
Serum Resistin Level and Progression of Atherosclerosis during Glucocorticoid Therapy for Systemic Autoimmune Diseases


Abstract
:1. Introduction
2. Results
2.1. Patient Profile
2.2. Serum Adipokines
2.3. Premature Atherosclerosis
2.4. Multivariate Analysis of Factors Associated with Progression of Premature Atherosclerosis
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Clinical and Laboratory Measurements
4.3. Mesurement of Serum Adipokines
4.4. Carotid Ultrasonography
4.5. Measument of CAVI and ABI
4.6. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lago, F.; Dieguez, C.; Gomez-Reino, J.; Gualillo, O. Adipokines as emerging mediators of immune response and inflammation. Nat. Clin. Pract. Rheumatol. 2007, 3, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Spiegelman, B.M. Adipocytes as regulators of energy balance and glucose homeostasis. Nature 2006, 444, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Filkova, M.; Haluzik, M.; Gay, S.; Senolt, L. The role of resistin as a regulator of inflammation: Implications for various human pathologies. Clin. Immunol. 2009, 133, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Nurmohamed, M.T.; Heslinga, M.; Kitas, G.D. Cardiovascular comorbidity in rheumatic diseases. Nat. Rev. Rheumatol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Avina-Zubieta, J.A.; Thomas, J.; Sadatsafavi, M.; Lehman, A.J.; Lacaille, D. Risk of incident cardiovascular events in patients with rheumatoid arthritis: A meta-analysis of observational studies. Ann. Rheum. Dis. 2012, 71, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.J.; van Halm, V.P.; Voskuyl, A.E.; Smulders, Y.M.; Boers, M.; Lems, W.F.; Visser, M.; Stehouwer, C.D.; Dekker, J.M.; Nijpels, G.; et al. Does rheumatoid arthritis equal diabetes mellitus as an independent risk factor for cardiovascular disease? A prospective study. Arthritis Rheum. 2009, 61, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Symmons, D.P.; Gabriel, S.E. Epidemiology of CVD in rheumatic disease, with a focus on RA and SLE. Nat. Rev. Rheumatol. 2011, 7, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Urowitz, M.B.; Bookman, A.A.; Koehler, B.E.; Gordon, D.A.; Smythe, H.A.; Ogryzlo, M.A. The bimodal mortality pattern of systemic lupus erythematosus. Am. J. Med. 1976, 60, 221–225. [Google Scholar] [CrossRef]
- Manzi, S.; Meilahn, E.N.; Rairie, J.E.; Conte, C.G.; Medsger, T.A., Jr.; Jansen-McWilliams, L.; D’Agostino, R.B.; Kuller, L.H. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: Comparison with the Framingham Study. Am. J. Epidemiol. 1997, 145, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Ungprasert, P.; Suksaranjit, P.; Spanuchart, I.; Leeaphorn, N.; Permpalung, N. Risk of coronary artery disease in patients with idiopathic inflammatory myopathies: A systematic review and meta-analysis of observational studies. Semin. Arthritis Rheum. 2014, 44, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Ungprasert, P.; Koster, M.J.; Warrington, K.J. Coronary artery disease in giant cell arteritis: A systematic review and meta-analysis. Semin. Arthritis Rheum. 2014, 44, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Esdaile, J.M.; Abrahamowicz, M.; Grodzicky, T.; Li, Y.; Panaritis, C.; du Berger, R.; Cote, R.; Grover, S.A.; Fortin, P.R.; Clarke, A.E.; et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum. 2001, 44, 2331–2337. [Google Scholar] [CrossRef]
