
Genome-Scale Metabolic Modeling Enables In-Depth Understanding of Big Data

 ,
,  , and
, and 
Abstract
1. Introduction
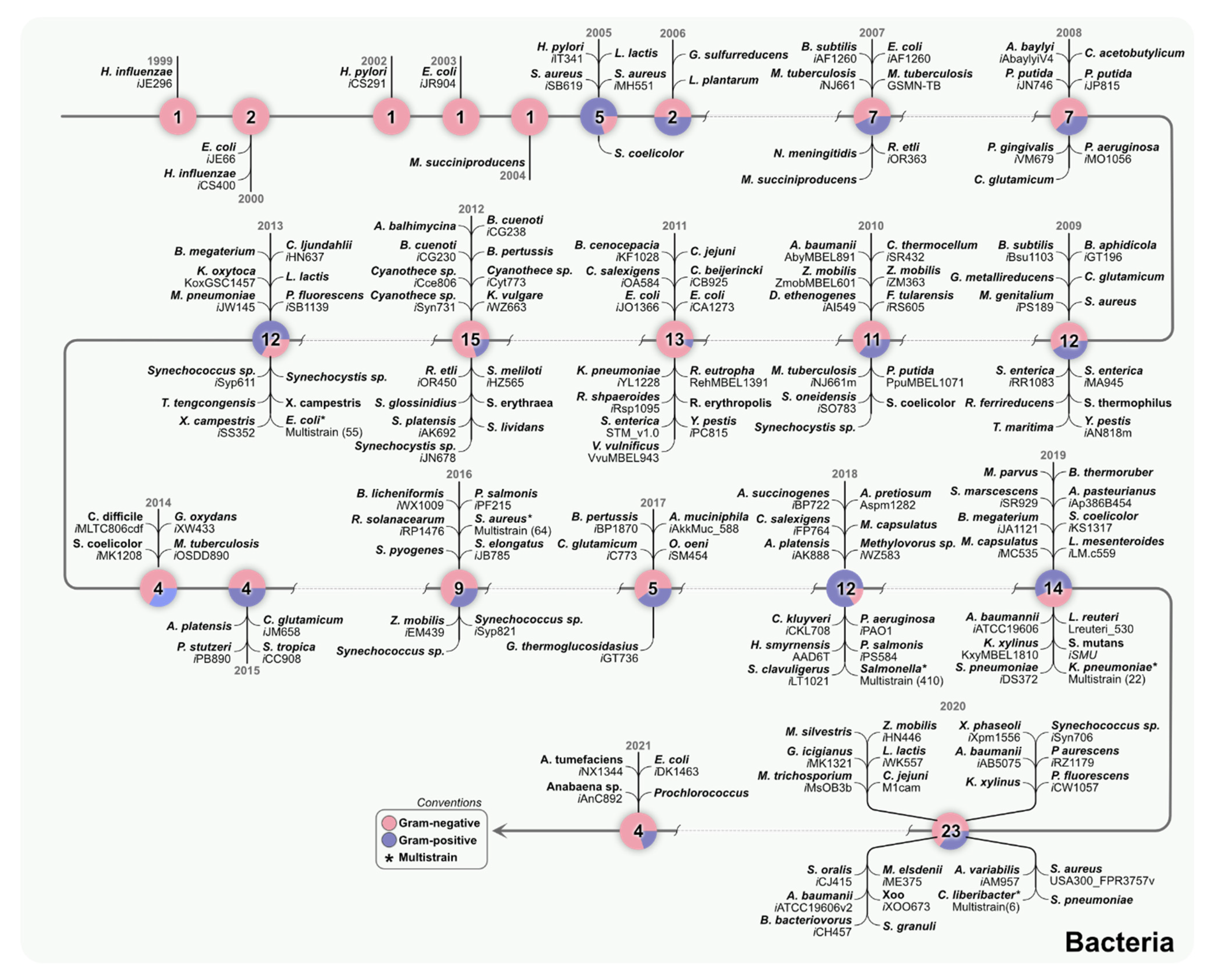
2. Individual and Multi-Strain GEMs Connect Genomics with Metabolism
3. Multi-Strain Reconstructions of Bacteria Can Help Understand Metabolic Diversity
4. Using GEMs to Understand the Metabolism of Archaea
5. The Metabolic Complexity of Eukaryotes Is Addressed in GEMs
6. A growing Branch of Big Data: GEM Reconstruction Tools and Datasets

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tool | Reaction Databases | Advantages/Limitations | Platform | Availability | Citations (Average/Year) | Reference |
|---|---|---|---|---|---|---|
| AuReMe | Available GEMs, MetaCyc, and BiGG | It stores the information at each step during the reconstruction process to maintain transparency and reproducibility. | Docker image | Public | 36 (13) | [71] |
| AutoKEGGRec | KEGG | It can be used to reconstruct models for a single organism and a given list of organisms. It generates an intermediate consolidated model that contains all the genes and reactions for all target organisms. Further, this consolidated model can be used to generate individual models. It does not incorporate transports, exchange, and biomass reactions to the draft model. Gap-filling is also not part of this reconstruction tool. | Matlab | Public | 22 (7.33) | [94] |
| CarveMe | BiGG | It is an automatic tool for reconstructing and gap-filling the draft model. CarveMe generates ready-to-use models for flux balance analysis. As a reaction database, manually curated BiGG models are used in the reconstruction process. | Python | Public | 151 (50.33) | [97] |
| COBRA toolbox, COBRApy, COBRA.ji | - | These tools do not provide any function to build the models based on annotated genomes. However, they provide the functions to incorporate all the components, such as genes, reactions, and metabolites into the model. In particular, these tools are useful for expanding upon existing draft models. | Matlab, Python, and Julia | Public | COBRA toolbox v.1-3.0—2733 (170) COBRApy—612 (76.50) COBRA.ji—25 (6.25) | [99,100,101] |
| COBRAme | Available GEMs | It is used to develop ME (Metabolism and Expression) models, which are the extended version of GEMs. In addition to a high-quality GEM, these models also contain transcription, translation, and tRNA charging reactions. | Python | Public | 73 (24.33) | [102] |
| CoReCo | Available GEMs, KEGG | It is a comparative reconstruction approach that uses available high-quality GEMs for comparison and reactions from the KEGG database to build models for closely related species. Its capability to compare models makes this tool useful for conducting evolutionary studies. | Python, R, Perl | Public | 68 (9.71) | [72] |
| FAME | KEGG | It only works on the organisms available in the KEGG database. It allows the visualization of FBA results on KEGG pathway maps. | Web-based | Public | 93 (10.33) | [95] |
| GEMsiRV | Available GEMs, BiGG, KEGG, MetaCyc, ModelSEED | It generates the model based on orthologous genes between the target and template model provided by the user. It can perform gap-filling using reference databases from BiGG, KEGG, MetaCyc, and ModelSEED. | Web-based | Public | 43 (4.78) | [93] |
| Merlin | KEGG, TCDB | It comprises several specific features, such as annotation of both enzymatic and transport genes, subcellular localization. Therefore, it can be used to reconstruct the models for both prokaryotes and eukaryotes. This tool also has a function to visualize all reactions in the model that can help users in the gap-filling process using the KEGG pathway browser. | Java | Public | 90 (15) | [73] |
| MetaDraft | Available GEMs | It uses available GEMs as templates to build models for a new organism. It contains internal template models (BiGG models) as reaction databases; however, users can create and use more templates. | Python GUI | Public | 28 (7) | [98] |
| ModelSEED/KBase | ModelSEED | In the first step, it uses RAST to annotate the genome of target organisms. This tool builds the models based on annotated genome and internal reaction databases. It performs gap-filling as a part of an algorithm based on user-provided media or complete media. It is a fully automated tool and does not allow users to customize any steps during reconstruction. It works on the assumption that all the reactions in the internal database are mass and charge-balanced. It also supports model reconstruction for plants. | Web-based | Public | 919 (83.55) | [92] |
| Pantograph | Available GEMs | It uses available models as a reaction database and orthology mappings between genomes of target and template organisms to reconstruct the GEM. It does not apply automatic gap-filling to the draft models. | Python | Public | 22 (3.67) | [96] |
| Pathway Tools | MetaCyc | It generates the model based on genes, reactions, and metabolites stored in organism-specific PGDB (pathway/genome database) and annotated genome. PGDB also helps in filling the gaps in the pathways. It contains 12 experimentally confirmed biomass reactions. Based on the taxonomy of the targeted organism, one biomass reaction is incorporated into the model. | Web-based, Python (via PythonCyc) | Free for academic and government researchers, a license fee applies for commercial use. | 216 (43.2) | [75] |
| RAVEN | Available GEMs, KEGG. MetaCyc | It provides a flexible environment to build a draft model. Users can employ multiple template models simultaneously. This tool can also be used to build the models using reaction databases like KEGG and MetaCyc. Additionally, networks built on different databases can be merged into one model. RAVEN also contains functions for gap-filling and subcellular localization (for eukaryotes). | Matlab | Public | 97 (32.33) | [76] |
