
The Emerging Roles of Intracellular PCSK9 and Their Implications in Endoplasmic Reticulum Stress and Metabolic Diseases


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
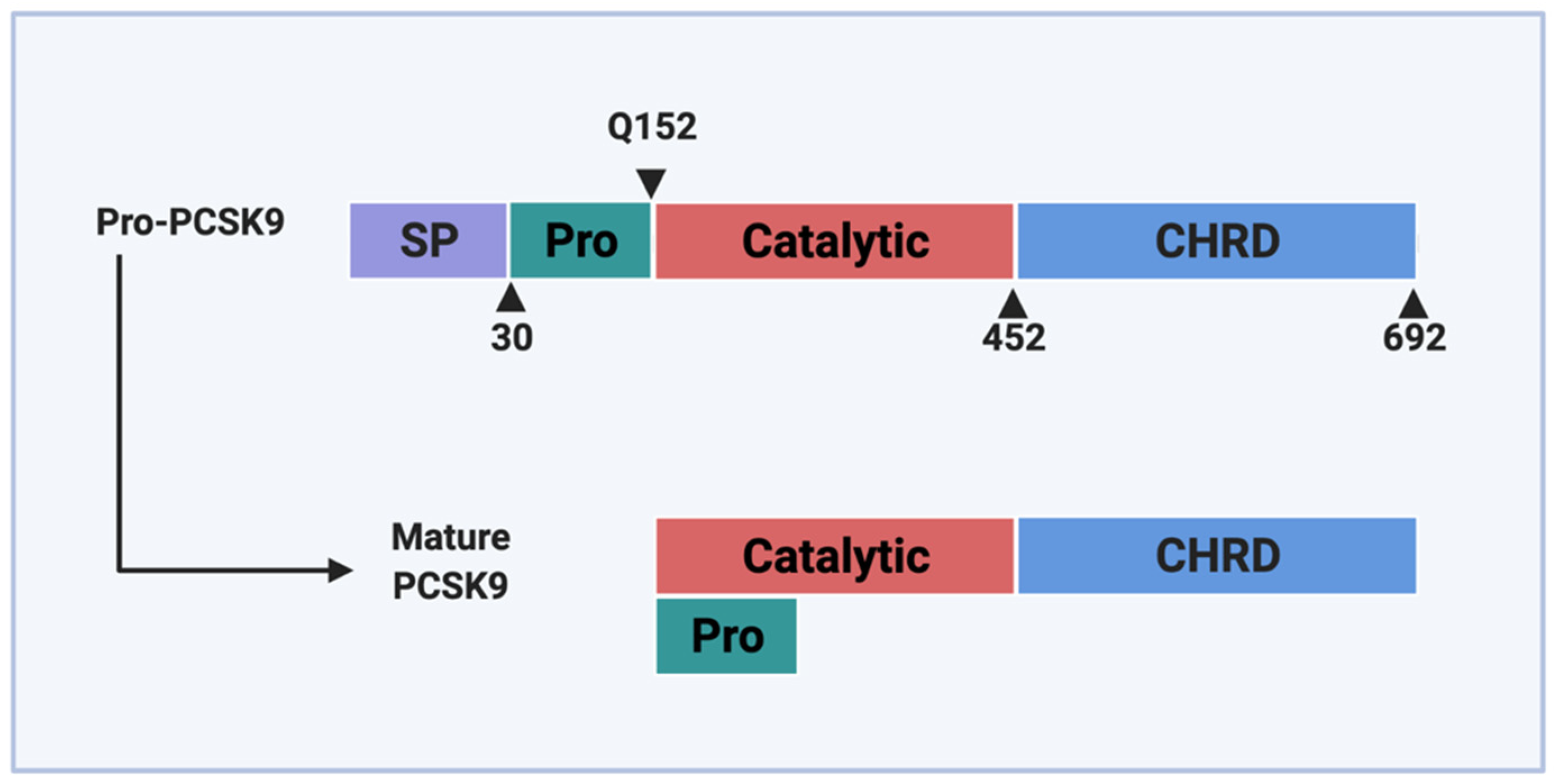
2. PCSK9 and Its Loss-of-Function Variants in the Endoplasmic Reticulum
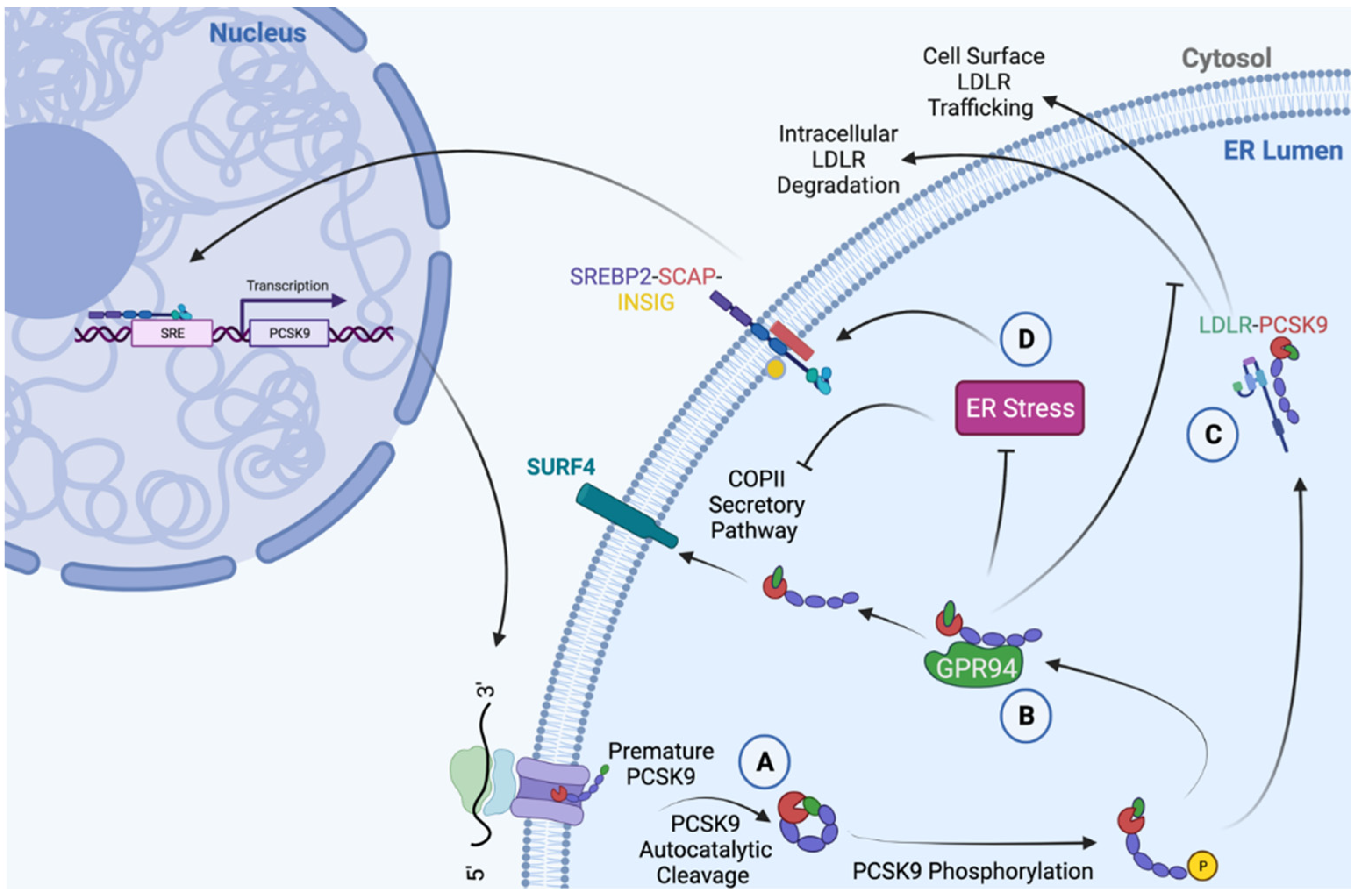
3. PCSK9 as a Putative Co-Chaperone of the ER
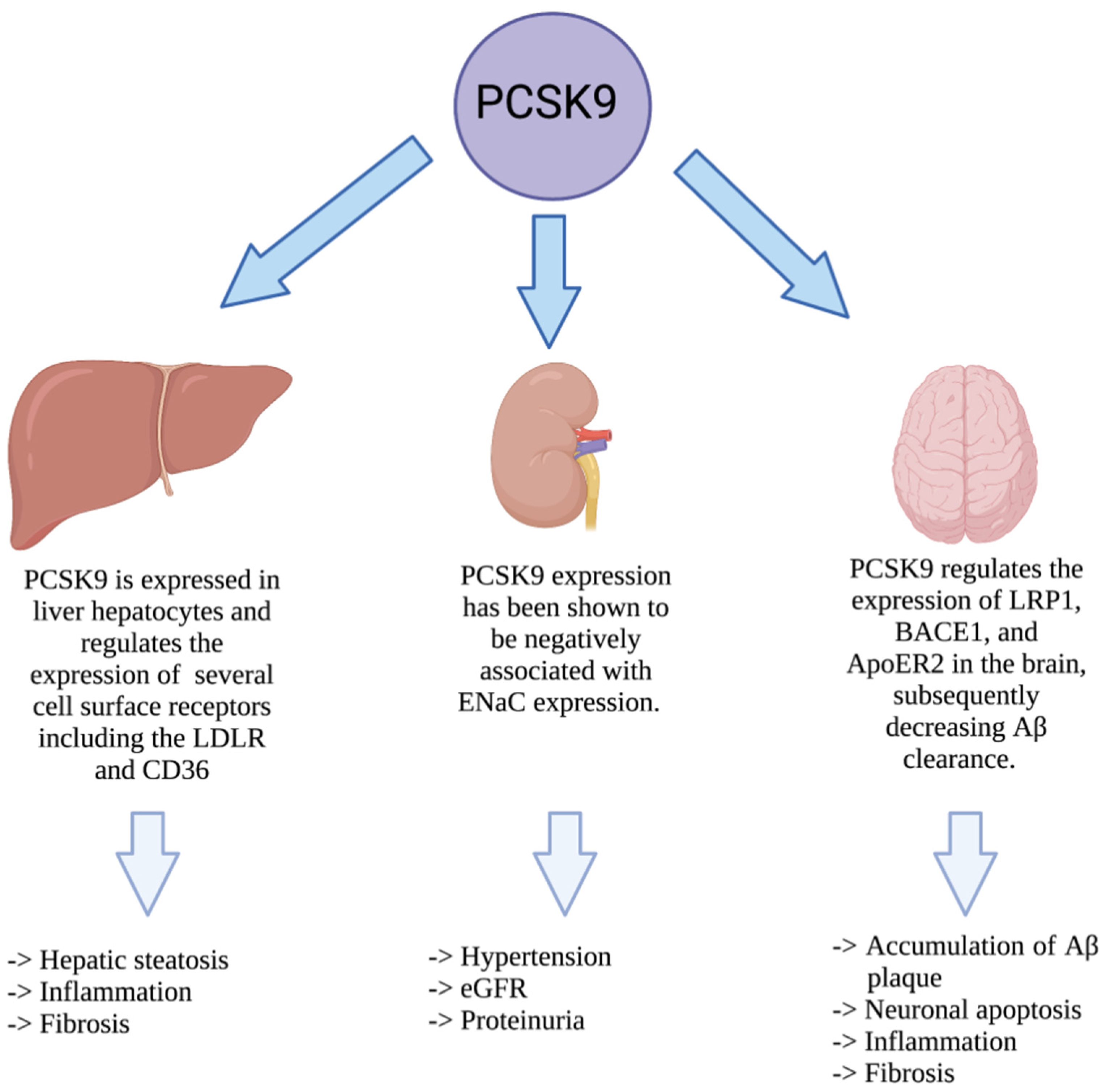
4. PCSK9 and Liver Disease
5. PCSK9 and Cardio-Renal Syndrome
6. PCSK9 and Neurodegenerative Disease
7. PCSK9 and Inflammatory Disease
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Abifadel, M.; Varret, M.; Rabes, J.P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef]
- Seidah, N.G.; Benjannet, S.; Wickham, L.; Marcinkiewicz, J.; Jasmin, S.B.; Stifani, S.; Basak, A.; Prat, A.; Chretien, M. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): Liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. USA 2003, 100, 928–933. [Google Scholar] [CrossRef]
- Cohen, J.; Pertsemlidis, A.; Kotowski, I.K.; Graham, R.; Garcia, C.K.; Hobbs, H.H. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 2005, 37, 161–165. [Google Scholar] [CrossRef]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H., Jr.; Hobbs, H.H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef]
- Mayne, J.; Dewpura, T.; Raymond, A.; Bernier, L.; Cousins, M.; Ooi, T.C.; Davignon, J.; Seidah, N.G.; Mbikay, M.; Chretien, M. Novel loss-of-function PCSK9 variant is associated with low plasma LDL cholesterol in a French-Canadian family and with impaired processing and secretion in cell culture. Clin. Chem. 2011, 57, 1415–1423. [Google Scholar] [CrossRef]
- Benjannet, S.; Hamelin, J.; Chretien, M.; Seidah, N.G. Loss- and gain-of-function PCSK9 variants: Cleavage specificity, dominant negative effects, and low density lipoprotein receptor (LDLR) degradation. J. Biol. Chem. 2012, 287, 33745–33755. [Google Scholar] [CrossRef]
- Chretien, M.; Mbikay, M. 60 YEARS OF POMC: From the prohormone theory to pro-opiomelanocortin and to proprotein convertases (PCSK1 to PCSK9). J. Mol. Endocrinol. 2016, 56, T49–T62. [Google Scholar] [CrossRef]
- Maxwell, K.N.; Breslow, J.L. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc. Natl. Acad. Sci. USA 2004, 101, 7100–7105. [Google Scholar] [CrossRef]
- Maxwell, K.N.; Fisher, E.A.; Breslow, J.L. Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. Proc. Natl. Acad. Sci. USA 2005, 102, 2069–2074. [Google Scholar] [CrossRef]
- Lagace, T.A.; Curtis, D.E.; Garuti, R.; McNutt, M.C.; Park, S.W.; Prather, H.B.; Anderson, N.N.; Ho, Y.K.; Hammer, R.E.; Horton, J.D. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J. Clin. Investig. 2006, 116, 2995–3005. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Prat, A. The multifaceted biology of PCSK9. Endocr. Rev. 2021, 1–25. [Google Scholar] [CrossRef]
- Benjannet, S.; Rhainds, D.; Essalmani, R.; Mayne, J.; Wickham, L.; Jin, W.; Asselin, M.C.; Hamelin, J.; Varret, M.; Allard, D.; et al. NARC-1/PCSK9 and its natural mutants: Zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J. Biol. Chem. 2004, 279, 48865–48875. [Google Scholar] [CrossRef]
- Seidah, N.G.; Mayer, G.; Zaid, A.; Rousselet, E.; Nassoury, N.; Poirier, S.; Essalmani, R.; Prat, A. The activation and physiological functions of the proprotein convertases. Int. J. Biochem. Cell Biol. 2008, 40, 1111–1125. [Google Scholar] [CrossRef]
- Saavedra, Y.G.; Day, R.; Seidah, N.G. The M2 module of the Cys-His-rich domain (CHRD) of PCSK9 protein is needed for the extracellular low-density lipoprotein receptor (LDLR) degradation pathway. J. Biol. Chem. 2012, 287, 43492–43501. [Google Scholar] [CrossRef]