- Skaggs, B.J.; Hahn, B.H.; McMahon, M. Accelerated atherosclerosis in patients with SLE—mechanisms and management. Nat. Rev. Rheumatol. 2012, 8, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Kitas, G.D.; Gabriel, S.E. Cardiovascular disease in rheumatoid arthritis: State of the art and future perspectives. Ann. Rheum. Dis. 2011, 70, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, T.; Kusunoki, N.; Tanaka, N.; Kaneko, K.; Kusunoki, Y.; Endo, H.; Hasunuma, T.; Kawai, S. Elevated Serum Levels of Resistin, Leptin, and Adiponectin are Associated with C-reactive Protein and also Other Clinical Conditions in Rheumatoid Arthritis. Intern. Med. 2011, 50, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Kemmotsu, Y.; Saji, T.; Kusunoki, N.; Tanaka, N.; Nishimura, C.; Ishiguro, A.; Kawai, S. Serum adipokine profiles in Kawasaki disease. Mod. Rheumatol. 2012, 22, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Kusunoki, N.; Kusunoki, Y.; Hasunuma, T.; Kawai, S. Resistin is associated with the inflammation process in patients with systemic autoimmune diseases undergoing glucocorticoid therapy: Comparison with leptin and adiponectin. Mod. Rheumatol. 2013, 23, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Sacks, D.; Bakal, C.W.; Beatty, P.T.; Becker, G.J.; Cardella, J.F.; Raabe, R.D.; Wiener, H.M.; Lewis, C.A. Position statement on the use of the ankle-brachial index in the evaluation of patients with peripheral vascular disease: A consensus statement developed by the standards division of the society of cardiovascular & interventional radiology. J. Vasc. Interv. Radiol. 2002, 13. [Google Scholar] [CrossRef]
- Salmon, J.E.; Roman, M.J. Subclinical atherosclerosis in rheumatoid arthritis and systemic lupus erythematosus. Am. J. Med. 2008, 121 (Suppl. S1), S3–S8. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Juanatey, C.; Llorca, J.; Martin, J.; Gonzalez-Gay, M.A. Carotid intima-media thickness predicts the development of cardiovascular events in patients with rheumatoid arthritis. Semin. Arthritis Rheum. 2009, 38, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Kao, A.H.; Lertratanakul, A.; Elliott, J.R.; Sattar, A.; Santelices, L.; Shaw, P.; Birru, M.; Avram, Z.; Thompson, T.; Sutton-Tyrrell, K.; et al. Relation of carotid intima-media thickness and plaque with incident cardiovascular events in women with systemic lupus erythematosus. Am. J. Cardiol. 2013, 112, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Zureik, M.; Ducimetiere, P.; Touboul, P.J.; Courbon, D.; Bonithon-Kopp, C.; Berr, C.; Magne, C. Common carotid intima-media thickness predicts occurrence of carotid atherosclerotic plaques: Longitudinal results from the Aging Vascular Study (EVA) study. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1622–1629. [Google Scholar] [CrossRef] [PubMed]