| rBioNet | - | This is a part of COBRA Toolbox. It is not an automatic tool to populate the reactions in a draft model from any reaction database. Users need to provide manually or automatically created reaction databases as input for this tool. It comprises the functions to check the quality of newly added reactions such as duplication, charge, and mass balances. | Matlab | Public | 71 (7.1) | [103] |
| SuBliMinal Toolbox | KEGG, MetaCyc | It provides the modules to extract the reactions from KEGG and MetaCyc and merge both versions into a single network. This tool creates biomass reactions based on the biomass precursor present in the draft model. It also has a module to perform subcellular compartmentalization for reactions in the network. | Java | Public | 103 (10.3) | [104] |
7. Integrating Big Data and Machine Learning to Improve Manual Curation of GEMs
8. Systems Applications of GEMs Enable a Better Understanding of Big Data
9. Elucidation of Underground Metabolic Mechanisms of Well-Studied Organisms
10. Simulation of Phenotypic Traits Depends on the Medium Conditions
11. Utilization of GEMs in Drug Target Identification
12. Contextualization of Disease-Associated Big Data—Systems Medicine
13. Multi-Level Integration of Big Data in Emergent Modeling Approaches
14. Adding Macromolecular Resolution—Proteometrics
15. Simulating Gene Expression of Cells
16. Overcoming the Steady-State Assumption in Genome-Scale Metabolic Models
17. Challenges Associated with Reconstruction of GEM and Omics Data Integration
18. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GEM | Genome-Scale Metabolic Model |
| GSMMs | Genome-scale metabolic models |
| NCBI | National Centre for Biotechnology Information |
| ENCODE | Encyclopedia of DNA Elements |
| DGV | Database of Genomic Variants |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| RAVEN | Reconstruction, Analysis and Visualization of Metabolic Networks |
| COBRA | Constraint-Based Reconstruction and Analysis Toolbox |
| TCDB | Transporter Classification Database |
| FBA | Flux Balance Analysis |
| MFI | Methanobacterium formicicum |
References
- O’Driscoll, A.; Daugelaite, J.; Sleator, R.D. ‘Big data’, Hadoop and cloud computing in genomics. J. Biomed. Inform. 2013, 46, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Alyass, A.; Turcotte, M.; Meyre, D. From big data analysis to personalized medicine for all: Challenges and opportunities. BMC Med. Genom. 2015, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- McCue, M.E.; McCoy, A.M. The Scope of Big Data in One Medicine: Unprecedented Opportunities and Challenges. Front. Vet. Sci. 2017, 4, 194. [Google Scholar] [CrossRef]
- Sayers, E.W.; Beck, J.; Bolton, E.E.; Bourexis, D.; Brister, J.R.; Canese, K.; Comeau, D.C.; Funk, K.; Kim, S.; Klimke, W.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2021, 49, D10. [Google Scholar] [CrossRef]
- Nielsen, J. Systems Biology of Metabolism: A Driver for Developing Personalized and Precision Medicine. Cell Metab. 2017, 25, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Ebrahim, A.; Brunk, E.; Tan, J.; O’Brien, E.J.; Kim, D.; Szubin, R.; Lerman, J.A.; Lechner, A.; Sastry, A.; Bordbar, A.; et al. Multi-omic data integration enables discovery of hidden biological regularities. Nat. Commun. 2016, 7, 13091. [Google Scholar] [CrossRef] [PubMed]
- Blais, A.; Dynlacht, B.D. Constructing transcriptional regulatory networks. Genes Dev. 2005, 19, 1499–1511. [Google Scholar] [CrossRef]
- Safari-Alighiarloo, N.; Taghizadeh, M.; Rezaei-Tavirani, M.; Goliaei, B.; Peyvandi, A.A. Protein-protein interaction networks (PPI) and complex diseases. Gastroenterol. Hepatol. Bed Bench 2014, 7, 17–31. [Google Scholar]
- García del Valle, E.P.; Lagunes García, G.; Prieto Santamaría, L.; Zanin, M.; Menasalvas Ruiz, E.; Rodríguez-González, A. Disease networks and their contribution to disease understanding: A review of their evolution, techniques and data sources. J. Biomed. Inform. 2019, 94, 103206. [Google Scholar] [CrossRef]
- Wagner, A. Metabolic networks and their evolution. Adv. Exp. Med. Biol. 2012, 751, 29–52. [Google Scholar] [CrossRef]
- Vidal, M.; Cusick, M.E.; Barabási, A.-L. Interactome networks and human disease. Cell 2011, 144, 986–998. [Google Scholar] [CrossRef]
- Antonakoudis, A.; Barbosa, R.; Kotidis, P.; Kontoravdi, C. The era of big data: Genome-scale modelling meets machine learning. Comput. Struct. Biotechnol. J. 2020, 18, 3287–3300. [Google Scholar] [CrossRef]
- Friboulet, A.; Thomas, D. Systems Biology—An interdisciplinary approach. Biosens. Bioelectron. 2005, 20, 2404–2407. [Google Scholar] [CrossRef] [PubMed]
- Zuñiga, C.; Peacock, B.; Liang, B.; McCollum, G.; Irigoyen, S.C.; Tec-Campos, D.; Marotz, C.; Weng, N.C.; Zepeda, A.; Vidalakis, G.; et al. Linking metabolic phenotypes to pathogenic traits among “Candidatus Liberibacter asiaticus” and its hosts. NPJ Syst. Biol. Appl. 2020, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Bintener, T.; Pacheco, M.P.; Sauter, T. Towards the routine use of in silico screenings for drug discovery using metabolic modelling. Biochem. Soc. Trans. 2020, 48, 955–969. [Google Scholar] [CrossRef]
- Zielinski, D.C.; Patel, A.; Palsson, B.O. The Expanding Computational Toolbox for Engineering Microbial Phenotypes at the Genome Scale. Microorganisms 2020, 8, 2050. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Hua, Q. Applications of genome-scale metabolic models in biotechnology and systems medicine. Front. Physiol. 2016, 6, 413. [Google Scholar] [CrossRef]