- Cunningham, D.; Danley, D.E.; Geoghegan, K.F.; Griffor, M.C.; Hawkins, J.L.; Subashi, T.A.; Varghese, A.H.; Ammirati, M.J.; Culp, J.S.; Hoth, L.R.; et al. Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat. Struct. Mol. Biol. 2007, 14, 413–419. [Google Scholar] [CrossRef]
- Piper, D.E.; Romanow, W.G.; Gunawardane, R.N.; Fordstrom, P.; Masterman, S.; Pan, O.; Thibault, S.T.; Zhang, R.; Meininger, D.; Schwarz, M.; et al. The high-resolution crystal structure of human LCAT. J. Lipid Res. 2015, 56, 1711–1719. [Google Scholar] [CrossRef]
- Deng, S.J.; Shen, Y.; Gu, H.M.; Guo, S.; Wu, S.R.; Zhang, D.W. The role of the C-terminal domain of PCSK9 and SEC24 isoforms in PCSK9 secretion. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158660. [Google Scholar] [CrossRef]
- Emmer, B.T.; Hesketh, G.G.; Kotnik, E.; Tang, V.T.; Lascuna, P.J.; Xiang, J.; Gingras, A.C.; Chen, X.W.; Ginsburg, D. The cargo receptor SURF4 promotes the efficient cellular secretion of PCSK9. eLife 2018, 7, e38839. [Google Scholar] [CrossRef]
- Wang, B.; Shen, Y.; Zhai, L.; Xia, X.; Gu, H.M.; Wang, M.; Zhao, Y.; Chang, X.; Alabi, A.; Xing, S.; et al. Atherosclerosis-associated hepatic secretion of VLDL but not PCSK9 is dependent on cargo receptor protein Surf4. J. Lipid Res. 2021, 62, 100091. [Google Scholar] [CrossRef]
- Shen, Y.; Wang, B.; Deng, S.; Zhai, L.; Gu, H.M.; Alabi, A.; Xia, X.; Zhao, Y.; Chang, X.; Qin, S.; et al. Surf4 regulates expression of proprotein convertase subtilisin/kexin type 9 (PCSK9) but is not required for PCSK9 secretion in cultured human hepatocytes. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158555. [Google Scholar] [CrossRef]
- Rogers, M.A.; Hutcheson, J.D.; Okui, T.; Goettsch, C.; Singh, S.A.; Halu, A.; Schlotter, F.; Higashi, H.; Wang, L.; Whelan, M.C.; et al. Dynamin-related protein 1 inhibition reduces hepatic PCSK9 secretion. Cardiovasc. Res. 2021, 117, 2340–2353. [Google Scholar] [CrossRef]
- Dewpura, T.; Raymond, A.; Hamelin, J.; Seidah, N.G.; Mbikay, M.; Chretien, M.; Mayne, J. PCSK9 is phosphorylated by a Golgi casein kinase-like kinase ex vivo and circulates as a phosphoprotein in humans. FEBS J. 2008, 275, 3480–3493. [Google Scholar] [CrossRef] [PubMed]
- Ben Djoudi Ouadda, A.; Gauthier, M.S.; Susan-Resiga, D.; Girard, E.; Essalmani, R.; Black, M.; Marcinkiewicz, J.; Forget, D.; Hamelin, J.; Evagelidis, A.; et al. Ser-Phosphorylation of PCSK9 (Proprotein Convertase Subtilisin-Kexin 9) by Fam20C (Family with Sequence Similarity 20, Member C) Kinase Enhances Its Ability to Degrade the LDLR (Low-Density Lipoprotein Receptor). Arter. Thromb. Vasc. Biol. 2019, 39, 1996–2013. [Google Scholar] [CrossRef]
- Benjannet, S.; Rhainds, D.; Hamelin, J.; Nassoury, N.; Seidah, N.G. The proprotein convertase (PC) PCSK9 is inactivated by furin and/or PC5/6A: Functional consequences of natural mutations and post-translational modifications. J. Biol. Chem. 2006, 281, 30561–30572. [Google Scholar] [CrossRef]
- Jeong, H.J.; Lee, H.S.; Kim, K.S.; Kim, Y.K.; Yoon, D.; Park, S.W. Sterol-dependent regulation of proprotein convertase subtilisin/kexin type 9 expression by sterol-regulatory element binding protein-2. J. Lipid Res. 2008, 49, 399–409. [Google Scholar] [CrossRef]
- Dong, B.; Wu, M.; Li, H.; Kraemer, F.B.; Adeli, K.; Seidah, N.G.; Park, S.W.; Liu, J. Strong induction of PCSK9 gene expression through HNF1alpha and SREBP2: Mechanism for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters. J. Lipid Res. 2010, 51, 1486–1495. [Google Scholar] [CrossRef]
- Dubuc, G.; Chamberland, A.; Wassef, H.; Davignon, J.; Seidah, N.G.; Bernier, L.; Prat, A. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arter. Thromb. Vasc. Biol. 2004, 24, 1454–1459. [Google Scholar] [CrossRef]