- Svenungsson, E.; Jensen-Urstad, K.; Heimburger, M.; Silveira, A.; Hamsten, A.; de Faire, U.; Witztum, J.L.; Frostegard, J. Risk factors for cardiovascular disease in systemic lupus erythematosus. Circulation 2001, 104, 1887–1893. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, P.N.; Beyene, J.; Feldman, B.M.; McCrindle, B.W.; Silverman, E.D.; Bradley, T.J. Rheumatic disease and carotid intima-media thickness: A systematic review and meta-analysis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Galkina, E.; Ley, K. Immune and inflammatory mechanisms of atherosclerosis. Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.J.; Symmons, D.P.; McCarey, D.; Dijkmans, B.A.; Nicola, P.; Kvien, T.K.; McInnes, I.B.; Haentzschel, H.; Gonzalez-Gay, M.A.; Provan, S.; et al. EULAR evidence-based recommendations for cardiovascular risk management in patients with rheumatoid arthritis and other forms of inflammatory arthritis. Ann. Rheum. Dis. 2010, 69, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Urowitz, M.B.; Gladman, D.D.; Anderson, N.M.; Su, J.; Romero-Diaz, J.; Bae, S.C.; Fortin, P.R.; Sanchez-Guerrero, J.; Clarke, A.; Bernatsky, S.; et al. Cardiovascular events prior to or early after diagnosis of systemic lupus erythematosus in the systemic lupus international collaborating clinics cohort. Lupus Sci. Med. 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Maradit-Kremers, H.; Crowson, C.S.; Nicola, P.J.; Ballman, K.V.; Roger, V.L.; Jacobsen, S.J.; Gabriel, S.E. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: A population-based cohort study. Arthritis Rheum. 2005, 52, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Majka, D.S.; Chang, R.W. Is preclinical autoimmunity benign?: The case of cardiovascular disease. Rheum. Dis. Clin. North Am. 2014, 40, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Vogt, M.T.; McKenna, M.; Wolfson, S.K.; Kuller, L.H. The relationship between ankle brachial index, other atherosclerotic disease, diabetes, smoking and mortality in older men and women. Atherosclerosis 1993, 101, 191–202. [Google Scholar] [CrossRef]
- Newman, A.B.; Siscovick, D.S.; Manolio, T.A.; Polak, J.; Fried, L.P.; Borhani, N.O.; Wolfson, S.K. Ankle-Arm Index as a Marker of Atherosclerosis in the Cardiovascular Health Study. Cardiovascular Heart Study (CHS) Collaborative Research Group. Circulation 1993, 88, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Abbott, R.D.; Petrovitch, H.; Rodriguez, B.L.; Yano, K.; Schatz, I.J.; Popper, J.S.; Masaki, K.H.; Ross, G.W.; Curb, J.D. Ankle/brachial blood pressure in men >70 years of age and the risk of coronary heart disease. Am. J. Cardiol. 2000, 86, 280–284. [Google Scholar] [CrossRef]
- Zheng, Z.J.; Sharrett, A.R.; Chambless, L.E.; Rosamond, W.D.; Nieto, F.J.; Sheps, D.S.; Dobs, A.; Evans, G.W.; Heiss, G. Associations of ankle-brachial index with clinical coronary heart disease, stroke and preclinical carotid and popliteal atherosclerosis: The Atherosclerosis Risk in Communities (ARIC) Study. Atherosclerosis 1997, 131, 115–125. [Google Scholar] [CrossRef]