- Jeon, J.; Kim, H.U. Setup of a scientific computing environment for computational biology: Simulation of a genome-scale metabolic model of Escherichia coli as an example. J. Microbiol. 2020, 58, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Zuñiga, C.; Li, T.; Guarnieri, M.T.; Jenkins, J.P.; Li, C.-T.; Bingol, K.; Kim, Y.-M.; Betenbaugh, M.J.; Zengler, K. Synthetic microbial communities of heterotrophs and phototrophs facilitate sustainable growth. Nat. Commun. 2020, 11, 3803. [Google Scholar] [CrossRef] [PubMed]
- Zuñiga, C.; Li, C.T.; Yu, G.; Al-Bassam, M.M.; Li, T.; Jiang, L.; Zaramela, L.S.; Guarnieri, M.; Betenbaugh, M.J.; Zengler, K. Environmental stimuli drive a transition from cooperation to competition in synthetic phototrophic communities. Nat. Microbiol. 2019, 4, 2184–2191. [Google Scholar] [CrossRef]
- Zuniga, C.; Tibocha-Bonilla, J.D.; Betenbaugh, M.J. Kinetic, metabolic, and statistical analytics: Addressing metabolic transport limitations among organelles and microbial communities. Curr. Opin. Biotechnol. 2021, 71, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Zaramela, L.S.; Moyne, O.; Kumar, M.; Zuniga, C.; Tibocha-Bonilla, J.D.; Zengler, K. The sum is greater than the parts: Exploiting microbial communities to achieve complex functions. Curr. Opin. Biotechnol. 2021, 67, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Whon, T.W.; Shin, N.R.; Kim, J.Y.; Roh, S.W. Omics in gut microbiome analysis. J. Microbiol. 2021, 59, 292–297. [Google Scholar] [CrossRef]
- Proctor, L.M.; Creasy, H.H.; Fettweis, J.M.; Lloyd-Price, J.; Mahurkar, A.; Zhou, W.; Buck, G.A.; Snyder, M.P.; Strauss, J.F.; Weinstock, G.M.; et al. The Integrative Human Microbiome Project. Nature 2019, 569, 641–648. [Google Scholar] [CrossRef]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackermann, G.; et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef]
- Rhie, A.; McCarthy, S.A.; Fedrigo, O.; Damas, J.; Formenti, G.; Koren, S.; Uliano-Silva, M.; Chow, W.; Fungtammasan, A.; Kim, J.; et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature 2021, 592, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Fremin, B.J.; Sberro, H.; Bhatt, A.S. MetaRibo-Seq measures translation in microbiomes. Nat. Commun. 2020, 11, 3268. [Google Scholar] [CrossRef]
- Richelle, A.; Chiang, A.W.T.; Kuo, C.-C.; Lewis, N.E. Increasing consensus of context-specific metabolic models by integrating data-inferred cell functions. PLoS Comput. Biol. 2019, 15, e1006867. [Google Scholar] [CrossRef]
- Gu, C.; Kim, G.B.; Kim, W.J.; Kim, H.U.; Lee, S.Y. Current status and applications of genome-scale metabolic models. Genome Biol. 2019, 20, 1–18. [Google Scholar] [CrossRef]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef]
- Mahadevan, R.; Edwards, J.S.; Doyle, F.J. Dynamic flux balance analysis of diauxic growth in Escherichia coli. Biophys. J. 2002, 83, 1331–1340. [Google Scholar] [CrossRef]
- Rasko, D.A.; Rosovitz, M.J.; Myers, G.S.A.; Mongodin, E.F.; Fricke, W.F.; Gajer, P.; Crabtree, J.; Sebaihia, M.; Thomson, N.R.; Chaudhuri, R.; et al. The pangenome structure of Escherichia coli: Comparative genomic analysis of E. coli commensal and pathogenic isolates. J. Bacteriol. 2008, 190, 6881–6893. [Google Scholar] [CrossRef]
- Yang, Z.K.; Luo, H.; Zhang, Y.; Wang, B.; Gao, F. Pan-genomic analysis provides novel insights into the association of E.coli with human host and its minimal genome. Bioinformatics 2019, 35, 1987–1991. [Google Scholar] [CrossRef]
- Norsigian, C.J.; Pusarla, N.; McConn, J.L.; Yurkovich, J.T.; Dräger, A.; Palsson, B.O.; King, Z. BiGG Models 2020: Multi-strain genome-scale models and expansion across the phylogenetic tree. Nucleic Acids Res. 2020, 48, D402–D406. [Google Scholar] [CrossRef]
- Monk, J.M.; Charusanti, P.; Aziz, R.K.; Lerman, J.A.; Premyodhin, N.; Orth, J.D.; Feist, A.M.; Palsson, B. Genome-scale metabolic reconstructions of multiple Escherichia coli strains highlight strain-specific adaptations to nutritional environments. Proc. Natl. Acad. Sci. USA 2013, 110, 20338–20343. [Google Scholar] [CrossRef] [PubMed]
- Seif, Y.; Kavvas, E.; Lachance, J.C.; Yurkovich, J.T.; Nuccio, S.P.; Fang, X.; Catoiu, E.; Raffatellu, M.; Palsson, B.O.; Monk, J.M. Genome-scale metabolic reconstructions of multiple Salmonella strains reveal serovar-specific metabolic traits. Nat. Commun. 2018, 9, 3771. [Google Scholar] [CrossRef]
- Bosi, E.; Monk, J.M.; Aziz, R.K.; Fondi, M.; Nizet, V.; Palsson, B. Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity. Proc. Natl. Acad. Sci. USA 2016, 113, E3801–E3809. [Google Scholar] [CrossRef]
- Norsigian, C.J.; Attia, H.; Szubin, R.; Yassin, A.S.; Palsson, B.Ø.; Aziz, R.K.; Monk, J.M. Comparative Genome-Scale Metabolic Modeling of Metallo-Beta-Lactamase–Producing Multidrug-Resistant Klebsiella pneumoniae Clinical Isolates. Front. Cell. Infect. Microbiol. 2019, 9, 161. [Google Scholar] [CrossRef] [PubMed]
- Rajput, A.; Seif, Y.; Choudhary, K.S.; Dalldorf, C.; Poudel, S.; Monk, J.M.; Palsson, B.O. Pangenome Analytics Reveal Two-Component Systems as Conserved Targets in ESKAPEE Pathogens. Msystems 2021, 6, e00981-20. [Google Scholar] [CrossRef]
- Jarrell, K.F.; Walters, A.D.; Bochiwal, C.; Borgia, J.M.; Dickinson, T.; Chong, J.P.J. Major players on the microbial stage: Why archaea are important. Microbiology 2011, 157, 919–936. [Google Scholar] [CrossRef] [PubMed]
- Albers, S.; Eichler, J.; Aebi, M. Archaea. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2015; pp. 283–292. [Google Scholar]
- Buan, N.R. Methanogens: Pushing the boundaries of biology. Emerg. Top. Life Sci. 2018, 2, 629–646. [Google Scholar] [CrossRef]
- Niehaus, F.; Bertoldo, C.; Kähler, M.; Antranikian, G. Extremophiles as a source of novel enzymes for industrial application. Appl. Microbiol. Biotechnol. 1999, 51, 711–729. [Google Scholar] [CrossRef] [PubMed]