- Sakai, J.; Rawson, R.B.; Espenshade, P.J.; Cheng, D.; Seegmiller, A.C.; Goldstein, J.L.; Brown, M.S. Molecular identification of the sterol-regulated luminal protease that cleaves SREBPs and controls lipid composition of animal cells. Mol. Cell 1998, 2, 505–514. [Google Scholar] [CrossRef]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein sensors for membrane sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Schuck, S.; Prinz, W.A.; Thorn, K.S.; Voss, C.; Walter, P. Correction: Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell Biol. 2021, 220. [Google Scholar] [CrossRef]
- Colgan, S.M.; Hashimi, A.A.; Austin, R.C. Endoplasmic reticulum stress and lipid dysregulation. Expert Rev. Mol. Med. 2011, 13, e4. [Google Scholar] [CrossRef]
- Colgan, S.M.; Tang, D.; Werstuck, G.H.; Austin, R.C. Endoplasmic reticulum stress causes the activation of sterol regulatory element binding protein-2. Int. J. Biochem. Cell Biol. 2007, 39, 1843–1851. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, P.F.; Byun, J.H.; Platko, K.; Saliba, P.; Sguazzin, M.; MacDonald, M.E.; Pare, G.; Steinberg, G.R.; Janssen, L.J.; Igdoura, S.A.; et al. Caffeine blocks SREBP2-induced hepatic PCSK9 expression to enhance LDLR-mediated cholesterol clearance. Nat. Commun. 2022, 13, 770. [Google Scholar] [CrossRef]
- Dong, B.; Singh, A.B.; Shende, V.R.; Liu, J. Hepatic HNF1 transcription factors control the induction of PCSK9 mediated by rosuvastatin in normolipidemic hamsters. Int. J. Mol. Med. 2017, 39, 749–756. [Google Scholar] [CrossRef]
- Costet, P.; Cariou, B.; Lambert, G.; Lalanne, F.; Lardeux, B.; Jarnoux, A.L.; Grefhorst, A.; Staels, B.; Krempf, M. Hepatic PCSK9 expression is regulated by nutritional status via insulin and sterol regulatory element-binding protein 1c. J. Biol. Chem. 2006, 281, 6211–6218. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, C.L.; Wiederkehr, A.; Baquie, M.; Akhmedov, D.; Wang, H.; Gauthier, B.R.; Akerman, I.; Ishihara, H.; Ferrer, J.; Wollheim, C.B. Hepatic nuclear factor 1alpha (HNF1alpha) dysfunction down-regulates X-box-binding protein 1 (XBP1) and sensitizes beta-cells to endoplasmic reticulum stress. J. Biol. Chem. 2011, 286, 32300–32312. [Google Scholar] [CrossRef]
- Naureckiene, S.; Ma, L.; Sreekumar, K.; Purandare, U.; Lo, C.F.; Huang, Y.; Chiang, L.W.; Grenier, J.M.; Ozenberger, B.A.; Jacobsen, J.S.; et al. Functional characterization of Narc 1, a novel proteinase related to proteinase K. Arch. Biochem. Biophys. 2003, 420, 55–67. [Google Scholar] [CrossRef]
- Chorba, J.S.; Shokat, K.M. The proprotein convertase subtilisin/kexin type 9 (PCSK9) active site and cleavage sequence differentially regulate protein secretion from proteolysis. J. Biol. Chem. 2014, 289, 29030–29043. [Google Scholar] [CrossRef]
- Dron, J.S.; Hegele, R.A. Complexity of mechanisms among human proprotein convertase subtilisin-kexin type 9 variants. Curr. Opin. Lipidol. 2017, 28, 161–169. [Google Scholar] [CrossRef]
- Cameron, J.; Holla, O.L.; Laerdahl, J.K.; Kulseth, M.A.; Ranheim, T.; Rognes, T.; Berge, K.E.; Leren, T.P. Characterization of novel mutations in the catalytic domain of the PCSK9 gene. J. Intern. Med. 2008, 263, 420–431. [Google Scholar] [CrossRef]
- Seidah, N.G.; Awan, Z.; Chretien, M.; Mbikay, M. PCSK9: A key modulator of cardiovascular health. Circ. Res. 2014, 114, 1022–1036. [Google Scholar] [CrossRef]
- Zhao, Z.; Tuakli-Wosornu, Y.; Lagace, T.A.; Kinch, L.; Grishin, N.V.; Horton, J.D.; Cohen, J.C.; Hobbs, H.H. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am. J. Hum. Genet. 2006, 79, 514–523. [Google Scholar] [CrossRef]
- Lebeau, P.F.; Wassef, H.; Byun, J.H.; Platko, K.; Ason, B.; Jackson, S.; Dobroff, J.; Shetterly, S.; Richards, W.G.; Al-Hashimi, A.A.; et al. The loss-of-function PCSK9Q152H variant increases ER chaperones GRP78 and GRP94 and protects against liver injury. J. Clin. Investig. 2021, 131, e128650. [Google Scholar] [CrossRef] [PubMed]