- Shirai, K.; Utino, J.; Otsuka, K.; Takata, M. A novel blood pressure-independent arterial wall stiffness parameter; cardio-ankle vascular index (CAVI). J. Atheroscler. Thromb. 2006, 13, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Laucevičius, A.; Ryliškytė, L.; Balsytė, J.; Badarienė, J.; Puronaitė, R.; Navickas, R.; Solovjova, S. Association of cardio-ankle vascular index with cardiovascular risk factors and cardiovascular events in metabolic syndrome patients. Medicina 2015, 51, 152–158. [Google Scholar] [CrossRef]
- Satoh-Asahara, N.; Kotani, K.; Yamakage, H.; Yamada, T.; Araki, R.; Okajima, T.; Adachi, M.; Oishi, M.; Shimatsu, A. Japan Obesity and Metabolic Syndrome Study (JOMS) Group. Cardio-ankle vascular index predicts for the incidence of cardiovascular events in obese patients: A multicenter prospective cohort study (Japan Obesity and Metabolic Syndrome Study: JOMS). Atherosclerosis 2015, 242, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Nagayama, D.; Saiki, A.; Watanabe, R.; Watanabe, Y.; Imamura, H.; Yamaguchi, T.; Ban, N.; Kawana, H.; Nagumo, A.; et al. Cardio-Ankle Vascular Index is Independently Associated with Future Cardiovascular Events in Outpatients with Metabolic Disorders. J. Atheroscler. Thromb. 2015. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Tomaru, T.; Yamamura, S.; Miyashita, Y.; Shirai, K.; Noike, H. Cardio-ankle vascular index is a candidate predictor of coronary atherosclerosis. Circ. J. 2008, 72, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Patel, L.; Buckels, A.C.; Kinghorn, I.J.; Murdock, P.R.; Holbrook, J.D.; Plumpton, C.; Macphee, C.H.; Smith, S.A. Resistin is expressed in human macrophages and directly regulated by PPAR γ activators. Biochem. Biophys. Res. Commun. 2003, 300, 472–476. [Google Scholar] [CrossRef]
- Nagaev, I.; Smith, U. Insulin resistance and type 2 diabetes are not related to resistin expression in human fat cells or skeletal muscle. Biochem. Biophys. Res. Commun. 2001, 285, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.B.; Sewter, C.P.; Klenk, E.S.; Segal, D.G.; Vidal-Puig, A.; Considine, R.V.; O’Rahilly, S. Resistin/Fizz3 expression in relation to obesity and peroxisome proliferator-activated receptor-γ action in humans. Diabetes 2001, 50, 2199–2202. [Google Scholar] [CrossRef] [PubMed]
- Kunnari, A.M.; Savolainen, E.R.; Ukkola, O.H.; Kesaniemi, Y.A.; Jokela, M.A. The expression of human resistin in different leucocyte lineages is modulated by LPS and TNFα. Regul. Pept. 2009, 157, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Silswal, N.; Singh, A.K.; Aruna, B.; Mukhopadhyay, S.; Ghosh, S.; Ehtesham, N.Z. Human resistin stimulates the pro-inflammatory cytokines TNF-α and IL-12 in macrophages by NF-κB-dependent pathway. Biochem. Biophys. Res. Commun. 2005, 334, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Bokarewa, M.; Nagaev, I.; Dahlberg, L.; Smith, U.; Tarkowski, A. Resistin, an adipokine with potent proinflammatory properties. J. Immunol. 2005, 174, 5789–5795. [Google Scholar] [CrossRef] [PubMed]