- Sirohi, S.K.; Goel, N.; Pandey, P. Efficacy of different methanolic plant extracts on anti-methanogenesis, rumen fermentation and gas production kinetics in vitro. Open Vet. J. 2012, 2, 72–77. [Google Scholar] [PubMed]
- Thorpe, A. Enteric fermentation and ruminant eructation: The role (and control?) of methane in the climate change debate. Clim. Change 2009, 93, 407–431. [Google Scholar] [CrossRef]
- Feist, A.M.; Scholten, J.C.M.; Palsson, B.Ø.; Brockman, F.J.; Ideker, T. Modeling methanogenesis with a genome-scale metabolic reconstruction of Methanosarcina barkeri. Mol. Syst. Biol. 2006, 2, 2006-0004. [Google Scholar] [CrossRef]
- Gonnerman, M.C.; Benedict, M.N.; Feist, A.M.; Metcalf, W.W.; Price, N.D. Genomically and biochemically accurate metabolic reconstruction of Methanosarcina barkeri Fusaro, iMG746. Biotechnol. J. 2013, 8, 1070–1079. [Google Scholar] [CrossRef]
- Benedict, M.N.; Gonnerman, M.C.; Metcalf, W.W.; Price, N.D. Genome-scale metabolic reconstruction and hypothesis testing in the methanogenic archaeon Methanosarcina acetivorans C2A. J. Bacteriol. 2012, 194, 855–865. [Google Scholar] [CrossRef]
- Satish Kumar, V.; Ferry, J.G.; Maranas, C.D. Metabolic reconstruction of the archaeon methanogen Methanosarcina Acetivorans. BMC Syst. Biol. 2011, 5, 28. [Google Scholar] [CrossRef]
- Goyal, N.; Widiastuti, H.; Karimi, I.A.; Zhou, Z. A genome-scale metabolic model of Methanococcus maripaludis S2 for CO 2 capture and conversion to methane. Mol. BioSyst. 2014, 10, 1043–1054. [Google Scholar] [CrossRef]
- Li, C.T.; Yelsky, J.; Chen, Y.; Zuñiga, C.; Eng, R.; Jiang, L.; Shapiro, A.; Huang, K.W.; Zengler, K.; Betenbaugh, M.J. Utilizing genome-scale models to optimize nutrient supply for sustained algal growth and lipid productivity. NPJ Syst. Biol. Appl. 2019, 5, 33. [Google Scholar] [CrossRef]
- Tibocha-Bonilla, J.D.; Kumar, M.; Richelle, A.; Godoy-Silva, R.D.; Zengler, K.; Zuñiga, C. Dynamic resource allocation drives growth under nitrogen starvation in eukaryotes. NPJ Syst. Biol. Appl. 2020, 6, 14. [Google Scholar] [CrossRef]
- Arnolds, K.L.; Dahlin, L.R.; Ding, L.; Wu, C.; Yu, J.; Xiong, W.; Zuniga, C.; Suzuki, Y.; Zengler, K.; Linger, J.G.; et al. Biotechnology for secure biocontainment designs in an emerging bioeconomy. Curr. Opin. Biotechnol. 2021, 71, 25–31. [Google Scholar] [CrossRef]
- Zaramela, L.S.; Martino, C.; Alisson-Silva, F.; Rees, S.D.; Diaz, S.L.; Chuzel, L.; Ganatra, M.B.; Taron, C.H.; Secrest, P.; Zuñiga, C.; et al. Gut bacteria responding to dietary change encode sialidases that exhibit preference for red meat-associated carbohydrates. Nat. Microbiol. 2019, 4, 2082–2089. [Google Scholar] [CrossRef]
- Tibocha-Bonilla, J.D.; Zuñiga, C.; Godoy-Silva, R.D.; Zengler, K. Advances in metabolic modeling of oleaginous microalgae. Biotechnol. Biofuels 2018, 11, 241. [Google Scholar] [CrossRef]
- Gruber, A.; Rocap, G.; Kroth, P.G.; Armbrust, E.V.; Mock, T. Plastid proteome prediction for diatoms and other algae with secondary plastids of the red lineage. Plant J. 2015, 81, 519–528. [Google Scholar] [CrossRef]
- Chou, K.-C.; Shen, H.-B. Cell-PLoc 2.0: An improved package of web-servers for predicting subcellular localization of proteins in various organisms. Nat. Sci. 2010, 2, 1090. [Google Scholar] [CrossRef]
- Chou, K.C.; Shen, H.B. Cell-PLoc: A package of Web servers for predicting subcellular localization of proteins in various organisms. Nat. Protoc. 2008, 3, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Gschloessl, B.; Guermeur, Y.; Cock, J.M. HECTAR: A method to predict subcellular targeting in heterokonts. BMC Bioinform. 2008, 9, 393. [Google Scholar] [CrossRef]
- Claros, M.G. Mitoprot, a macintosh application for studying mitochondrial proteins. Bioinformatics 1995, 11, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Cokol, M.; Nair, R.; Rost, B. Finding nuclear localization signals. EMBO Rep. 2000, 1, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Gardy, J.L.; Laird, M.R.; Chen, F.; Rey, S.; Walsh, C.J.; Ester, M.; Brinkman, F.S.L. PSORTb v.2.0: Expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis. Bioinformatics 2005, 21, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Mooney, C.; Wang, Y.H.; Pollastri, G. SCLpred: Protein subcellular localization prediction by N-to-1 neural networks. Bioinformatics 2011, 27, 2812–2819. [Google Scholar] [CrossRef] [PubMed]
- Briesemeister, S.; Blum, T.; Brady, S.; Lam, Y.; Kohlbacher, O.; Shatkay, H. SherLoc2: A high-accuracy hybrid method for predicting subcellular localization of proteins. J. Proteome Res. 2009, 8, 5363–5366. [Google Scholar] [CrossRef] [PubMed]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Emanuelsson, O.; Brunak, S.; von Heijne, G.; Nielsen, H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2007, 2, 953–971. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Horton, P.; Park, K.J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef]
- Levering, J.; Broddrick, J.; Dupont, C.L.; Peers, G.; Beeri, K.; Mayers, J.; Gallina, A.A.; Allen, A.E.; Palsson, B.O.; Zengler, K. Genome-scale model reveals metabolic basis of biomass partitioning in a model diatom. PLoS ONE 2016, 11, e0155038. [Google Scholar] [CrossRef]
- Sunaga, Y.; Maeda, Y.; Yabuuchi, T.; Muto, M.; Yoshino, T.; Tanaka, T. Chloroplast-targeting protein expression in the oleaginous diatom Fistulifera solaris JPCC DA0580 toward metabolic engineering. J. Biosci. Bioeng. 2015, 119, 28–34. [Google Scholar] [CrossRef]
