
Gene Therapy Targeting PCSK9


Abstract
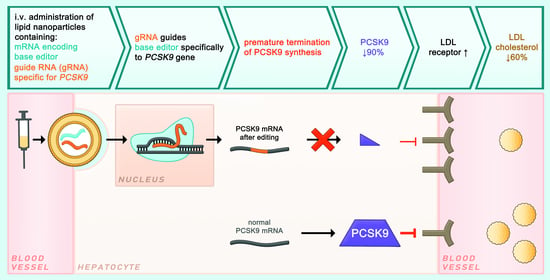
:
1. Introduction
2. PCSK9, LDL-C, and Atherosclerotic Cardiovascular Disease
3. Genome Editing Using the CRISPR/Cas System
3.1. The CRISPR/Cas System as Part of Adaptive Bacterial Immunity
3.2. Application of the CRISPR/Cas System for Genome Editing
3.2.1. Principle of Application
3.2.2. Advantages and Limitations of CRISPR/Cas Genome Editing
4. Editing of PCSK9 Using CRISPR/Cas
4.1. Initial In Vivo Studies in Mice
4.2. In Vivo Studies in Non-Human Primates
4.2.1. PCSK9 Editing Using a Meganuclease
4.2.2. PCSK9 Editing Using a Base Editor (I)
4.2.3. PCSK9 Editing Using a Base Editor (II)
5. Discussion
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Vallejo-Vaz, A.J.; Stevens, C.A.; Lyons, A.R.; Dharmayat, K.I.; Freiberger, T.; Hovingh, G.K.; Mata, P.; Raal, F.J.; Santos, R.D.; Soran, H.; et al. Global perspective of familial hypercholesterolaemia: A cross-sectional study from the EAS Familial Hypercholesterolaemia Studies Collaboration (FHSC). Lancet 2021, 398, 1713–1725. [Google Scholar] [CrossRef]
- Hu, P.; Dharmayat, K.I.; Stevens, C.A.T.; Sharabiani, M.T.A.; Jones, R.S.; Watts, G.F.; Genest, J.; Ray, K.K.; Vallejo-Vaz, A.J. Prevalence of Familial Hypercholesterolemia Among the General Population and Patients With Atherosclerotic Cardiovascular Disease: A Systematic Review and Meta-Analysis. Circulation 2020, 141, 1742–1759. [Google Scholar] [CrossRef] [PubMed]
- Abifadel, M.; Varret, M.; Rabès, J.-P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef]
- Banach, M.; Rizzo, M.; Obradovic, M.; Montalto, G.; Rysz, J.; Mikhailidis, D.P.; Isenovic, E.R. PCSK9 inhibition-a novel mechanism to treat lipid disorders? Curr. Pharm. Des. 2013, 19, 3869–3877. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Prat, A.; Pirillo, A.; Catapano, A.L.; Norata, G.D. Novel strategies to target proprotein convertase subtilisin kexin 9: Beyond monoclonal antibodies. Cardiovasc. Res. 2019, 115, 510–518. [Google Scholar] [CrossRef] [Green Version]
- Musunuru, K.; Chadwick, A.C.; Mizoguchi, T.; Garcia, S.P.; DeNizio, J.E.; Reiss, C.W.; Wang, K.; Iyer, S.; Dutta, C.; Clendaniel, V.; et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature 2021, 593, 429–434. [Google Scholar] [CrossRef]
- Cameron, J.; Holla, Ø.L.; Ranheim, T.; Kulseth, M.A.; Berge, K.E.; Leren, T.P. Effect of mutations in the PCSK9 gene on the cell surface LDL receptors. Hum. Mol. Genet. 2006, 15, 1551–1558. [Google Scholar] [CrossRef] [Green Version]
- Lagace, T.A.; Curtis, D.E.; Garuti, R.; McNutt, M.C.; Park, S.W.; Prather, H.B.; Anderson, N.N.; Ho, Y.K.; Hammer, R.E.; Horton, J.D. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J. Clin. Investig. 2006, 116, 2995–3005. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H., Jr.; Hobbs, H.H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef]
