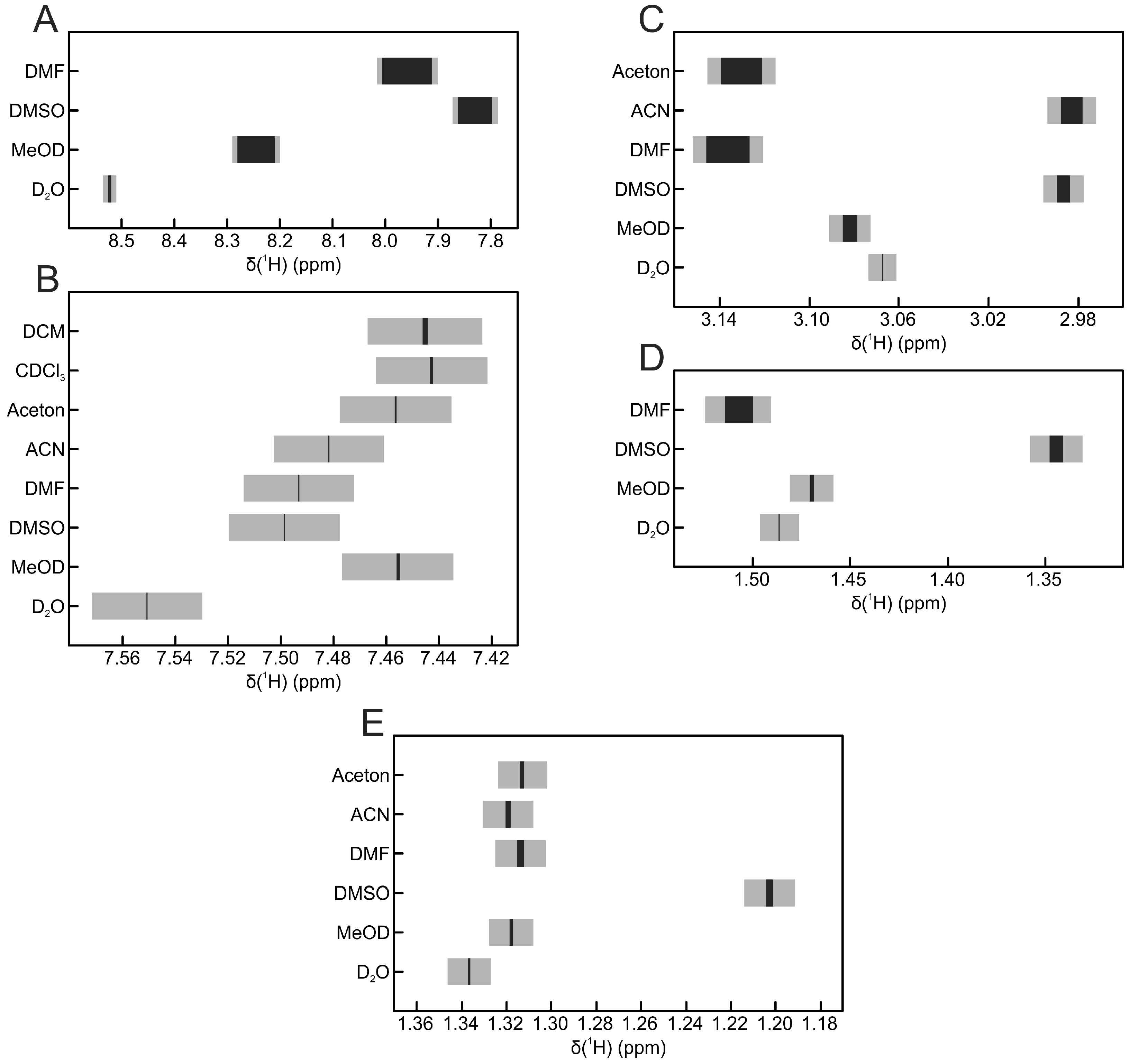
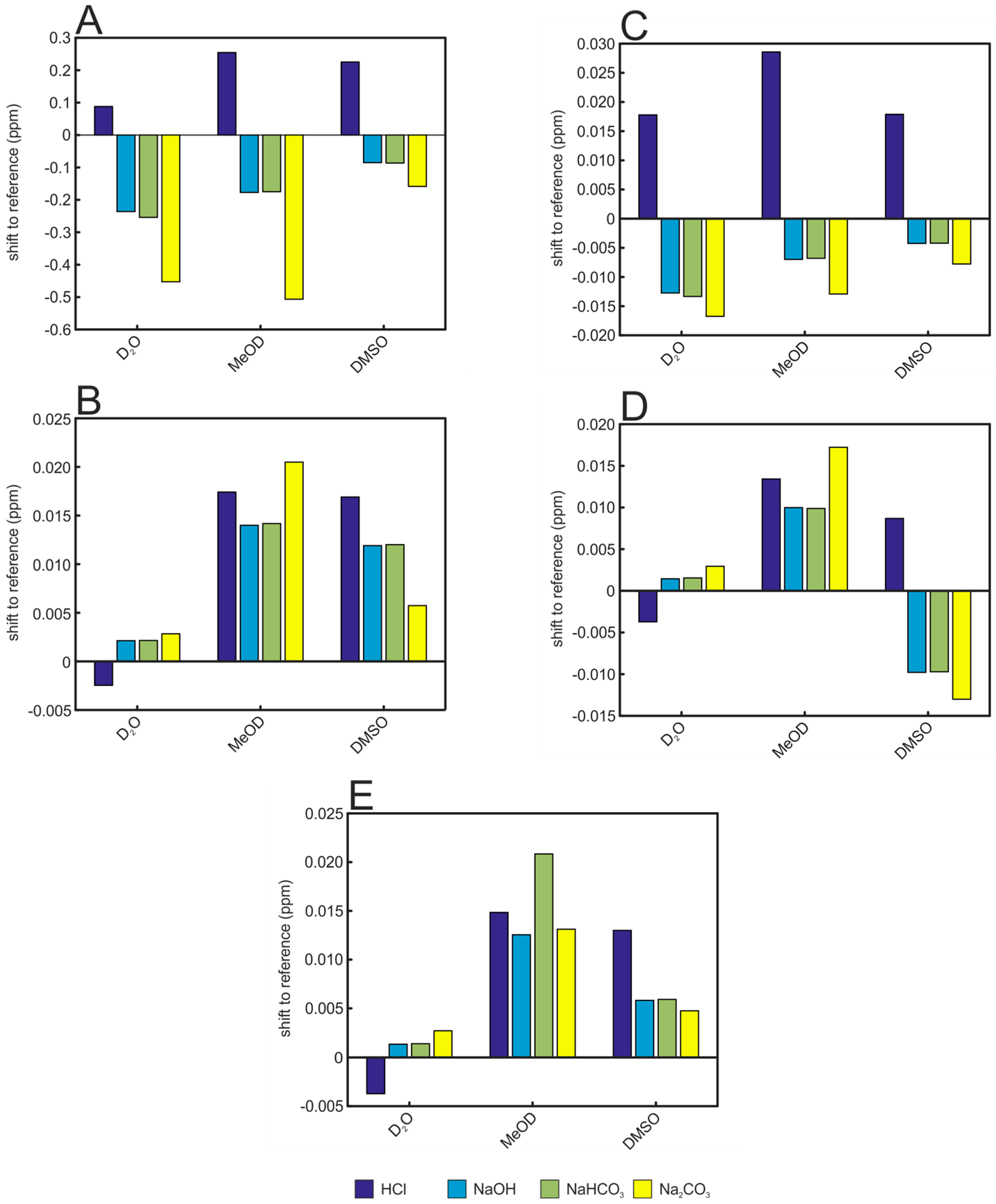
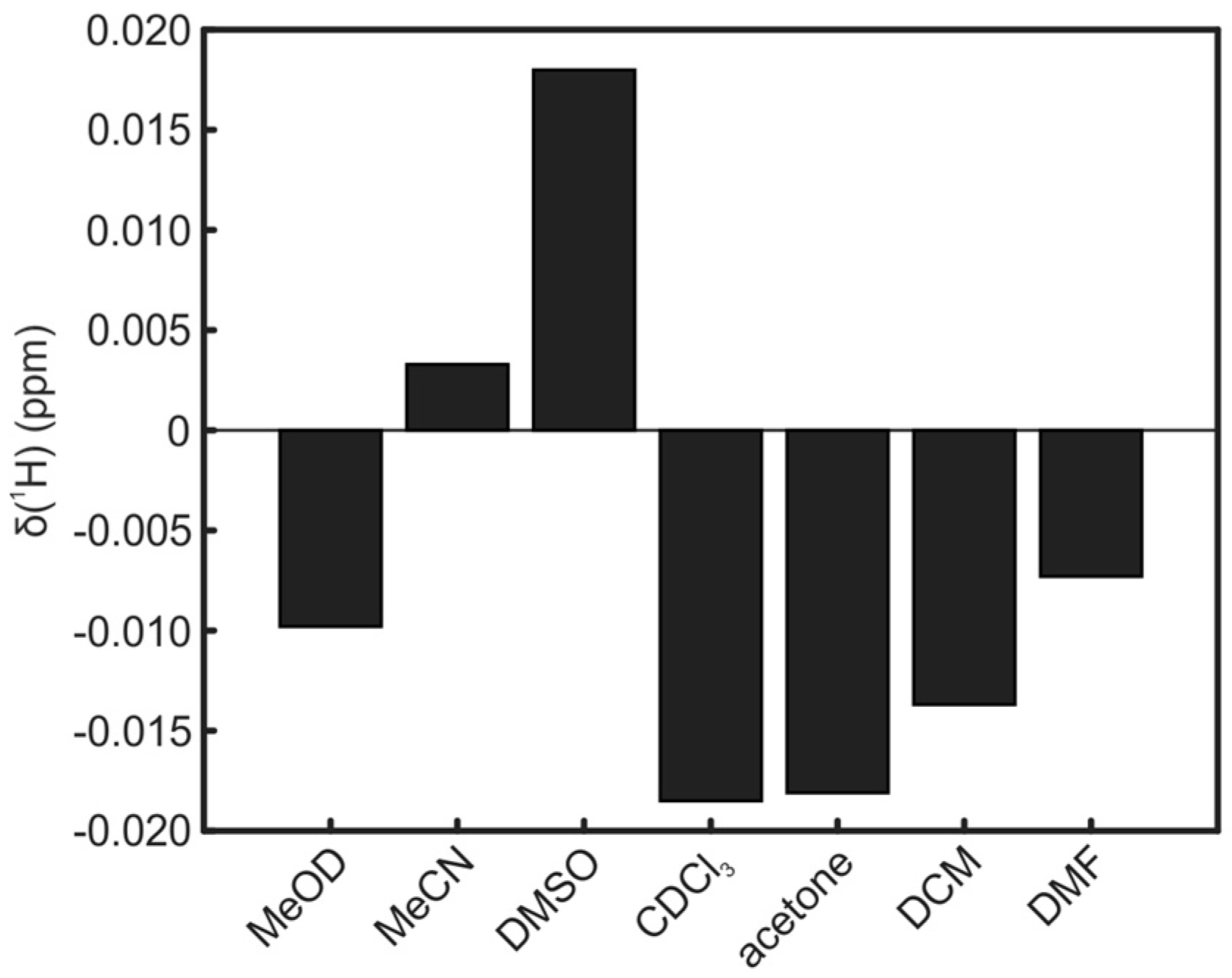
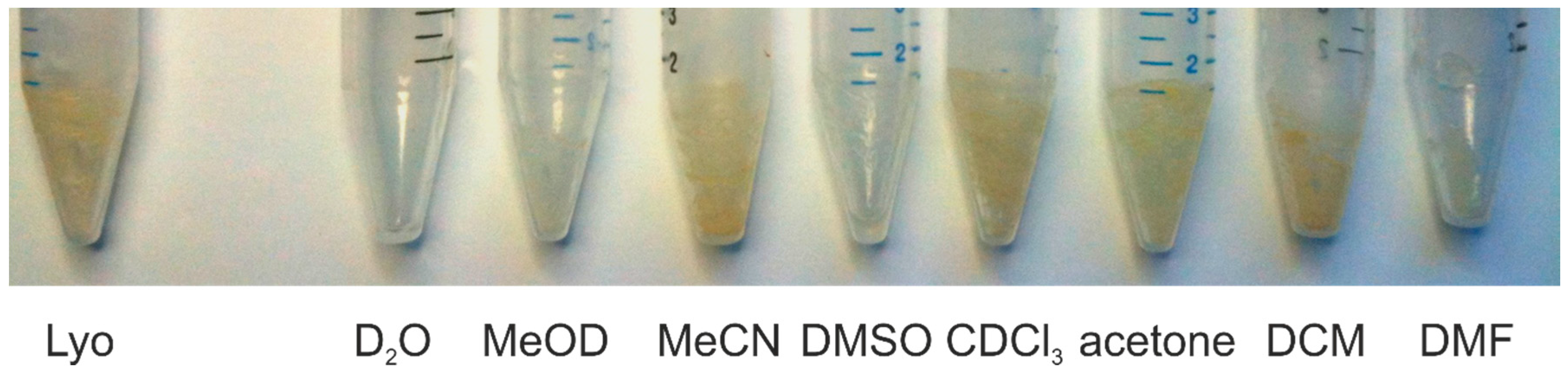
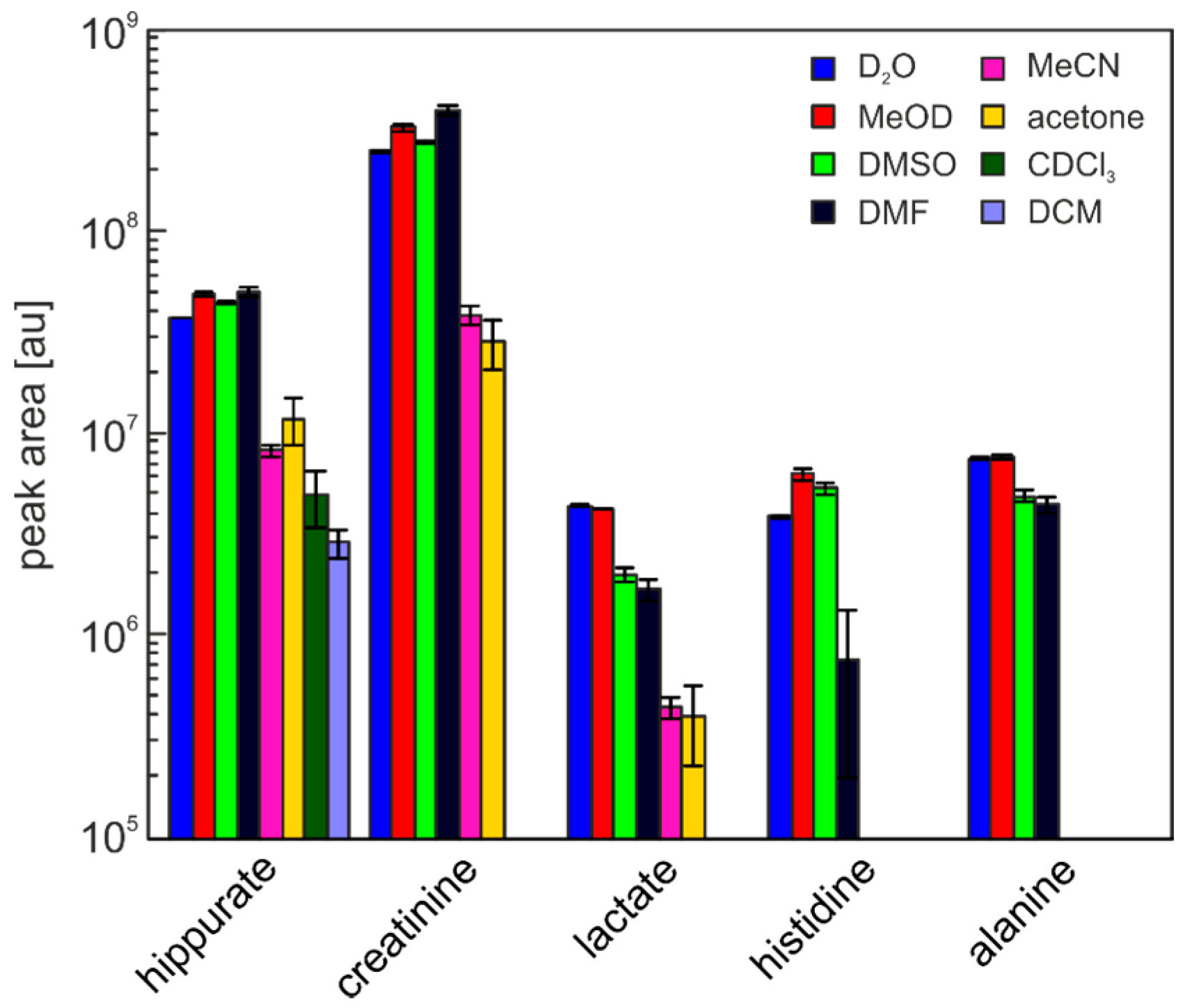
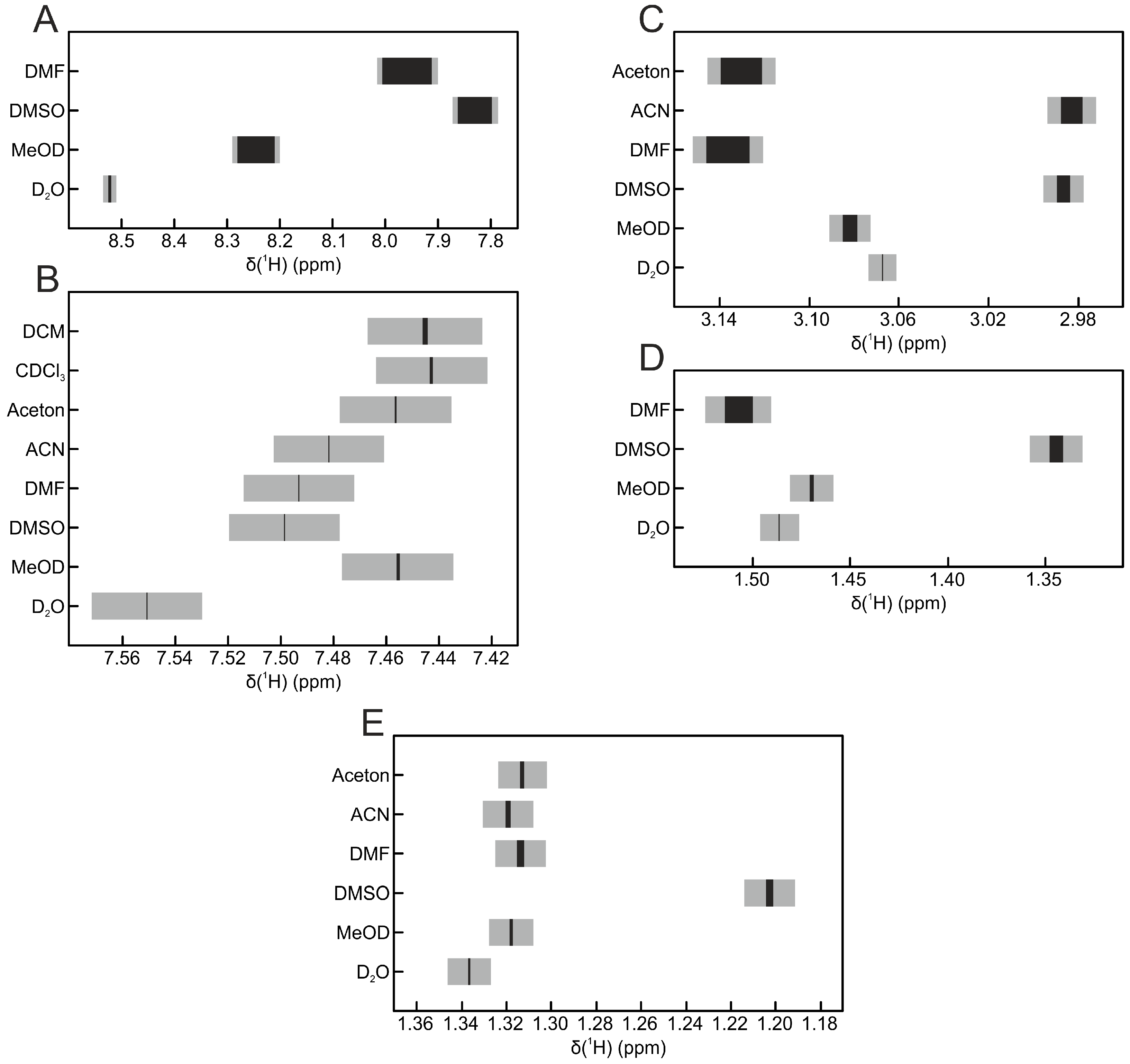
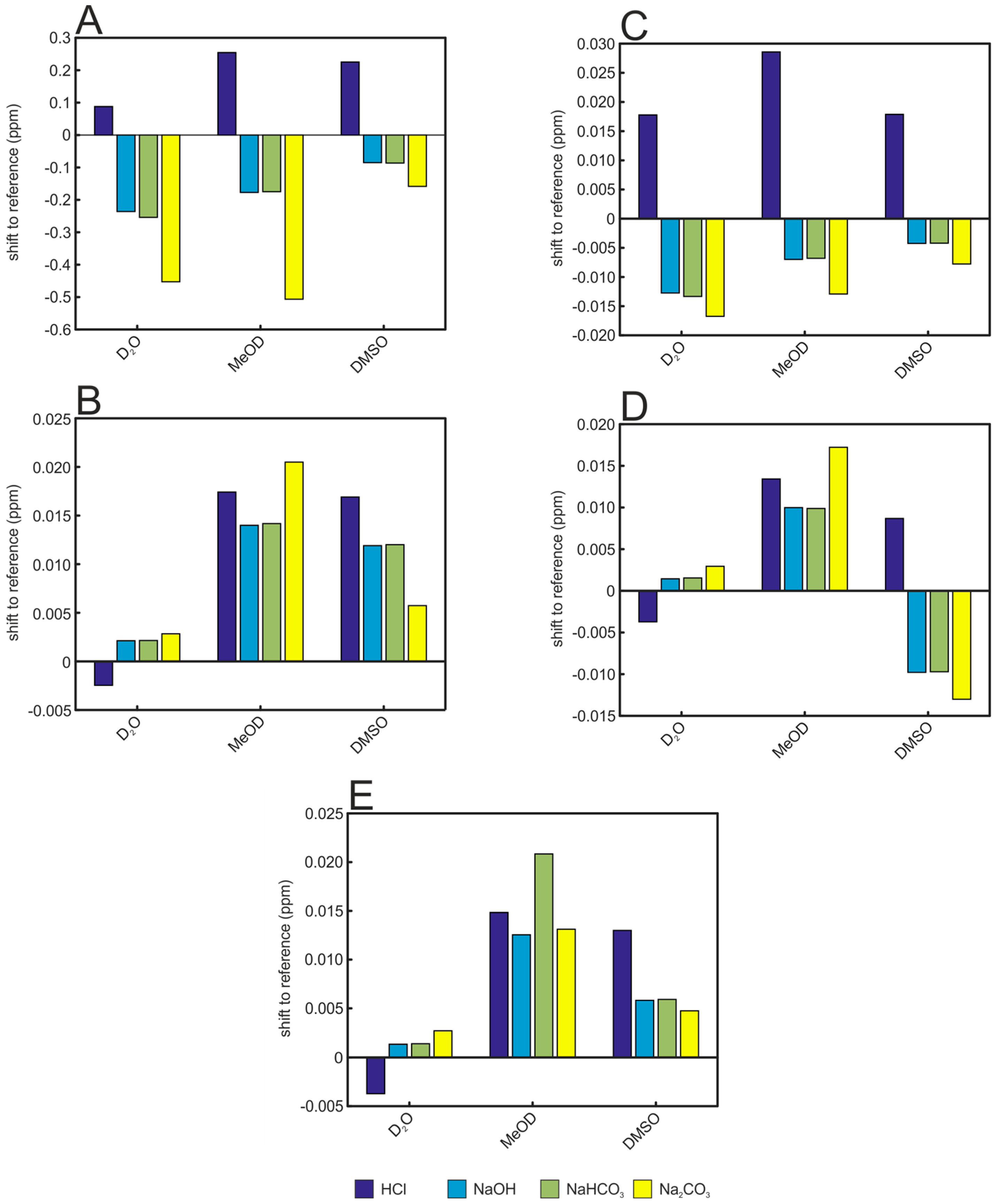
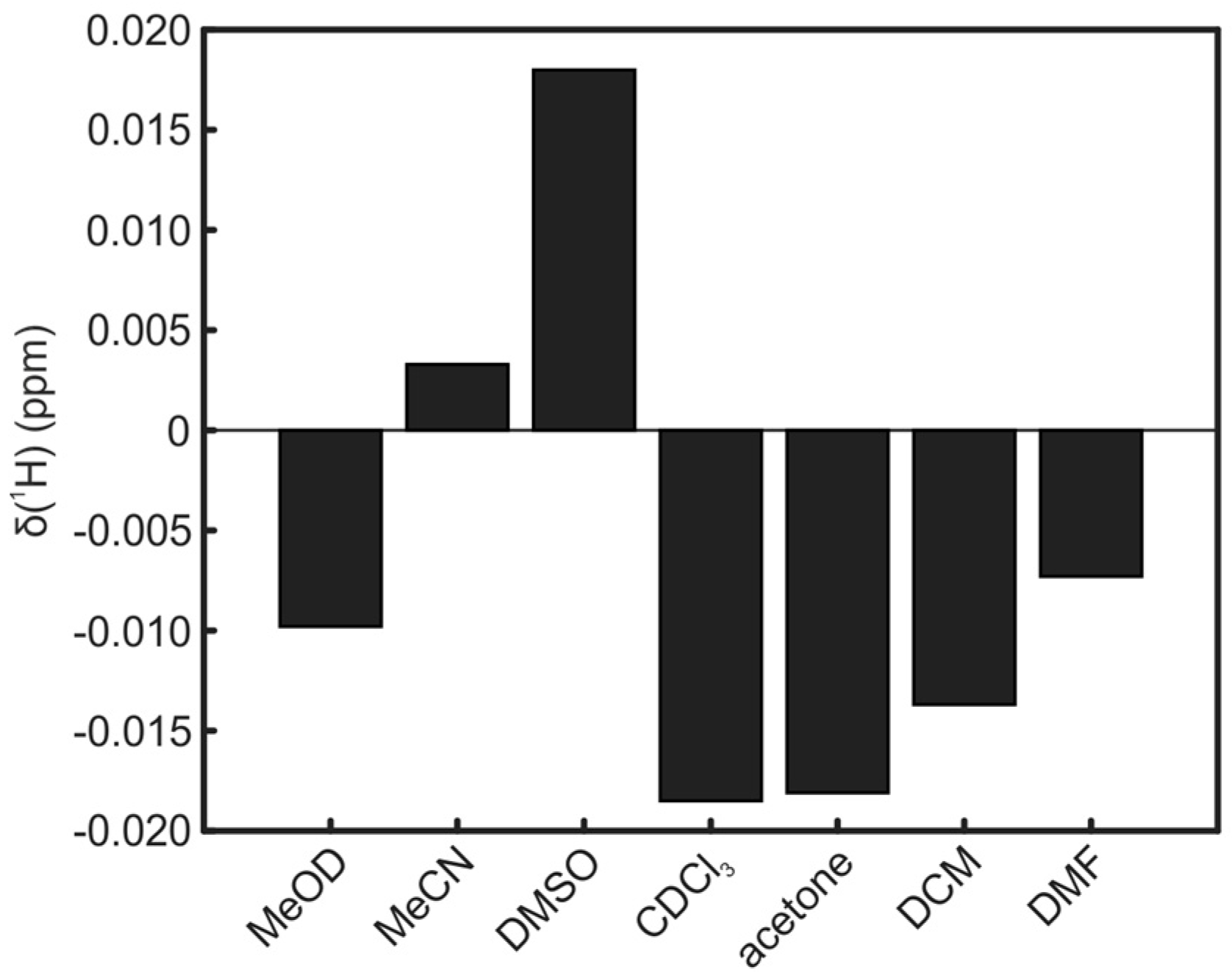
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lindon, J.C.; Nicholson, J.K.; Everett, J.R. NMR Spectroscopy of Biofluids. In Annual Reports on NMR Spectroscopy; Webb, G.A., Ed.; Academic Press: Cambridge, MA, USA, 1999; Volume 38, pp. 1–88. [Google Scholar]
- Reo, N.V. NMR-based metabolomics. Drug Chem. Toxicol. 2002, 25, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Lindon, J.C.; Keun, H.C.; Ebbels, T.M.D.; Pearce, J.M.T.; Holmes, E.; Nicholson, J.K. The Consortium for Metabonomic Toxicology (COMET): Aims, activities and achievements. Pharmacogenomics 2005, 6, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.; Foxall, P.J.D.; Spraul, M.; Duncan Farrant, R.; Nicholson, J.K.; Lindon, J.C. 750 MHz 1H-NMR spectroscopy characterisation of the complex metabolic pattern of urine from patients with inborn errors of metabolism: 2-Hydroxyglutaric aciduria and maple syrup urine disease. J. Pharm. Biomed. Anal. 1997, 15, 1647–1659. [Google Scholar] [CrossRef]
- Rist, M.; Muhle-Goll, C.; Görling, B.; Bub, A.; Heissler, S.; Watzl, B.; Luy, B. Influence of Freezing and Storage Procedure on Human Urine Samples in NMR-Based Metabolomics. Metabolites 2013, 3, 243. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.M.; Klein, M.S.; Hochrein, J.; Oefner, P.J.; Spang, R.; Gronwald, W. State-of-the art data normalization methods improve NMR-based metabolomic analysis. Metabolomics 2012, 8, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Bales, J.R.; Higham, D.P.; Howe, I.; Nicholson, J.K.; Sadler, P.J. Use of High-Resolution Proton Nuclear Magnetic Resonance Spectroscopy for Rapid Multi-Component Analysis of Urine. Clin. Chem. 1984, 30, 426–432. [Google Scholar] [PubMed]
- Miyataka, H.; Ozaki, T.; Himeno, S. Effect of pH on 1H-NMR Spectroscopy of Mouse Urine. Biol. Pharm. Bull. 2007, 30, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Asiago, V.M.; Nagana Gowda, G.A.; Zhang, S.; Shanaiah, N.; Clark, J.; Raftery, D. Use of EDTA to minimize ionic strength dependent frequency shifts in the 1H-NMR spectra of urine. Metabolomics 2008, 4, 328–336. [Google Scholar] [CrossRef]
- Xiao, C.; Hao, F.; Qin, X.; Wang, Y.; Tang, H. An optimized buffer system for NMR-based urinary metabonomics with effective pH control, chemical shift consistency and dilution minimization. Analyst 2009, 134, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Huang, J.; Wang, Y.; Tang, H. Eliminating