- Slimani, A.; Hrira, M.Y.; Najah, M.; Jomaa, W.; Maatouk, F.; Hamda, K.B.; Abifadel, M.; Rabes, J.P.; Boileau, C.; Rouis, M.; et al. PCSK9 polymorphism in a Tunisian cohort: Identification of a new allele, L8, and association of allele L10 with reduced coronary heart disease risk. Mol. Cell Probes 2015, 29, 1–6. [Google Scholar] [CrossRef]
- Pisciotta, L.; Sallo, R.; Rabacchi, C.; Wunsch, A.; Calandra, S.; Bertolini, S. Leucine 10 allelic variant in signal peptide of PCSK9 increases the LDL cholesterol-lowering effect of statins in patients with familial hypercholesterolaemia. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 831–835. [Google Scholar] [CrossRef]
- Cameron, J.; Holla, O.L.; Laerdahl, J.K.; Kulseth, M.A.; Berge, K.E.; Leren, T.P. Mutation S462P in the PCSK9 gene reduces secretion of mutant PCSK9 without affecting the autocatalytic cleavage. Atherosclerosis 2009, 203, 161–165. [Google Scholar] [CrossRef]
- Cariou, B.; Ouguerram, K.; Zair, Y.; Guerois, R.; Langhi, C.; Kourimate, S.; Benoit, I.; Le May, C.; Gayet, C.; Belabbas, K.; et al. PCSK9 dominant negative mutant results in increased LDL catabolic rate and familial hypobetalipoproteinemia. Arter. Thromb. Vasc. Biol. 2009, 29, 2191–2197. [Google Scholar] [CrossRef]
- Abifadel, M.; Elbitar, S.; El Khoury, P.; Ghaleb, Y.; Chemaly, M.; Moussalli, M.L.; Rabes, J.P.; Varret, M.; Boileau, C. Living the PCSK9 adventure: From the identification of a new gene in familial hypercholesterolemia towards a potential new class of anticholesterol drugs. Curr. Atheroscler. Rep. 2014, 16, 439. [Google Scholar] [CrossRef] [PubMed]
- Dubuc, G.; Tremblay, M.; Pare, G.; Jacques, H.; Hamelin, J.; Benjannet, S.; Boulet, L.; Genest, J.; Bernier, L.; Seidah, N.G.; et al. A new method for measurement of total plasma PCSK9: Clinical applications. J. Lipid Res. 2010, 51, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Teckman, J.H.; Perlmutter, D.H. Retention of mutant alpha(1)-antitrypsin Z in endoplasmic reticulum is associated with an autophagic response. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G961–G974. [Google Scholar] [CrossRef]
- Lin, L.; Schmidt, B.; Teckman, J.; Perlmutter, D.H. A naturally occurring nonpolymerogenic mutant of alpha 1-antitrypsin characterized by prolonged retention in the endoplasmic reticulum. J. Biol. Chem. 2001, 276, 33893–33898. [Google Scholar] [CrossRef]
- Beuret, N.; Rutishauser, J.; Bider, M.D.; Spiess, M. Mechanism of endoplasmic reticulum retention of mutant vasopressin precursor caused by a signal peptide truncation associated with diabetes insipidus. J. Biol. Chem. 1999, 274, 18965–18972. [Google Scholar] [CrossRef]
- Birk, J.; Friberg, M.A.; Prescianotto-Baschong, C.; Spiess, M.; Rutishauser, J. Dominant pro-vasopressin mutants that cause diabetes insipidus form disulfide-linked fibrillar aggregates in the endoplasmic reticulum. J. Cell Sci. 2009, 122, 3994–4002. [Google Scholar] [CrossRef]
- Gilbert, A.; Jadot, M.; Leontieva, E.; Wattiaux-De Coninck, S.; Wattiaux, R. Delta F508 CFTR localizes in the endoplasmic reticulum-Golgi intermediate compartment in cystic fibrosis cells. Exp. Cell Res. 1998, 242, 144–152. [Google Scholar] [CrossRef]
- Kim, P.S.; Lee, J.; Jongsamak, P.; Menon, S.; Li, B.; Hossain, S.A.; Bae, J.H.; Panijpan, B.; Arvan, P. Defective protein folding and intracellular retention of thyroglobulin-R19K mutant as a cause of human congenital goiter. Mol. Endocrinol. 2008, 22, 477–484. [Google Scholar] [CrossRef]
- Takahashi, K.; Adachi, K.; Yoshizaki, K.; Kunimoto, S.; Kalaria, R.N.; Watanabe, A. Mutations in NOTCH3 cause the formation and retention of aggregates in the endoplasmic reticulum, leading to impaired cell proliferation. Hum. Mol. Genet. 2010, 19, 79–89. [Google Scholar] [CrossRef]