- Hooper, A.J.; Marais, A.D.; Tanyanyiwa, D.M.; Burnett, J.R. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis 2007, 193, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Tuakli-Wosornu, Y.; Lagace, T.A.; Kinch, L.; Grishin, N.V.; Horton, J.D.; Cohen, J.C.; Hobbs, H.H. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am. J. Hum. Genet. 2006, 79, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Macchi, C.; Ferri, N.; Sirtori, C.R.; Corsini, A.; Banach, M.; Ruscica, M. Proprotein Convertase Subtilisin/Kexin Type 9: A View beyond the Canonical Cholesterol-Lowering Impact. Am. J. Pathol. 2021, 191, 1385–1397. [Google Scholar] [CrossRef]
- Seidah, N.G.; Prat, A. The multifaceted biology of PCSK9; Oxford University Press: Oxford, UK, 2021. [Google Scholar] [CrossRef]
- Katzmann, J.L.; Gouni-Berthold, I.; Laufs, U. PCSK9 Inhibition: Insights from Clinical Trials and Future Prospects. Front. Physiol. 2020, 11, 595819. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Macchi, C.; Sirtori, C.R.; Corsini, A.; Santos, R.D.; Watts, G.F.; Ruscica, M. A new dawn for managing dyslipidemias: The era of rna-based therapies. Pharmacol. Res. 2019, 150, 104413. [Google Scholar] [CrossRef]
- Katzmann, J.L.; Packard, C.J.; Chapman, M.J.; Katzmann, I.; Laufs, U. Targeting RNA with antisense oligonucleotides and small interfering RNA in dyslipidemias. J. Am. Coll. Cardiol. 2020, 76, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, Y.; Stoian, A.P.; Cicero, A.F.G.; Fogacci, F.; Nikolic, D.; Sachinidis, A.; Rizvi, A.A.; Janez, A.; Rizzo, M. Inclisiran: A small interfering RNA strategy targeting PCSK9 to treat hypercholesterolemia. Expert Opin. Drug Saf. 2021, 21, 1988568. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.J.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the treatment of heterozygous familial hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two phase 3 trials of inclisiran in patients with elevated LDL cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- A Double-Blind Randomized Placebo-Controlled Trial Assessing the Effects of Inclisiran on Clinical Outcomes among People with Atherosclerotic Cardiovascular Disease (ORION-4). Available online: https://clinicaltrials.gov/ct2/show/NCT03705234 (accessed on 3 January 2022).
- Wang, H.; La Russa, M.; Qi, L.S. CRISPR/Cas9 in Genome Editing and Beyond. Annu. Rev. Biochem. 2016, 85, 227–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, A.C.; Musunuru, K. Treatment of Dyslipidemia Using CRISPR/Cas9 Genome Editing. Curr. Atheroscler. Rep. 2017, 19, 32. [Google Scholar] [CrossRef] [PubMed]
- Porto, E.M.; Komor, A.C.; Slaymaker, I.M.; Yeo, G.W. Base editing: Advances and therapeutic opportunities. Nat. Rev. Drug Discov. 2020, 19, 839–859. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Lam, D.K.; Rees, H.A.; Solá-Esteves, N.M.; Barrera, L.A.; Born, D.A.; Edwards, A.; Gehrke, J.M.; Lee, S.-J.; Liquori, A.J.; et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 2020, 38, 892–900. [Google Scholar] [CrossRef]
- Walker, H.E.; Rizzo, M.; Fras, Z.; Jug, B.; Banach, M.; Penson, P.E. CRISPR Gene Editing in Lipid Disorders and Atherosclerosis: Mechanisms and Opportunities. Metabolites 2021, 11, 857. [Google Scholar] [CrossRef]