the dication-induced intersample chemical-shift variations for NMR-based biofluid metabonomic analysis. Analyst 2012, 137, 4209–4219. [Google Scholar] [CrossRef] [PubMed]
- Lauridsen, M.; Hansen, S.H.; Jaroszewski, J.W.; Cornett, C. Human Urine as Test Material in 1H-NMR-Based Metabonomics: Recommendations for Sample Preparation and Storage. Anal. Chem. 2007, 79, 1181–1186. [Google Scholar] [CrossRef] [PubMed]
- Lindon, J.C.; Nicholson, J.K.; Holmes, E.; Everett, J.R. Metabonomics: Metabolic processes studied by NMR spectroscopy of biofluids. Concepts Magn. Reson. 2000, 12, 289–320. [Google Scholar] [CrossRef]
- Schreier, C.; Kremer, W.; Huber, F.; Neumann, S.; Pagel, P.; Lienemann, K.; Pestel, S. Reproducibility of NMR Analysis of Urine Samples: Impact of Sample Preparation, Storage Conditions, and Animal Health Status. BioMed Res. Int. 2013, 2013, 19. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Song, Y.P.; Tang, H.; Wang, Y. Recent developments in sample preparation and data pre-treatment in metabonomics research. Arch. Biochem. Biophys. 2016, 589, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Belle, J.E.L.; Harris, N.G.; Williams, S.R.; Bhakoo, K.K. A comparison of cell and tissue extraction techniques using high-resolution 1H-NMR spectroscopy. NMR Biomed. 2002, 15, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Wu, H.; Tjeerdema, R.S.; Viant, M.R. Evaluation of metabolite extraction strategies from tissue samples using NMR metabolomics. Metabolomics 2007, 3, 55–67. [Google Scholar] [CrossRef]
- Beckonert, O.; Keun, H.C.; Ebbels, T.M.D.; Bundy, J.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat. Protoc. 2007, 2, 2692–2703. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, G.; GÖrling, B.; Luy, B.; Du, J.; Yan, J. Integrative Analysis of Circadian Transcriptome and Metabolic Network Reveals the Role of De Novo Purine Synthesis in Circadian Control of Cell Cycle. PLoS Comput. Biol. 2015, 11, e1004086. [Google Scholar] [CrossRef]
- Needham, T.E.; Paruta, A.N.; Gerraughty, R.J. Solubility of amino acids in pure solvent systems. J. Pharm. Sci. 1971, 60, 565–567. [Google Scholar] [CrossRef] [PubMed]
- Bouatra, S.; Aziat, F.; Mandal, R.; Guo, A.C.; Wilson, M.R.; Knox, C.; Bjorndahl, T.C.; Krishnamurthy, R.; Saleem, F.; Liu, P.; et al. The Human Urine Metabolome. PLoS ONE 2013, 8, e73076. [Google Scholar] [CrossRef] [PubMed]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Reichardt, C.; Welton, T. Empirical Parameters of Solvent Polarity. In Solvents and Solvent Effects in Organic Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 425–508. [Google Scholar]
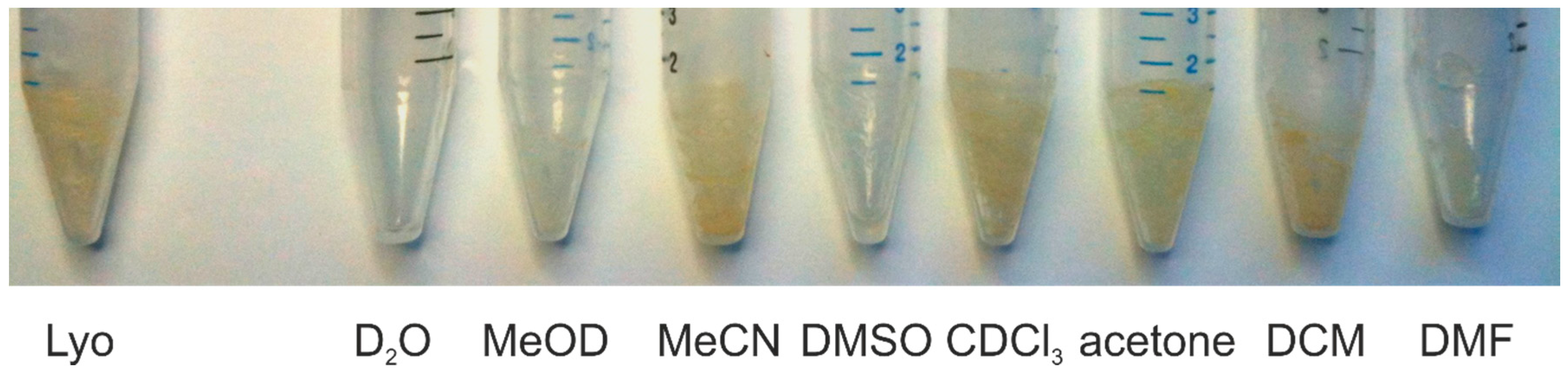