- Reilly, M.P.; Lehrke, M.; Wolfe, M.L.; Rohatgi, A.; Lazar, M.A.; Rader, D.J. Resistin is an inflammatory marker of atherosclerosis in humans. Circulation 2005, 111, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Gencer, B.; Auer, R.; de Rekeneire, N.; Butler, J.; Kalogeropoulos, A.; Bauer, D.C.; Kritchevsky, S.B.; Miljkovic, I.; Vittinghoff, E.; Harris, T.; et al. Association between resistin levels and cardiovascular disease events in older adults: The health, aging and body composition study. Atherosclerosis 2016, 245, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Muse, E.D.; Feldman, D.I.; Blaha, M.J.; Dardari, Z.A.; Blumenthal, R.S.; Budoff, M.J.; Nasir, K.; Criqui, M.H.; Cushman, M.; McClelland, R.L.; et al. The association of resistin with cardiovascular disease in the Multi-Ethnic Study of Atherosclerosis. Atherosclerosis 2015, 239, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Weikert, C.; Westphal, S.; Berger, K.; Dierkes, J.; Mohlig, M.; Spranger, J.; Rimm, E.B.; Willich, S.N.; Boeing, H.; Pischon, T. Plasma resistin levels and risk of myocardial infarction and ischemic stroke. J. Clin. Endocrinol. Metable 2008, 93, 2647–2653. [Google Scholar] [CrossRef] [PubMed]
- Moya, F.B.; Pineda Galindo, L.F.; Garcia de la Pena, M. Impact of Chronic Glucocorticoid Treatment on Cardiovascular Risk Profile in Patients with Systemic Lupus Erythematosus. J. Clin. Rheumatol. 2016, 22, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Roubille, C.; Richer, V.; Starnino, T.; McCourt, C.; McFarlane, A.; Fleming, P.; Siu, S.; Kraft, J.; Lynde, C.; Pope, J.; et al. The effects of tumour necrosis factor inhibitors, methotrexate, non-steroidal anti-inflammatory drugs and corticosteroids on cardiovascular events in rheumatoid arthritis, psoriasis and psoriatic arthritis: A systematic review and meta-analysis. Ann. Rheum. Dis. 2015, 74, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, Y.; Oeser, A.; Shintani, A.K.; Turner, E.; Olsen, N.; Fazio, S.; Linton, M.F.; Raggi, P.; Stein, C.M. Premature coronary-artery atherosclerosis in systemic lupus erythematosus. N. Engl. J. Med. 2003, 349, 2407–2415. [Google Scholar] [CrossRef] [PubMed]
- Roman, M.J.; Shanker, B.A.; Davis, A.; Lockshin, M.D.; Sammaritano, L.; Simantov, R.; Crow, M.K.; Schwartz, J.E.; Paget, S.A.; Devereux, R.B.; et al. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N. Engl. J. Med. 2003, 349, 2399–2406. [Google Scholar] [CrossRef] [PubMed]
- Roman, M.J.; Crow, M.K.; Lockshin, M.D.; Devereux, R.B.; Paget, S.A.; Sammaritano, L.; Levine, D.M.; Davis, A.; Salmon, J.E. Rate and determinants of progression of atherosclerosis in systemic lupus erythematosus. Arthritis Rheum. 2007, 56, 3412–3419. [Google Scholar] [CrossRef] [PubMed]
- Kiani, A.N.; Post, W.S.; Magder, L.S.; Petri, M. Predictors of progression in atherosclerosis over 2 years in systemic lupus erythematosus. Rheumatology 2011, 50, 2071–2079. [Google Scholar] [CrossRef] [PubMed]
- Toloza, S.M.; Uribe, A.G.; McGwin, G., Jr.; Alarcon, G.S.; Fessler, B.J.; Bastian, H.M.; Vila, L.M.; Wu, R.; Shoenfeld, Y.; Roseman, J.M.; et al. Systemic lupus erythematosus in a multiethnic US cohort (LUMINA). XXIII. Baseline predictors of vascular events. Arthritis Rheum. 2004, 50, 3947–3957. [Google Scholar] [CrossRef] [PubMed]
- Urowitz, M.B.; Ibanez, D.; Gladman, D.D. Atherosclerotic vascular events in a single large lupus cohort: Prevalence and risk factors. J. Rheumatol. 2007, 34, 70–75. [Google Scholar] [PubMed]