- Austin, R.C. The unfolded protein response in health and disease. Antioxid Redox Signal 2009, 11, 2279–2287. [Google Scholar] [CrossRef]
- Ruggiano, A.; Foresti, O.; Carvalho, P. Quality control: ER-associated degradation: Protein quality control and beyond. J. Cell Biol. 2014, 204, 869–879. [Google Scholar] [CrossRef]
- Lin, J.H.; Walter, P.; Yen, T.S. Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425. [Google Scholar] [CrossRef]
- Schonthal, A.H. Endoplasmic reticulum stress: Its role in disease and novel prospects for therapy. Scientifica 2012, 2012, 857516. [Google Scholar] [CrossRef]
- Ghemrawi, R.; Battaglia-Hsu, S.F.; Arnold, C. Endoplasmic Reticulum Stress in Metabolic Disorders. Cells 2018, 7, 63. [Google Scholar] [CrossRef]
- Poirier, S.; Mamarbachi, M.; Chen, W.T.; Lee, A.S.; Mayer, G. GRP94 Regulates Circulating Cholesterol Levels through Blockade of PCSK9-Induced LDLR Degradation. Cell Rep. 2015, 13, 2064–2071. [Google Scholar] [CrossRef]
- Lebeau, P.; Platko, K.; Al-Hashimi, A.A.; Byun, J.H.; Lhoták, Š.; Holzapfel, N.; Gyulay, G.; Igdoura, S.A.; Cool, D.R.; Trigatti, B.; et al. Loss-of-function PCSK9 mutants evade the unfolded protein response sensor GRP78 and fail to induce endoplasmic reticulum stress when retained. J. Biol. Chem. 2018, 293, 7329–7343. [Google Scholar] [CrossRef]
- Lebeau, P.; Al-Hashimi, A.; Sood, S.; Lhotak, S.; Yu, P.; Gyulay, G.; Pare, G.; Chen, S.R.; Trigatti, B.; Prat, A.; et al. Endoplasmic Reticulum Stress and Ca2+ Depletion Differentially Modulate the Sterol Regulatory Protein PCSK9 to Control Lipid Metabolism. J. Biol. Chem. 2017, 292, 1510–1523. [Google Scholar] [CrossRef]
- Strøm, T.B.; Tveten, K.; Leren, T.P. PCSK9 acts as a chaperone for the LDL receptor in the endoplasmic reticulum. Biochem. J. 2014, 457, 99–105. [Google Scholar] [CrossRef]
- Lin, J.; Chung, S.; Ueda, K.; Matsuda, K.; Nakamura, Y.; Park, J.H. GALNT6 Stabilizes GRP78 Protein by O-glycosylation and Enhances its Activity to Suppress Apoptosis Under Stress Condition. Neoplasia 2017, 19, 43–53. [Google Scholar] [CrossRef]
- Gupta, M.K.; Tahrir, F.G.; Knezevic, T.; White, M.K.; Gordon, J.; Cheung, J.Y.; Khalili, K.; Feldman, A.M. GRP78 Interacting Partner Bag5 Responds to ER Stress and Protects Cardiomyocytes From ER Stress-Induced Apoptosis. J. Cell Biochem. 2016, 117, 1813–1821. [Google Scholar] [CrossRef]
- Bellentani, S. The epidemiology of non-alcoholic fatty liver disease. Liver Int. 2017, 37 (Suppl. 1), 81–84. [Google Scholar] [CrossRef]
- Brunt, E.M.; Wong, V.W.; Nobili, V.; Day, C.P.; Sookoian, S.; Maher, J.J.; Bugianesi, E.; Sirlin, C.B.; Neuschwander-Tetri, B.A.; Rinella, M.E. Nonalcoholic fatty liver disease. Nat. Rev. Dis. Primers 2015, 1, 15080. [Google Scholar] [CrossRef]
- Gruben, N.; Shiri-Sverdlov, R.; Koonen, D.P.; Hofker, M.H. Nonalcoholic fatty liver disease: A main driver of insulin resistance or a dangerous liaison? Biochim. Biophys. Acta 2014, 1842, 2329–2343. [Google Scholar] [CrossRef]
- Deprince, A.; Haas, J.T.; Staels, B. Dysregulated lipid metabolism links NAFLD to cardiovascular disease. Mol. Metab. 2020, 42, 101092. [Google Scholar] [CrossRef]
- Targher, G.; Byrne, C.D.; Lonardo, A.; Zoppini, G.; Barbui, C. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: A meta-analysis. J. Hepatol. 2016, 65, 589–600. [Google Scholar] [CrossRef]
- Mantovani, A.; Zaza, G.; Byrne, C.D.; Lonardo, A.; Zoppini, G.; Bonora, E.; Targher, G. Nonalcoholic fatty liver disease increases risk of incident chronic kidney disease: A systematic review and meta-analysis. Metabolism 2018, 79, 64–76. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Lebeau, P.F.; Byun, J.H.; Platko, K.; MacDonald, M.E.; Poon, S.V.; Faiyaz, M.; Seidah, N.G.; Austin, R.C. Diet-induced hepatic steatosis abrogates cell-surface LDLR by inducing de novo PCSK9 expression in mice. J. Biol. Chem. 2019, 294, 9037–9047. [Google Scholar] [CrossRef]