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | Hippurate | Creatinine | Lactate | Histidine | Alanine |
|---|---|---|---|---|---|
| D2O | 0.86% | 1.15% | 1.20% | 1.70% | 1.35% |
| MeOD | 2.76% | 3.29% | 0.60% | 7.01% | 1.99% |
| DMSO | 6.92% | 11.16% | 12.01% | 7.30% | 6.64% |
| DMF | 1.10% | 1.60% | 7.03% | 74.12% | 9.37% |
| MeCN | 31.83% | 27.97% | 43.27% | - | - |
| Acetone | 26.84% | 4.66% | 10.74% | - | - |
| CDCl3 | 15.65% | - | - | - | - |
| DCM | 4.83% | - | - | - | - |
| Solvent | Hippurate | Creatinine | Lactate | Histidine | Alanine |
|---|---|---|---|---|---|
| MeOD | 1.30 | 1.30 | 0.96 | 1.64 | 1.01 |
| DMSO | 1.18 | 1.10 | 0.46 | 1.39 | 0.65 |
| DMF | 1.34 | 1.59 | 0.38 | 0.20 | 0.59 |
| MeCN | 0.22 | 0.15 | 0.10 | - | - |
| Acetone | 0.32 | 0.11 | 0.09 | - | - |
| CDCl3 | 0.13 | - | - | - | - |
| DCM | 0.08 | - | - | - | - |
| H2O | MeOD | MeCN | DMSO | DMF | Acetone | DCM | CHCl3 | |
|---|---|---|---|---|---|---|---|---|
| ETN | 1.000 | 0.762 | 0.460 | 0.444 | 0.386 | 0.355 | 0.309 | 0.259 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Görling, B.; Bräse, S.; Luy, B. NMR Chemical Shift Ranges of Urine Metabolites in Various Organic Solvents. Metabolites 2016, 6, 27. https://doi.org/10.3390/metabo6030027
Görling B, Bräse S, Luy B. NMR Chemical Shift Ranges of Urine Metabolites in Various Organic Solvents. Metabolites. 2016; 6(3):27. https://doi.org/10.3390/metabo6030027
Chicago/Turabian StyleGörling, Benjamin, Stefan Bräse, and Burkhard Luy. 2016. "NMR Chemical Shift Ranges of Urine Metabolites in Various Organic Solvents" Metabolites 6, no. 3: 27. https://doi.org/10.3390/metabo6030027
APA StyleGörling, B., Bräse, S., & Luy, B. (2016). NMR Chemical Shift Ranges of Urine Metabolites in Various Organic Solvents. Metabolites, 6(3), 27. https://doi.org/10.3390/metabo6030027