- Aite, M.; Chevallier, M.; Frioux, C.; Trottier, C.; Got, J.; Cortés, M.P.; Mendoza, S.N.; Carrier, G.; Dameron, O.; Guillaudeux, N.; et al. Traceability, reproducibility and wiki-exploration for “à-la-carte” reconstructions of genome-scale metabolic models. PLoS Comput. Biol. 2018, 14, e1006146. [Google Scholar] [CrossRef]
- Pitkänen, E.; Jouhten, P.; Hou, J.; Syed, M.F.; Blomberg, P.; Kludas, J.; Oja, M.; Holm, L.; Penttilä, M.; Rousu, J.; et al. Comparative Genome-Scale Reconstruction of Gapless Metabolic Networks for Present and Ancestral Species. PLoS Comput. Biol. 2014, 10, e1003465. [Google Scholar] [CrossRef] [PubMed]
- Dias, O.; Rocha, M.; Ferreira, E.C.; Rocha, I. Reconstructing genome-scale metabolic models with merlin. Nucleic Acids Res. 2015, 43, 3899–3910. [Google Scholar] [CrossRef] [PubMed]
- Saier, M.H.; Reddy, V.S.; Moreno-Hagelsieb, G.; Hendargo, K.J.; Zhang, Y.; Iddamsetty, V.; Lam, K.J.K.; Tian, N.; Russum, S.; Wang, J.; et al. The transporter classification database (TCDB): 2021 update. Nucleic Acids Res. 2021, 49, D461–D467. [Google Scholar] [CrossRef]
- Karp, P.D.; Midford, P.E.; Billington, R.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Ong, W.K.; Subhraveti, P.; Caspi, R.; Fulcher, C.; et al. Pathway Tools version 23.0 update: Software for pathway/genome informatics and systems biology. Brief. Bioinform. 2021, 22, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Marcišauskas, S.; Sánchez, B.J.; Domenzain, I.; Hermansson, D.; Agren, R.; Nielsen, J.; Kerkhoven, E.J. RAVEN 2.0: A versatile toolbox for metabolic network reconstruction and a case study on Streptomyces coelicolor. PLoS Comput. Biol. 2018, 14, e1006541. [Google Scholar] [CrossRef] [PubMed]
- Seaver, S.M.D.; Lerma-Ortiz, C.; Conrad, N.; Mikaili, A.; Sreedasyam, A.; Hanson, A.D.; Henry, C.S. PlantSEED enables automated annotation and reconstruction of plant primary metabolism with improved compartmentalization and comparative consistency. Plant J. 2018, 95, 1102–1113. [Google Scholar] [CrossRef]
- Agarwala, R.; Barrett, T.; Beck, J.; Benson, D.A.; Bollin, C.; Bolton, E.; Bourexis, D.; Brister, J.R.; Bryant, S.H.; Canese, K.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D7–D19. [Google Scholar] [CrossRef]
- Kersey, P.J.; Allen, J.E.; Allot, A.; Barba, M.; Boddu, S.; Bolt, B.J.; Carvalho-Silva, D.; Christensen, M.; Davis, P.; Grabmueller, C.; et al. Ensembl Genomes 2018: An integrated omics infrastructure for non-vertebrate species. Nucleic Acids Res. 2018, 46, D802–D808. [Google Scholar] [CrossRef]
- Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Malladi, V.S.; Strattan, J.S.; Hitz, B.C.; Gabdank, I.; Narayanan, A.K.; Ho, M.; Lee, B.T.; et al. ENCODE data at the ENCODE portal. Nucleic Acids Res. 2016, 44, D726–D732. [Google Scholar] [CrossRef]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef]
- Clarke, L.; Fairley, S.; Zheng-Bradley, X.; Streeter, I.; Perry, E.; Lowy, E.; Tassé, A.-M.; Flicek, P. The international Genome sample resource (IGSR): A worldwide collection of genome variation incorporating the 1000 Genomes Project data. Nucleic Acids Res. 2017, 45, D854–D859. [Google Scholar] [CrossRef]
- MacDonald, J.R.; Ziman, R.; Yuen, R.K.C.; Feuk, L.; Scherer, S.W. The Database of Genomic Variants: A curated collection of structural variation in the human genome. Nucleic Acids Res. 2014, 42, D986–D992. [Google Scholar] [CrossRef]
- King, Z.A.; Lu, J.; Dräger, A.; Miller, P.; Federowicz, S.; Lerman, J.A.; Ebrahim, A.; Palsson, B.O.; Lewis, N.E. BiGG Models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 2016, 44, D515–D522. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc database of metabolic pathways and ENZYMES—A 2019 update. Nucleic Acids Res. 2020, 48, D445–D453. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Ferrer, L.; Foerster, H.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016, 44, D459–D471. [Google Scholar] [CrossRef]
- Moretti, S.; Martin, O.; Van Du Tran, T.; Bridge, A.; Morgat, A.; Pagni, M. MetaNetX/MNXref—Reconciliation of metabolites and biochemical reactions to bring together genome-scale metabolic networks. Nucleic Acids Res. 2016, 44, D523–D526. [Google Scholar] [CrossRef]
- Morgat, A.; Lombardot, T.; Axelsen, K.B.; Aimo, L.; Niknejad, A.; Hyka-Nouspikel, N.; Coudert, E.; Pozzato, M.; Pagni, M.; Moretti, S.; et al. Updates in Rhea—An expert curated resource of biochemical reactions. Nucleic Acids Res. 2017, 45, D415–D418. [Google Scholar] [CrossRef] [PubMed]
- Aimo, L.; Liechti, R.; Hyka-Nouspikel, N.; Niknejad, A.; Gleizes, A.; Götz, L.; Kuznetsov, D.; David, F.P.A.; Van Der Goot, F.G.; Riezman, H.; et al. The SwissLipids knowledgebase for lipid biology. Bioinformatics 2015, 31, 2860–2866. [Google Scholar] [CrossRef] [PubMed]
- Elbourne, L.D.H.; Tetu, S.G.; Hassan, K.A.; Paulsen, I.T. TransportDB 2.0: A database for exploring membrane transporters in sequenced genomes from all domains of life. Nucleic Acids Res. 2017, 45, D320–D324. [Google Scholar] [CrossRef]
- Seaver, S.M.D.; Liu, F.; Zhang, Q.; Jeffryes, J.; Faria, J.P.; Edirisinghe, J.N.; Mundy, M.; Chia, N.; Noor, E.; Beber, M.E.; et al. The ModelSEED Biochemistry Database for the integration of metabolic annotations and the reconstruction, comparison and analysis of metabolic models for plants, fungi and microbes. Nucleic Acids Res. 2021, 49, D575–D588. [Google Scholar] [CrossRef]
- Liao, Y.C.; Tsai, M.H.; Chen, F.C.; Hsiung, C.A. GEMSiRV: A software platform for GEnome-scale metabolic model simulation, reconstruction and visualization. Bioinformatics 2012, 28, 1752–1758. [Google Scholar] [CrossRef] [PubMed]
- Karlsen, E.; Schulz, C.; Almaas, E. Automated generation of genome-scale metabolic draft reconstructions based on KEGG. BMC Bioinform. 2018, 19, 467. [Google Scholar] [CrossRef]
- Boele, J.; Olivier, B.G.; Teusink, B. FAME, the Flux Analysis and Modeling Environment. BMC Syst. Biol. 2012, 6, 8. [Google Scholar] [CrossRef]