- Demers, A.; Samami, S.; Lauzier, B.; Des Rosiers, C.; Ngo Sock, E.T.; Ong, H.; Mayer, G. PCSK9 Induces CD36 Degradation and Affects Long-Chain Fatty Acid Uptake and Triglyceride Metabolism in Adipocytes and in Mouse Liver. Arter. Thromb. Vasc. Biol. 2015, 35, 2517–2525. [Google Scholar] [CrossRef]
- Lebeau, P.F.; Byun, J.H.; Platko, K.; Al-Hashimi, A.A.; Lhotak, S.; MacDonald, M.E.; Mejia-Benitez, A.; Prat, A.; Igdoura, S.A.; Trigatti, B.; et al. Pcsk9 knockout exacerbates diet-induced non-alcoholic steatohepatitis, fibrosis and liver injury in mice. JHEP Rep. 2019, 1, 418–429. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Lee, S.P.; Linsley, P.S.; Gersuk, V.; Yeh, M.M.; Chen, Y.Y.; Peng, Y.J.; Dutta, M.; Mascarinas, G.; Molla, B.; et al. Pcsk9 Deletion Promotes Murine Nonalcoholic Steatohepatitis and Hepatic Carcinogenesis: Role of Cholesterol. Hepatol. Commun. 2021. [Google Scholar] [CrossRef]
- Shapiro, M.D.; Miles, J.; Tavori, H.; Fazio, S. Diagnosing Resistance to a Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitor. Ann. Intern. Med. 2018, 168, 376–379. [Google Scholar] [CrossRef]
- Grimaudo, S.; Bartesaghi, S.; Rametta, R.; Marra, F.; Margherita Mancina, R.; Pihlajamaki, J.; Kakol-Palm, D.; Andreasson, A.C.; Dongiovanni, P.; Ludovica Fracanzani, A.; et al. PCSK9 rs11591147 R46L loss-of-function variant protects against liver damage in individuals with NAFLD. Liver Int. 2021, 41, 321–332. [Google Scholar] [CrossRef]
- Lai, Q.; Giralt, A.; Le May, C.; Zhang, L.; Cariou, B.; Denechaud, P.D.; Fajas, L. E2F1 inhibits circulating cholesterol clearance by regulating Pcsk9 expression in the liver. JCI Insight 2017, 2, e89729. [Google Scholar] [CrossRef]
- Emma, M.R.; Giannitrapani, L.; Cabibi, D.; Porcasi, R.; Pantuso, G.; Augello, G.; Giglio, R.V.; Re, N.L.; Capitano, A.R.; Montalto, G.; et al. Hepatic and circulating levels of PCSK9 in morbidly obese patients: Relation with severity of liver steatosis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158792. [Google Scholar] [CrossRef]
- Ruscica, M.; Ferri, N.; Macchi, C.; Meroni, M.; Lanti, C.; Ricci, C.; Maggioni, M.; Fracanzani, A.L.; Badiali, S.; Fargion, S.; et al. Liver fat accumulation is associated with circulating PCSK9. Ann. Med. 2016, 48, 384–391. [Google Scholar] [CrossRef]
- Wargny, M.; Ducluzeau, P.H.; Petit, J.M.; Le May, C.; Smati, S.; Arnaud, L.; Pichelin, M.; Bouillet, B.; Lannes, A.; Blanchet, O.; et al. Circulating PCSK9 levels are not associated with the severity of hepatic steatosis and NASH in a high-risk population. Atherosclerosis 2018, 278, 82–90. [Google Scholar] [CrossRef]
- Baragetti, A.; Balzarotti, G.; Grigore, L.; Pellegatta, F.; Guerrini, U.; Pisano, G.; Fracanzani, A.L.; Fargion, S.; Norata, G.D.; Catapano, A.L. PCSK9 deficiency results in increased ectopic fat accumulation in experimental models and in humans. Eur. J. Prev. Cardiol. 2017, 24, 1870–1877. [Google Scholar] [CrossRef]
- Walley, K.R.; Thain, K.R.; Russell, J.A.; Reilly, M.P.; Meyer, N.J.; Ferguson, J.F.; Christie, J.D.; Nakada, T.A.; Fjell, C.D.; Thair, S.A.; et al. PCSK9 is a critical regulator of the innate immune response and septic shock outcome. Sci. Transl. Med. 2014, 6, 258ra143. [Google Scholar] [CrossRef]
- Konarzewski, M.; Szolkiewicz, M.; Sucajtys-Szulc, E.; Blaszak, J.; Lizakowski, S.; Swierczynski, J.; Rutkowski, B. Elevated circulating PCSK-9 concentration in renal failure patients is corrected by renal replacement therapy. Am. J. Nephrol. 2014, 40, 157–163. [Google Scholar] [CrossRef]