- Bertoli, A.M.; Vila, L.M.; Alarcon, G.S.; McGwin, G.; Edberg, J.C.; Petri, M.; Ramsey-Goldman, R.; Reveille, J.D.; Kimberly, R.P. Factors Associated with Arterial Vascular Events in PROFILE: A Multiethnic Lupus Cohort. Lupus 2009, 18, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Urowitz, M.B.; Gladman, D.; Ibanez, D.; Bae, S.C.; Sanchez-Guerrero, J.; Gordon, C.; Clarke, A.; Bernatsky, S.; Fortin, P.R.; Hanly, J.G.; et al. Systemic Lupus International Collaborating Clinics Atherosclerotic vascular events in a multinational inception cohort of systemic lupus erythematosus. Arthritis Care. Res. 2010, 62, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, C.; Ohman, M.L.; Nived, O.; Rantapaa Dahlqvist, S. Cardiovascular event in systemic lupus erythematosus in northern Sweden: Incidence and predictors in a 7-year follow-up study. Lupus 2012, 21, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Nikpour, M.; Urowitz, M.B.; Ibanez, D.; Harvey, P.J.; Gladman, D.D. Importance of cumulative exposure to elevated cholesterol and blood pressure in development of atherosclerotic coronary artery disease in systemic lupus erythematosus: A prospective proof-of-concept cohort study. Arthritis Res. Ther. 2011, 13. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.; Perez-Gutthann, S.; Spence, D.; Hochberg, M.C. Risk factors for coronary artery disease in patients with systemic lupus erythematosus. Am. J. Med. 1992, 93, 513–519. [Google Scholar] [CrossRef]
- Bailey, J.M.; Butler, J. Anti-inflammatory drugs in experimental atherosclerosis. Part 6. Combination therapy with steroid and non-steroid agents. Atherosclerosis 1985, 54, 205–212. [Google Scholar] [CrossRef]
- Asai, K.; Funaki, C.; Hayashi, T.; Yamada, K.; Naito, M.; Kuzuya, M.; Yoshida, F.; Yoshimine, N.; Kuzuya, F. Dexamethasone-induced suppression of aortic atherosclerosis in cholesterol-fed rabbits. Possible mechanisms. Arterioscler. Thromb. 1993, 13, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Voisard, R.; Seitzer, U.; Baur, R.; Dartsch, P.C.; Osterhues, H.; Hoher, M.; Hombach, V. Corticosteroid agents inhibit proliferation of smooth muscle cells from human atherosclerotic arteries in vitro. Int. J. Cardiol. 1994, 43, 257–267. [Google Scholar] [CrossRef]
- Yamada, K.; Naito, M.; Hayashi, T.; Asai, K.; Yoshimine, N.; Iguchi, A. Effects of dexamethasone on migration of human monocytes in response to oxidized beta-very low density lipoprotein. Artery 1993, 20, 253–267. [Google Scholar] [PubMed]
- Mackinnon, A.D.; Jerrard-Dunne, P.; Sitzer, M.; Buehler, A.; von Kegler, S.; Markus, H.S. Rates and determinants of site-specific progression of carotid artery intima-media thickness: The carotid atherosclerosis progression study. Stroke 2004, 35, 2150–2154. [Google Scholar] [CrossRef] [PubMed]
- Howard, G.; Sharrett, A.R.; Heiss, G.; Evans, G.W.; Chambless, L.E.; Riley, W.A.; Burke, G.L. Carotid artery intimal-medial thickness distribution in general populations as evaluated by B-mode ultrasound. ARIC Investigators. Stroke 1993, 24, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Battu, A.; Mohareer, K.; Hasnain, S.E.; Ehtesham, N.Z. Transcription of human resistin gene involves an interaction of Sp1 with peroxisome proliferator-activating receptor gamma (PPARγ). PLoS One 