- Loira, N.; Zhukova, A.; Sherman, D.J. Pantograph: A template-based method for genome-scale metabolic model reconstruction. J. Bioinform. Comput. Biol. 2015, 13, 1550006. [Google Scholar] [CrossRef]
- Machado, D.; Andrejev, S.; Tramontano, M.; Patil, K.R. Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 2018, 46, 7542–7553. [Google Scholar] [CrossRef]
- Hanemaaijer, M.; Olivier, B.G.; Röling, W.F.M.; Bruggeman, F.J.; Teusink, B. Model-based quantification of metabolic interactions from dynamic microbial-community data. PLoS ONE 2017, 12, e0173183. [Google Scholar] [CrossRef] [PubMed]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef]
- Ebrahim, A.; Lerman, J.A.; Palsson, B.O.; Hyduke, D.R. COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Syst. Biol. 2013, 7, 74. [Google Scholar] [CrossRef]
- Heirendt, L.; Thiele, I.; Fleming, R.M.T. DistributedFBA. jl: High-level, high-performance flux balance analysis in Julia. Bioinformatics 2017, 33, 1421–1423. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, C.J.; Ebrahim, A.; Yang, L.; King, Z.A.; Catoiu, E.; O’Brien, E.J.; Liu, J.K.; Palsson, B.O. COBRAme: A computational framework for genome-scale models of metabolism and gene expression. PLoS Comput. Biol. 2018, 14, e1006302. [Google Scholar] [CrossRef] [PubMed]
- Thorleifsson, S.G.; Thiele, I. rBioNet: A COBRA toolbox extension for reconstructing high-quality biochemical networks. Bioinformatics 2011, 27, 2009–2010. [Google Scholar] [CrossRef] [PubMed]
- Swainston, N.; Smallbone, K.; Mendes, P.; Kell, D.; Paton, N. The SuBliMinaL Toolbox: Automating steps in the reconstruction of metabolic networks. J. Integr. Bioinform. 2011, 8, 187–203. [Google Scholar] [CrossRef]
- Norena-Caro, D.A.; Zuniga, C.; Pete, A.J.; Saemundsson, S.A.; Donaldson, M.R.; Adams, A.J.; Dooley, K.M.; Zengler, K.; Benton, M.G. Analysis of the cyanobacterial amino acid metabolism with a precise genome-scale metabolic reconstruction of Anabaena sp. UTEX 2576. Biochem. Eng. J. 2021, 171, 108008. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, G.B.; Lee, S.Y. Machine learning applications in genome-scale metabolic modeling. Curr. Opin. Syst. Biol. 2021, 25, 42–49. [Google Scholar] [CrossRef]
- Ryu, J.Y.; Kim, H.U.; Lee, S.Y. Deep learning enables high-quality and high-throughput prediction of enzyme commission numbers. Proc. Natl. Acad. Sci. USA 2019, 116, 13996–14001. [Google Scholar] [CrossRef]
- Schinn, S.M.; Morrison, C.; Wei, W.; Zhang, L.; Lewis, N.E. A genome-scale metabolic network model and machine learning predict amino acid concentrations in Chinese Hamster Ovary cell cultures. Biotechnol. Bioeng. 2021, 118, 2118–2123. [Google Scholar] [CrossRef]
- Barrett, C.L.; Herrgard, M.J.; Palsson, B. Decomposing complex reaction networks using random sampling, principal component analysis and basis rotation. BMC Syst. Biol. 2009, 3, 30. [Google Scholar] [CrossRef]
- Plaimas, K.; Mallm, J.-P.; Oswald, M.; Svara, F.; Sourjik, V.; Eils, R.; König, R. Machine learning based analyses on metabolic networks supports high-throughput knockout screens. BMC Syst. Biol. 2008, 2, 67. [Google Scholar] [CrossRef]
- Acencio, M.L.; Lemke, N. Towards the prediction of essential genes by integration of network topology, cellular localization and biological process information. BMC Bioinform. 2009, 10, 290. [Google Scholar] [CrossRef]
- Sridhara, V.; Meyer, A.G.; Rai, P.; Barrick, J.E.; Ravikumar, P.; Segrè, D.; Wilke, C.O. Predicting growth conditions from internal metabolic fluxes in an in-silico model of E. coli. PLoS ONE 2014, 9, e114608. [Google Scholar] [CrossRef]
- Zuñiga, C.; Levering, J.; Antoniewicz, M.R.; Guarnieri, M.T.; Betenbaugh, M.J.; Zengler, K. Predicting Dynamic Metabolic Demands in the Photosynthetic Eukaryote Chlorella vulgaris. Plant Physiol. 2018, 176, 450–462. [Google Scholar] [CrossRef]
- Kavvas, E.S.; Yang, L.; Monk, J.M.; Heckmann, D.; Palsson, B.O. A biochemically-interpretable machine learning classifier for microbial GWAS. Nat. Commun. 2020, 11, 2580. [Google Scholar] [CrossRef] [PubMed]
- Medlock, G.L.; Papin, J.A. Guiding the Refinement of Biochemical Knowledgebases with Ensembles of Metabolic Networks and Machine Learning. Cell Syst. 2020, 10, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Oyetunde, T.; Zhang, M.; Chen, Y.; Tang, Y.; Lo, C. BoostGAPFILL: Improving the fidelity of metabolic network reconstructions through integrated constraint and pattern-based methods. Bioinformatics 2017, 33, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, T.J.B.; Campani, G.; Giordano, R.C.; Zangirolami, T.C.; Horta, A.C.L. Machine learning applied for metabolic flux-based control of micro-aerated fermentations in bioreactors. Biotechnol. Bioeng. 2021, 118, 2076–2091. [Google Scholar] [CrossRef]
- Culley, C.; Vijayakumar, S.; Zampieri, G.; Angione, C. A mechanism-aware and multiomic machine-learning pipeline characterizes yeast cell growth. Proc. Natl. Acad. Sci. USA 2020, 117, 18869–18879. [Google Scholar] [CrossRef]
- Mahood, E.H.; Kruse, L.H.; Moghe, G.D. Machine learning: A powerful tool for gene function prediction in plants. Appl. Plant Sci. 2020, 8, e11376. [Google Scholar] [CrossRef]
- Stiehler, F.; Steinborn, M.; Scholz, S.; Dey, D.; Weber, A.P.M.; Denton, A.K. Helixer: Cross-species gene annotation of large eukaryotic genomes using deep learning. Bioinformatics 2021, 36, 5291–5298. [Google Scholar] [CrossRef]
- Nachtweide, S.; Stanke, M. Multi-Genome Annotation with AUGUSTUS. Methods Mol. Biol. 2019, 1962, 139–160. [Google Scholar]
- Cai, Y.; Wang, J.; Deng, L. SDN2GO: An integrated deep learning model for protein function prediction. Front. Bioeng. Biotechnol. 2020, 8, 391. [Google Scholar] [CrossRef] [PubMed]