- Zhang, F.; Wen, Y.; Guo, X. CRISPR/Cas9 for genome editing: Progress, implications and challenges. Hum. Mol. Genet. 2014, 23, R40–R46. [Google Scholar] [CrossRef] [Green Version]
- Naeem, M.; Majeed, S.; Hoque, M.Z.; Ahmad, I. Latest Developed Strategies to Minimize the Off-Target Effects in CRISPR-Cas-Mediated Genome Editing. Cells 2020, 9, 1608. [Google Scholar] [CrossRef]
- Lu, X.; Xu, H.; Ke, Z.; Chen, J.; Ji, L. CRISPR-Cas9: A new and promising player in gene therapy. J. Med. Genet. 2015, 52, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Scholefield, J.; Harrison, P.T. Prime editing-an update on the field. Gene Ther. 2021, 28, 396–401. [Google Scholar] [CrossRef]
- Kantor, A.; McClements, M.E.; MacLaren, R.E. CRISPR-Cas9 DNA Base-Editing and Prime-Editing. Int. J. Mol. Sci. 2020, 21, 6240. [Google Scholar] [CrossRef]
- Chadwick, A.C.; Musunuru, K. CRISPR-Cas9 Genome Editing for Treatment of Atherogenic Dyslipidemia. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 12–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Q.; Strong, A.; Patel, K.M.; Ng, S.-L.; Gosis, B.S.; Regan, S.N.; Cowan, C.A.; Rader, D.J.; Musunuru, K. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ. Res. 2014, 115, 488–492. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Raghavan, A.; Chen, T.; Qiao, L.; Zhang, Y.; Ding, Q.; Musunuru, K. CRISPR-Cas9 Targeting of PCSK9 in Human Hepatocytes In Vivo-Brief Report. Arter. Thromb. Vasc. Biol. 2016, 36, 783–786. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.; Le, C.; Yan, W.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Wang, L.; Smith, J.; Breton, C.; Clark, P.; Zhang, J.; Ying, L.; Che, Y.; Lape, J.; Bell, P.; Calcedo, R.; et al. Meganuclease targeting of PCSK9 in macaque liver leads to stable reduction in serum cholesterol. Nat. Biotechnol. 2018, 36, 717–725. [Google Scholar] [CrossRef]
- Wang, L.; Breton, C.; Warzecha, C.C.; Bell, P.; Yan, H.; He, Z.; White, J.; Zhu, Y.; Li, M.; Buza, E.L.; et al. Long-term Stable Reduction of Low-density Lipoprotein in Nonhuman Primates Following In Vivo Genome Editing of PCSK9. Mol. Ther. 2021, 29, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadwick, A.C.; Wang, X.; Musunuru, K. In Vivo Base Editing of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) as a Therapeutic Alternative to Genome Editing. Arter. Thromb. Vasc. Biol. 2017, 37, 1741–1747. [Google Scholar] [CrossRef] [Green Version]
- Rothgangl, T.; Dennis, M.K.; Lin, P.J.C.; Oka, R.; Witzigmann, D.; Villiger, L.; Qi, W.; Hruzova, M.; Kissling, L.; Lenggenhager, D.; et al. In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nat. Biotechnol. 2021, 39, 949–957. [Google Scholar] [CrossRef]
- Rodriguez, F.; Maron, D.J.; Knowles, J.W.; Virani, S.S.; Lin, S.; Heidenreich, P.A. Association of Statin Adherence With Mortality in Patients With Atherosclerotic Cardiovascular Disease. JAMA Cardiol. 2019, 4, 206–213. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; de Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadwick, A.C.; Evitt, N.H.; Lv, W.; Musunuru, K. Reduced Blood Lipid Levels With In Vivo CRISPR-Cas9 Base Editing of ANGPTL3. Circulation 2018, 137, 975–977. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, P.F.; Byun, J.H.; Platko, K.; Al-Hashimi, A.A.; Lhoták, Š.; MacDonald, M.E.; Mejia-Benitez, A.; Prat, A.; Igdoura, S.A.; Trigatti, B.; et al. Pcsk9 knockout exacerbates diet-induced non-alcoholic steatohepatitis, fibrosis and liver injury in mice. JHEP Rep. 2019, 1, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Hajighasemi, S.; Gorabi, A.M.; Bianconi, V.; Pirro, M.; Banach, M.; Tafti, H.A.; Reiner, Ž.; Sahebkar, A. A review of gene- and cell-based therapies for familial hypercholesterolemia. Pharmacol. Res. 2019, 143, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Janik, E.; Niemcewicz, M.; Ceremuga, M.; Krzowski, L.; Saluk-Bijak, J.; Bijak, M. Various Aspects of a Gene Editing System-CRISPR-Cas9. Int. J. Mol. Sci. 2020, 21, 9604. [Google Scholar] [CrossRef]
- Strong, A.; Musunuru, K. Genome editing in cardiovascular diseases. Nat. Rev. Cardiol. 2017, 14, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Laufs, U.; Ference, B.A. Vaccination to prevent atherosclerotic cardiovascular diseases. Eur. Heart J. 2017, 38, 2508–2510. [Google Scholar] [CrossRef] [PubMed] [Green Version]

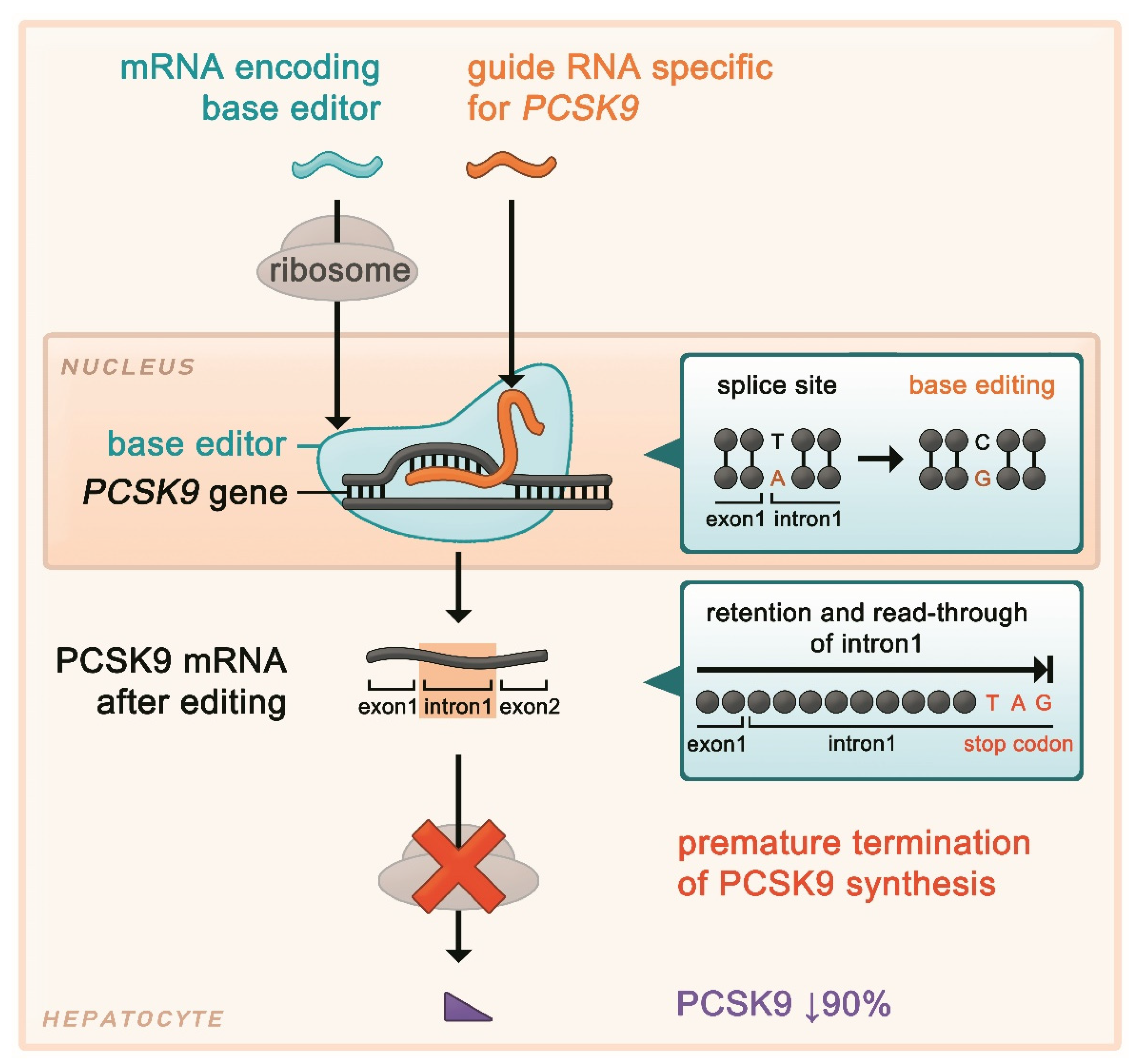

{kind=link}
{kind=link}
{kind=link}
| First Author, Journal (Year of Publication) [Reference] | Wang, Nature Biotechnology (2018/2021) [40,41] | Musunuru, Nature (2021) [6] | Rothgangl, Nature Biotechnology (2021) [44] |
|---|---|---|---|
| Non-human primate | Rhesus macaques (Macaca mulatta) | Cynomolgus macaques (Macaca fascicularis) | Cynomolgus macaques (Macaca fascicularis) |
| Delivery method | Adeno-associated virus transducing genetic information for the meganuclease | Lipid nanoparticle containing gRNA and base editor mRNA | Lipid nanoparticle containing gRNA and base editor mRNA |
| Genome editing technology | Meganuclease, induction of double strand breaks at specific site, random deletions/insertions after repair | Adenine base editor, specific single-nucleotide exchange | Adenine base editor, specific single-nucleotide exchange |
| Length of follow-up | Up to 3 years | 8 months | 29 days |
| PCSK9 editing * | Up to 46% | 66% | Max. 34%, mean 26% |
| Reduction PCSK9 | Up to 84% | 90% | 32% |
| Reduction LDL-C | Up to 60% | 60% | 14% |
| Off-target effects | Up to 629 detected off-target DNA cleavages | None detected | None detected |
| Immune response | T cell response | None detected | Humoral immune response against base editor |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katzmann, J.L.; Cupido, A.J.; Laufs, U. Gene Therapy Targeting PCSK9. Metabolites 2022, 12, 70. https://doi.org/10.3390/metabo12010070
Katzmann JL, Cupido AJ, Laufs U. Gene Therapy Targeting PCSK9. Metabolites. 2022; 12(1):70. https://doi.org/10.3390/metabo12010070
Chicago/Turabian StyleKatzmann, Julius L., Arjen J. Cupido, and Ulrich Laufs. 2022. "Gene Therapy Targeting PCSK9" Metabolites 12, no. 1: 70. https://doi.org/10.3390/metabo12010070
APA StyleKatzmann, J. L., Cupido, A. J., & Laufs, U. (2022). Gene Therapy Targeting PCSK9. Metabolites, 12(1), 70. https://doi.org/10.3390/metabo12010070