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids—New mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Sasayama, D.; Hori, H.; Nakamura, S.; Yamamoto, N.; Hattori, K.; Teraishi, T.; Ota, M.; Kunugi, H. Increased protein and mRNA expression of resistin after dexamethasone administration. Horm Metab Res. 2015, 47, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Jennette, J.C.; Falk, R.J.; Bacon, P.A.; Basu, N.; Cid, M.C.; Ferrario, F.; Flores-Suarez, L.F.; Gross, W.L.; Guillevin, L.; Hagen, E.C.; et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013, 65. [Google Scholar] [CrossRef] [PubMed]
- Friedewald, W.T.; Levy, R.I.; Fredrickson, D.S. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin. Chem. 1972, 18, 499–502. [Google Scholar] [PubMed]
- Kuzuya, T.; Nakagawa, S.; Satoh, J.; Kanazawa, Y.; Iwamoto, Y.; Kobayashi, M.; Nanjo, K.; Sasaki, A.; Seino, Y.; Ito, C.; et al. Report of the Committee on the classification and diagnostic criteria of diabetes mellitus. Diabetes Res. Clin. Pract. 2002, 55, 65–85. [Google Scholar] [CrossRef]
- Kumeda, Y.; Inaba, M.; Goto, H.; Nagata, M.; Henmi, Y.; Furumitsu, Y.; Ishimura, E.; Inui, K.; Yutani, Y.; Miki, T.; et al. Increased thickness of the arterial intima-media detected by ultrasonography in patients with rheumatoid arthritis. Arthritis Rheum. 2002, 46, 1489–1497. [Google Scholar] [CrossRef] [PubMed]
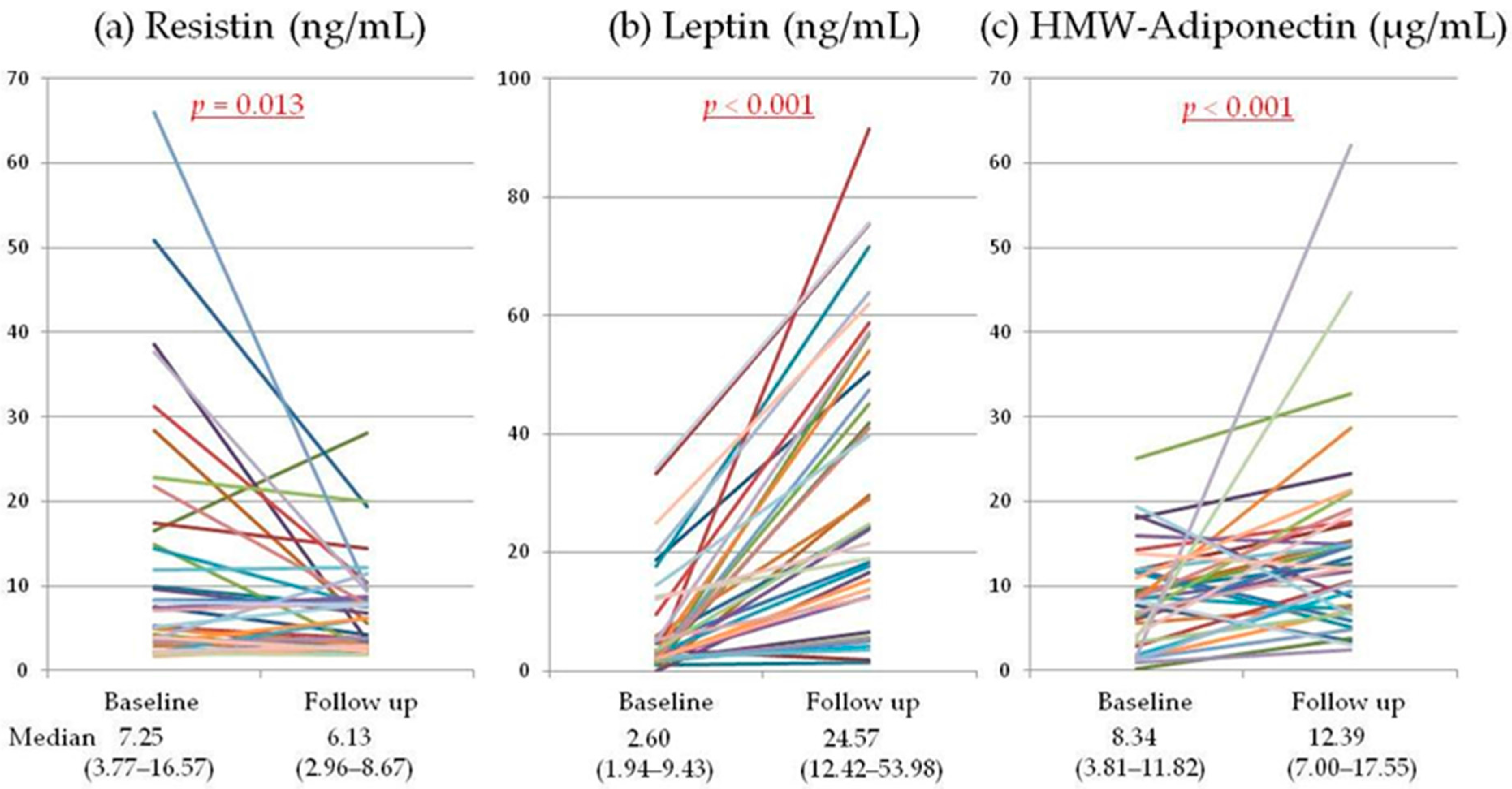

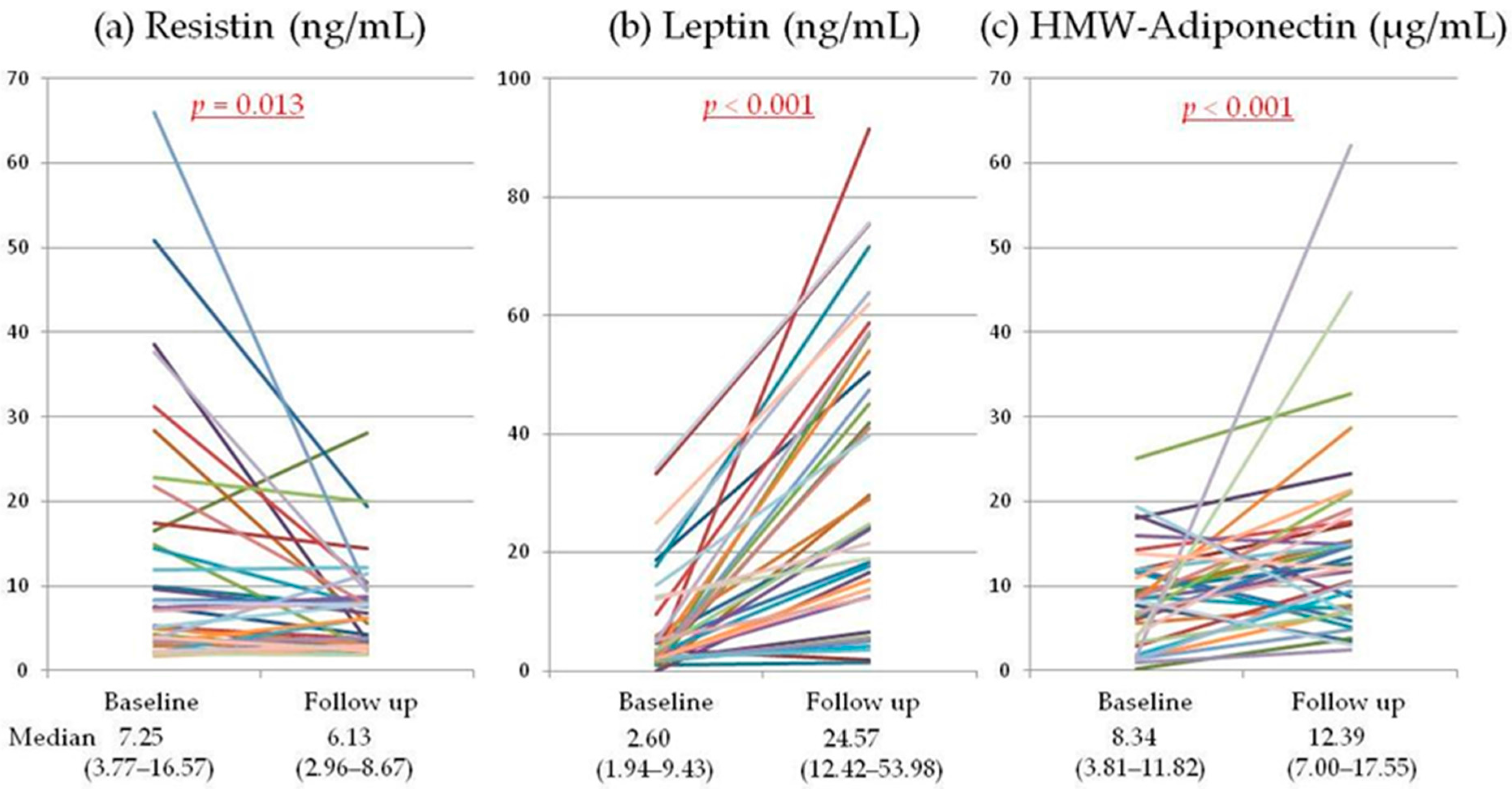{kind=link}
| Sex, Male: Female (% female) | 11:27 (71.1%) |
| Age (years) | 49.3 ± 15.2 |
| Height (m) | 1.61 ± 9.46 |
| Weight (kg) | 56.3 ± 11.5 |
| BMI (kg/m2) | 21.7 ± 3.5 |
| Systemic Autoimmune Diseases: | |
| SLE (%) | 16 (42.1%) |
| PM/DM (%) | 14 (36.8%) |
| Vasculitis Syndrome (%) | 6 (15.8%) |
| AOSD (%) | 2 (5.3%) |
| Disease Duration (weeks) | 4.4 (3.4–12.9) |
| Baseline (n = 38) | Follow-up (n = 38) | p Value | |
|---|---|---|---|
| Follow-up period (years) | - | 3.3 ± 0.9 | - |
| Comorbidities | |||
| Systolic blood pressure (mmHg) | 122 ± 15 | 124 ± 20 | 0.399 |
| Diastolic blood pressure (mmHg) | 73 ± 10 | 78 ± 11 | 0.043 |
| Hypertension (%) | 10 (26.3%) | 18 (47.4%) | 0.185 |
| Diabetes mellitus (%) | 4 (10.5%) | 9 (23.7%) | < 0.001 |
| Current smoking (%) | 11 (28.9%) | 4 (10.5%) | < 0.001 |
| Ever smoked (%) | 19 (50.0%) | 19 (50.0%) | 0.607 |
| History of CVD (%) | 4 (10.5%) | 4 (10.5%) | - |
| Carotid artery plaque (%) | 23 (60.5%) | 25 (65.7%) | 0.155 |
| Maximum IMT (mm) | 0.68 (0.50–0.81) | 0.73 (0.59–0.96) | 0.043 |
| CAVI | - | 7.8 ± 1.5 | - |
| ABI | - | 1.2 ± 0.1 | - |
| Laboratory Data | |||
| Total cholesterol (mg/dL) | 160 ± 43 | 198 ± 43 | < 0.001 |
| HDL cholesterol (mg/dL) | 34 ± 12 | 69 ± 20 | < 0.001 |
| LDL cholesterol (mg/dL) | 98 ± 38 | 105 ± 33 | 0.304 |
| Triglycerides (mg/dL) | 111 (79–186) | 102 (70–148) | 0.357 |
| CRP (mg/dL) | 0.9 (0.2–3.5) | 0.1 (0.0–0.33) | < 0.001 |
| Medications | |||
| Daily prednisolone dose (mg) | 48.2 ± 9.0 * | 7.9 ± 9.9 | - |
| Cumulative prednisolone dose (mg) | 0 | 18651 ± 8734 | - |
| Immunosuppressive agents (%) | 0 | 20 (52.6%) | - |
| Antihypertensive agents (%) | 7 (18.4%) | 14 (36.8%) | 0.017 |
| Antidiabetic agents (%) | 3 (7.9%) | 9 (23.7%) | < 0.001 |