- Toubiana, D.; Puzis, R.; Wen, L.; Sikron, N.; Kurmanbayeva, A.; Soltabayeva, A.; del Mar Rubio Wilhelmi, M.; Sade, N.; Fait, A.; Sagi, M.; et al. Combined network analysis and machine learning allows the prediction of metabolic pathways from tomato metabolomics data. Commun. Biol. 2019, 2, 214. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Lloyd, C.J.; Palsson, B.O. Reconstructing organisms in silico: Genome-scale models and their emerging applications. Nat. Rev. Microbiol. 2020, 18, 731–743. [Google Scholar] [CrossRef]
- Kumar, M.; Ji, B.; Zengler, K.; Nielsen, J. Modelling approaches for studying the microbiome. Nat. Microbiol. 2019, 4, 1253–1267. [Google Scholar] [CrossRef]
- Edwards, J.S.; Palsson, B.O. The Escherichia coli MG1655 in silico metabolic genotype: Its definition, characteristics, and capabilities. Proc. Natl. Acad. Sci. USA 2000, 97, 5528–5533. [Google Scholar] [CrossRef]
- Monk, J.M.; Lloyd, C.J.; Brunk, E.; Mih, N.; Sastry, A.; King, Z.; Takeuchi, R.; Nomura, W.; Zhang, Z.; Mori, H.; et al. iML1515, a knowledgebase that computes Escherichia coli traits. Nat. Biotechnol. 2017, 35, 904–908. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.J.; Lerman, J.A.; Chang, R.L.; Hyduke, D.R.; Palsson, B.Ø. Genome-scale models of metabolism and gene expression extend and refine growth phenotype prediction. Mol. Syst. Biol. 2013, 9, 693. [Google Scholar] [CrossRef]
- Mo, M.L.; Palsson, B.; Herrgård, M.J. Connecting extracellular metabolomic measurements to intracellular flux states in yeast. BMC Syst. Biol. 2009, 3, 1–17. [Google Scholar] [CrossRef]
- Zuñiga, C.; Li, C.T.; Huelsman, T.; Levering, J.; Zielinski, D.C.; McConnell, B.O.; Long, C.P.; Knoshaug, E.P.; Guarnieri, M.T.; Antoniewicz, M.R.; et al. Genome-scale metabolic model for the green alga chlorella vulgaris UTEX 395 accurately predicts phenotypes under autotrophic, heterotrophic, and mixotrophic growth conditions. Plant Physiol. 2016, 172, 589–602. [Google Scholar] [CrossRef]
- Islam, M.A.; Zengler, K.; Edwards, E.A.; Mahadevan, R.; Stephanopoulos, G. Investigating Moorella thermoacetica metabolism with a genome-scale constraint-based metabolic model. Integr. Biol. 2015, 7, 869–882. [Google Scholar] [CrossRef]
- Campos, D.T.; Zuñiga, C.; Passi, A.; Del Toro, J.; Tibocha-Bonilla, J.D.; Zepeda, A.; Betenbaugh, M.J.; Zengler, K. Modeling of nitrogen fixation and polymer production in the heterotrophic diazotroph Azotobacter vinelandii DJ: Genome-scale metabolic modeling of Azotobacter vinelandii DJ. Metab. Eng. Commun. 2020, 11, e00132. [Google Scholar] [CrossRef] [PubMed]
- Brunk, E.; Sahoo, S.; Zielinski, D.C.; Altunkaya, A.; Dräger, A.; Mih, N.; Gatto, F.; Nilsson, A.; Preciat Gonzalez, G.A.; Aurich, M.K.; et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 2018, 36, 272–281. [Google Scholar] [CrossRef]
- Hefzi, H.; Ang, K.S.; Hanscho, M.; Bordbar, A.; Ruckerbauer, D.; Lakshmanan, M.; Orellana, C.A.; Baycin-Hizal, D.; Huang, Y.; Ley, D.; et al. A Consensus Genome-scale Reconstruction of Chinese Hamster Ovary Cell Metabolism. Cell Syst. 2016, 3, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Larsson, I.; Uhlén, M.; Zhang, C.; Mardinoglu, A. Genome-Scale Metabolic Modeling of Glioblastoma Reveals Promising Targets for Drug Development. Front. Genet. 2020, 11, 381. [Google Scholar] [CrossRef]
- Raškevičius, V.; Mikalayeva, V.; Antanavičiūtė, I.; Ceslevičienė, I.; Skeberdis, V.A.; Kairys, V.; Bordel, S. Genome scale metabolic models as tools for drug design and personalized medicine. PLoS ONE 2018, 13, e0190636. [Google Scholar] [CrossRef]
- Jansma, J.; El Aidy, S. Understanding the host-microbe interactions using metabolic modeling. Microbiome 2021, 9, 16. [Google Scholar] [CrossRef]
- Nagarajan, H.; Sahin, M.; Nogales, J.; Latif, H.; Lovley, D.R.; Ebrahim, A.; Zengler, K. Characterizing acetogenic metabolism using a genome-scale metabolic reconstruction of Clostridium ljungdahlii. Microb. Cell Factories 2013, 12, 118. [Google Scholar] [CrossRef]
- Lu, H.; Li, F.; Sánchez, B.J.; Zhu, Z.; Li, G.; Domenzain, I.; Marcišauskas, S.; Anton, P.M.; Lappa, D.; Lieven, C.; et al. A consensus S. cerevisiae metabolic model Yeast8 and its ecosystem for comprehensively probing cellular metabolism. Nat. Commun. 2019, 10, 3586. [Google Scholar] [CrossRef]
- Chang, R.L.; Ghamsari, L.; Manichaikul, A.; Hom, E.F.Y.; Balaji, S.; Fu, W.; Shen, Y.; Hao, T.; Palsson, B.Ø.; Salehi-Ashtiani, K.; et al. Metabolic network reconstruction of Chlamydomonas offers insight into light-driven algal metabolism. Mol. Syst. Biol. 2011, 7, 518. [Google Scholar] [CrossRef]
- Sertbas, M.; Ulgen, K.O. Genome-Scale Metabolic Modeling for Unraveling Molecular Mechanisms of High Threat Pathogens. Front. Cell Dev. Biol. 2020, 8, 566702. [Google Scholar] [CrossRef]
- Viana, R.; Dias, O.; Lagoa, D.; Galocha, M.; Rocha, I.; Teixeira, M.C. Genome-scale metabolic model of the human pathogen candida albicans: A promising platform for drug target prediction. J. Fungi 2020, 6, 171. [Google Scholar] [CrossRef] [PubMed]
- Minato, Y.; Gohl, D.M.; Thiede, J.M.; Chacón, J.M.; Harcombe, W.R.; Maruyama, F.; Baughn, A.D. Genomewide Assessment of Mycobacterium tuberculosis Conditionally Essential Metabolic Pathways. mSystems 2019, 4, e00070-19. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Deng, Z.L.; Xie, Z.M.; Chu, X.Y.; Chang, J.W.; Kong, D.X.; Li, B.J.; Zhang, H.Y.; Chen, L.L. Construction of a genome-scale metabolic network of the plant pathogen Pectobacterium carotovorum provides new strategies for bactericide discovery. FEBS Lett. 2015, 589, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Haleem, A.M.; Hefzi, H.; Mineta, K.; Gao, X.; Gojobori, T.; Palsson, B.O.; Lewis, N.E.; Jamshidi, N. Functional interrogation of Plasmodium genus metabolism identifies species—And stage-specific differences in nutrient essentiality and drug targeting. PLoS Comput. Biol. 2018, 14, e1005895. [Google Scholar] [CrossRef]