- Rogacev, K.S.; Heine, G.H.; Silbernagel, G.; Kleber, M.E.; Seiler, S.; Emrich, I.; Lennartz, S.; Werner, C.; Zawada, A.M.; Fliser, D.; et al. PCSK9 Plasma Concentrations Are Independent of GFR and Do Not Predict Cardiovascular Events in Patients with Decreased GFR. PLoS ONE 2016, 11, e0146920. [Google Scholar] [CrossRef]
- Morena, M.; Le May, C.; Chenine, L.; Arnaud, L.; Dupuy, A.M.; Pichelin, M.; Leray-Moragues, H.; Chalabi, L.; Canaud, B.; Cristol, J.P.; et al. Plasma PCSK9 concentrations during the course of nondiabetic chronic kidney disease: Relationship with glomerular filtration rate and lipid metabolism. J. Clin. Lipidol. 2017, 11, 87–93. [Google Scholar] [CrossRef]
- Wu, D.; Zhou, Y.; Pan, Y.; Li, C.; Wang, Y.; Chen, F.; Chen, X.; Yang, S.; Zhou, Z.; Liao, Y.; et al. Vaccine against PCSK9 Improved Renal Fibrosis by Regulating Fatty Acid beta-Oxidation. J. Am. Heart Assoc. 2020, 9, e014358. [Google Scholar] [CrossRef]
- Zhang, G.; Li, Q. Inflammation Induces Lipid Deposition in Kidneys by Downregulating Renal PCSK9 in Mice with Adriamycin-Induced Nephropathy. Med. Sci. Monit. 2019, 25, 5327–5335. [Google Scholar] [CrossRef]
- Barisione, C.; Verzola, D.; Garibaldi, S.; Ferrari, P.F.; Garibotto, G.; Ameri, P.; Pane, B.; Spinella, G.; Pratesi, G.; Palombo, D. Renal Ischemia/Reperfusion Early Induces Myostatin and PCSK9 Expression in Rat Kidneys and HK-2 Cells. Int. J. Mol. Sci. 2021, 22, 9884. [Google Scholar] [CrossRef]
- Sharotri, V.; Collier, D.M.; Olson, D.R.; Zhou, R.; Snyder, P.M. Regulation of epithelial sodium channel trafficking by proprotein convertase subtilisin/kexin type 9 (PCSK9). J. Biol. Chem. 2012, 287, 19266–19274. [Google Scholar] [CrossRef]
- Berger, J.M.; Vaillant, N.; Le May, C.; Calderon, C.; Bregeon, J.; Prieur, X.; Hadchouel, J.; Loirand, G.; Cariou, B. PCSK9-deficiency does not alter blood pressure and sodium balance in mouse models of hypertension. Atherosclerosis 2015, 239, 252–259. [Google Scholar] [CrossRef]
- Bjorkhem, I.; Meaney, S. Brain cholesterol: Long secret life behind a barrier. Arter. Thromb. Vasc. Biol. 2004, 24, 806–815. [Google Scholar] [CrossRef]
- Tracey, T.J.; Steyn, F.J.; Wolvetang, E.J.; Ngo, S.T. Neuronal Lipid Metabolism: Multiple Pathways Driving Functional Outcomes in Health and Disease. Front. Mol. Neurosci. 2018, 11, 10. [Google Scholar] [CrossRef]
- Wu, Q.; Tang, Z.H.; Peng, J.; Liao, L.; Pan, L.H.; Wu, C.Y.; Jiang, Z.S.; Wang, G.X.; Liu, L.S. The dual behavior of PCSK9 in the regulation of apoptosis is crucial in Alzheimer’s disease progression (Review). Biomed. Rep. 2014, 2, 167–171. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Z.; Shi, J.; Jiang, Q.; Wang, H.; Li, X.; Hao, D. Inhibition of proprotein convertase subtilisin/kexin type 9 attenuates neuronal apoptosis following focal cerebral ischemia via apolipoprotein E receptor 2 downregulation in hyperlipidemic mice. Int. J. Mol. Med. 2018, 42, 2098–2106. [Google Scholar] [CrossRef]
- Abuelezz, S.A.; Hendawy, N. HMGB1/RAGE/TLR4 axis and glutamate as novel targets for PCSK9 inhibitor in high fat cholesterol diet induced cognitive impairment and amyloidosis. Life Sci. 2021, 273, 119310. [Google Scholar] [CrossRef]
- Adorni, M.P.; Ruscica, M.; Ferri, N.; Bernini, F.; Zimetti, F. Proprotein Convertase Subtilisin/Kexin Type 9, Brain Cholesterol Homeostasis and Potential Implication for Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 120. [Google Scholar] [CrossRef]
- Zimetti, F.; Caffarra, P.; Ronda, N.; Favari, E.; Adorni, M.P.; Zanotti, I.; Bernini, F.; Barocco, F.; Spallazzi, M.; Galimberti, D.; et al. Increased PCSK9 Cerebrospinal Fluid Concentrations in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 55, 315–320. [Google Scholar] [CrossRef]