| Statins (%) | 4 (10.5%) | 10 (26.3%) | < 0.001 |
| ΔCarotid Artery IMT/Year | |||||
|---|---|---|---|---|---|
| Univariate Model | R2 | Multivariate Model | |||
| β | p value | β | p value | ||
| Age | 0.001348 | 0.195 | 0.046 | −0.000477 | 0.608 |
| Female sex | −0.059111 | 0.032 | 0.121 | 0.034276 | 0.413 |
| BMI | −0.000186 | 0.662 | 0.005 | −0.000984 | 0.790 |
| Cumulative prednisolone dose | 0.000003 | 0.040 | 0.111 | −0.000004 | 0.011 |
| ΔCRP/year | 0.006042 | 0.233 | 0.039 | −0.014445 | 0.195 |
| ΔHDL cholesterol/year | 0.000069 | 0.647 | 0.006 | 0.000576 | 0.769 |
| ΔTriglycerides/year | −0.000122 | 0.123 | 0.065 | −0.000145 | 0.321 |
| Hypertension | 0.028606 | 0.378 | 0.022 | 0.038420 | 0.153 |
| Diabetes mellitus | 0.028536 | 0.027 | 0.325 | 0.074380 | 0.055 |
| History of CVD | 0.034926 | 0.037 | 0.022 | 0.051526 | 0.186 |
| Ever smoked | 0.040737 | 0.084 | 0.079 | 0.009914 | 0.743 |
| ΔLP/year | −0.027277 | 0.063 | 0.093 | −0.034033 | 0.132 |
| ΔRS/year | 0.004251 | 0.157 | 0.054 | 0.006317 | 0.046 |
| ΔHMW-AD/year | 0.017032 | 0.155 | 0.055 | 0.008543 | 0.609 |
| R2 | 0.234 | ||||
| CAVI (Follow up) | |||||
|---|---|---|---|---|---|
| Univariate Model | R2 | Multivariate Model | |||
| β | p Value | β | p Value | ||
| Age | 0.064309 | < 0.001 | 0.554 | 0.059720 | < 0.001 |
| Female sex | −0.582343 | 0.132 | 0.064 | 0.131749 | 0.733 |
| BMI | −0.015023 | 0.966 | 0.000 | −0.098105 | 0.054 |
| Cumulative prednisolone dose | −0.000006 | 0.718 | 0.004 | - | - |
| ΔCRP/year | −0.172600 | 0.551 | 0.010 | - | - |
| ΔHDL cholesterol/year | 0.003258 | 0.598 | 0.008 | - | - |
| ΔTriglycerides/year | −0.002384 | 0.237 | 0.040 | - | - |
| Hypertension | 0.319883 | 0.446 | 0.017 | - | - |
| Diabetes mellitus | 1.156746 | 0.038 | 0.117 | 1.154566 | 0.019 |
| History of CVD | 0.363258 | 0.792 | 0.002 | - | - |
| Ever smoked | 0.417251 | 0.147 | 0.059 | - | - |
| ΔLP/year | −0.224163 | 0.321 | 0.028 | - | - |
| ΔRS/year | 0.053863 | 0.969 | 0.000 | 0.088717 | 0.048 |
| ΔHMW-AD/year | 0.339796 | 0.073 | 0.089 | 0.366996 | 0.122 |
| R2 | 0.539 | ||||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanaka, N.; Masuoka, S.; Kusunoki, N.; Nanki, T.; Kawai, S. Serum Resistin Level and Progression of Atherosclerosis during Glucocorticoid Therapy for Systemic Autoimmune Diseases. Metabolites 2016, 6, 28. https://doi.org/10.3390/metabo6030028
Tanaka N, Masuoka S, Kusunoki N, Nanki T, Kawai S. Serum Resistin Level and Progression of Atherosclerosis during Glucocorticoid Therapy for Systemic Autoimmune Diseases. Metabolites. 2016; 6(3):28. https://doi.org/10.3390/metabo6030028
Chicago/Turabian StyleTanaka, Nahoko, Shotaro Masuoka, Natsuko Kusunoki, Toshihiro Nanki, and Shinichi Kawai. 2016. "Serum Resistin Level and Progression of Atherosclerosis during Glucocorticoid Therapy for Systemic Autoimmune Diseases" Metabolites 6, no. 3: 28. https://doi.org/10.3390/metabo6030028
APA StyleTanaka, N., Masuoka, S., Kusunoki, N., Nanki, T., & Kawai, S. (2016). Serum Resistin Level and Progression of Atherosclerosis during Glucocorticoid Therapy for Systemic Autoimmune Diseases. Metabolites, 6(3), 28. https://doi.org/10.3390/metabo6030028