- Jonas, M.C.; Costantini, C.; Puglielli, L. PCSK9 is required for the disposal of non-acetylated intermediates of the nascent membrane protein BACE1. EMBO Rep. 2008, 9, 916–922. [Google Scholar] [CrossRef]
- Apaijai, N.; Moisescu, D.M.; Palee, S.; McSweeney, C.M.; Saiyasit, N.; Maneechote, C.; Boonnag, C.; Chattipakorn, N.; Chattipakorn, S.C. Pretreatment With PCSK9 Inhibitor Protects the Brain Against Cardiac Ischemia/Reperfusion Injury Through a Reduction of Neuronal Inflammation and Amyloid Beta Aggregation. J. Am. Heart Assoc. 2019, 8, e010838. [Google Scholar] [CrossRef]
- Liu, M.; Wu, G.; Baysarowich, J.; Kavana, M.; Addona, G.H.; Bierilo, K.K.; Mudgett, J.S.; Pavlovic, G.; Sitlani, A.; Renger, J.J.; et al. PCSK9 is not involved in the degradation of LDL receptors and BACE1 in the adult mouse brain. J. Lipid Res. 2010, 51, 2611–2618. [Google Scholar] [CrossRef]
- Skalen, K.; Gustafsson, M.; Rydberg, E.K.; Hulten, L.M.; Wiklund, O.; Innerarity, T.L.; Boren, J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 2002, 417, 750–754. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Y.; Xu, R.X.; Guo, Y.L.; Zhu, C.G.; Wu, N.Q.; Qing, P.; Liu, G.; Dong, Q.; Li, J.J. Proprotein convertase subtilisin-kexin type 9 as a biomarker for the severity of coronary artery disease. Ann. Med. 2015, 47, 386–393. [Google Scholar] [CrossRef]
- Li, S.; Guo, Y.L.; Xu, R.X.; Zhang, Y.; Zhu, C.G.; Sun, J.; Qing, P.; Wu, N.Q.; Jiang, L.X.; Li, J.J. Association of plasma PCSK9 levels with white blood cell count and its subsets in patients with stable coronary artery disease. Atherosclerosis 2014, 234, 441–445. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhu, C.G.; Xu, R.X.; Li, S.; Guo, Y.L.; Sun, J.; Li, J.J. Relation of circulating PCSK9 concentration to fibrinogen in patients with stable coronary artery disease. J. Clin. Lipidol. 2014, 8, 494–500. [Google Scholar] [CrossRef]
- Tang, Z.H.; Peng, J.; Ren, Z.; Yang, J.; Li, T.T.; Li, T.H.; Wang, Z.; Wei, D.H.; Liu, L.S.; Zheng, X.L.; et al. New role of PCSK9 in atherosclerotic inflammation promotion involving the TLR4/NF-kappaB pathway. Atherosclerosis 2017, 262, 113–122. [Google Scholar] [CrossRef]
- Dwivedi, D.J.; Grin, P.M.; Khan, M.; Prat, A.; Zhou, J.; Fox-Robichaud, A.E.; Seidah, N.G.; Liaw, P.C. Differential Expression of PCSK9 Modulates Infection, Inflammation, and Coagulation in a Murine Model of Sepsis. Shock 2016, 46, 672–680. [Google Scholar] [CrossRef]
- Feingold, K.R.; Moser, A.H.; Shigenaga, J.K.; Patzek, S.M.; Grunfeld, C. Inflammation stimulates the expression of PCSK9. Biochem. Biophys. Res. Commun. 2008, 374, 341–344. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lebeau, P.F.; Platko, K.; Byun, J.H.; Makda, Y.; Austin, R.C. The Emerging Roles of Intracellular PCSK9 and Their Implications in Endoplasmic Reticulum Stress and Metabolic Diseases. Metabolites 2022, 12, 215. https://doi.org/10.3390/metabo12030215
Lebeau PF, Platko K, Byun JH, Makda Y, Austin RC. The Emerging Roles of Intracellular PCSK9 and Their Implications in Endoplasmic Reticulum Stress and Metabolic Diseases. Metabolites. 2022; 12(3):215. https://doi.org/10.3390/metabo12030215
Chicago/Turabian StyleLebeau, Paul F., Khrystyna Platko, Jae Hyun Byun, Yumna Makda, and Richard C. Austin. 2022. "The Emerging Roles of Intracellular PCSK9 and Their Implications in Endoplasmic Reticulum Stress and Metabolic Diseases" Metabolites 12, no. 3: 215. https://doi.org/10.3390/metabo12030215
APA StyleLebeau, P. F., Platko, K., Byun, J. H., Makda, Y., & Austin, R. C. (2022). The Emerging Roles of Intracellular PCSK9 and Their Implications in Endoplasmic Reticulum Stress and Metabolic Diseases. Metabolites, 12(3), 215. https://doi.org/10.3390/metabo12030215