- Weglarz-Tomczak, E.; Mondeel, T.D.G.A.; Piebes, D.G.E.; Westerhoff, H.V. Simultaneous Integration of Gene Expression and Nutrient Availability for Studying the Metabolism of Hepatocellular Carcinoma Cell Lines. Biomolecules 2021, 11, 490. [Google Scholar] [CrossRef]
- Puniya, B.L.; Amin, R.; Lichter, B.; Moore, R.; Ciurej, A.; Bennett, S.J.; Shah, A.R.; Barberis, M.; Helikar, T. Integrative computational approach identifies drug targets in CD4(+) T-cell-mediated immune disorders. NPJ Syst. Biol. Appl. 2021, 7, 4. [Google Scholar] [CrossRef]
- Barabási, A.-L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef]
- Nilsson, A.; Nielsen, J. Genome scale metabolic modeling of cancer. Metab. Eng. 2017, 43, 103–112. [Google Scholar] [CrossRef]
- Pandey, N.; Lanke, V.; Vinod, P.K. Network-based metabolic characterization of renal cell carcinoma. Sci. Rep. 2020, 10, 5955. [Google Scholar] [CrossRef]
- Gatto, F.; Ferreira, R.; Nielsen, J. Pan-cancer analysis of the metabolic reaction network. Metab. Eng. 2020, 57, 51–62. [Google Scholar] [CrossRef]
- Jerby, L.; Ruppin, E. Predicting drug targets and biomarkers of cancer via genome-scale metabolic modeling. Clin. Cancer Res. 2012, 18, 5572–5584. [Google Scholar] [CrossRef]
- Moolamalla, S.T.R.; Vinod, P.K. Genome-scale metabolic modelling predicts biomarkers and therapeutic targets for neuropsychiatric disorders. Comput. Biol. Med. 2020, 125, 103994. [Google Scholar] [CrossRef]
- Zhang, Y.; Thiele, I.; Weekes, D.; Li, Z.; Jaroszewski, L.; Ginalski, K.; Deacon, A.M.; Wooley, J.; Lesley, S.A.; Wilson, I.A.; et al. Three-dimensional structural view of the central metabolic network of Thermotoga maritima. Science 2009, 325, 1544–1549. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.L.; Xie, L.; Bourne, P.E.; Palsson, B.O. Antibacterial mechanisms identified through structural systems pharmacology. BMC Syst. Biol. 2013, 7, 102. [Google Scholar] [CrossRef]
- Chang, R.L.; Andrews, K.; Kim, D.; Li, Z.; Godzik, A.; Palsson, B.O. Structural systems biology evaluation of metabolic thermotolerance in Escherichia coli. Science 2013, 340, 1220–1223. [Google Scholar] [CrossRef]
- Marcão, A.; Azevedo, J.E.; Gieselmann, V.; Sá Miranda, M.C. Oligomerization capacity of two arylsulfatase A mutants: C300F and P425T. Biochem. Biophys. Res. Commun. 2003, 306, 293–297. [Google Scholar] [CrossRef]
- Seif, Y.; Monk, J.M.; Mih, N.; Tsunemoto, H.; Poudel, S.; Zuniga, C.; Broddrick, J.; Zengler, K.; Palsson, B.O. A computational knowledge-base elucidates the response of Staphylococcus aureus to different media types. PLoS Comput. Biol. 2019, 15, e1006644. [Google Scholar] [CrossRef] [PubMed]
- Lerman, J.A.; Hyduke, D.R.; Latif, H.; Portnoy, V.A.; Lewis, N.E.; Orth, J.D.; Schrimpe-Rutledge, A.C.; Smith, R.D.; Adkins, J.N.; Zengler, K.; et al. In silico method for modelling metabolism and gene product expression at genome scale. Nat. Commun. 2012, 3, 929. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Fleming, R.M.T.; Que, R.; Bordbar, A.; Diep, D.; Palsson, B.O. Multiscale modeling of metabolism and macromolecular synthesis in E. coli and its application to the evolution of codon usage. PLoS ONE 2012, 7, e45635. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.K.; O’Brien, E.J.; Lerman, J.A.; Zengler, K.; Palsson, B.O.; Feist, A.M. Reconstruction and modeling protein translocation and compartmentalization in Escherichia coli at the genome-scale. BMC Syst. Biol. 2014, 8, 110. [Google Scholar] [CrossRef]
- Yang, L.; Ma, D.; Ebrahim, A.; Lloyd, C.J.; Saunders, M.A.; Palsson, B.O. solveME: Fast and reliable solution of nonlinear ME models. BMC Bioinform. 2016, 17, 391. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.K.; Lloyd, C.; Al-Bassam, M.M.; Ebrahim, A.; Kim, J.-N.; Olson, C.; Aksenov, A.; Dorrestein, P.; Zengler, K. Predicting proteome allocation, overflow metabolism, and metal requirements in a model acetogen. PLoS Comput. Biol. 2019, 15, e1006848. [Google Scholar] [CrossRef]
- Chen, K.; Gao, Y.; Mih, N.; O’Brien, E.J.; Yang, L.; Palsson, B.O. Thermosensitivity of growth is determined by chaperone-mediated proteome reallocation. Proc. Natl. Acad. Sci. USA 2017, 114, 11548–11553. [Google Scholar] [CrossRef]
- Du, B.; Yang, L.; Lloyd, C.J.; Fang, X.; Palsson, B.O. Genome-scale model of metabolism and gene expression provides a multi-scale description of acid stress responses in Escherichia coli. PLoS Comput. Biol. 2019, 15, e1007525. [Google Scholar] [CrossRef]
- Yang, L.; Mih, N.; Anand, A.; Park, J.H.; Tan, J.; Yurkovich, J.T.; Monk, J.M.; Lloyd, C.J.; Sandberg, T.E.; Seo, S.W.; et al. Cellular responses to reactive oxygen species are predicted from molecular mechanisms. Proc. Natl. Acad. Sci. USA 2019, 116, 14368–14373. [Google Scholar] [CrossRef]
- Zhao, X.; Noack, S.; Wiechert, W.; Lieres, E.v. Dynamic flux balance analysis with nonlinear objective function. J. Math. Biol. 2017, 75, 1487–1515. [Google Scholar] [CrossRef] [PubMed]
- Qinghua, Z.; Dan, W.; Momiao, X. Dynamic flux balance analysis of metabolic networks using the penalty function methods. In Proceedings of the 2007 IEEE International Conference on Systems, Man and Cybernetics, Montreal, QC, Canada, 7–10 October 2007; pp. 3594–3599. [Google Scholar]
- Höffner, K.; Harwood, S.M.; Barton, P.I. A reliable simulator for dynamic flux balance analysis. Biotechnol. Bioeng. 2013, 110, 792–802. [Google Scholar] [CrossRef]
- Gomez, J.A.; Höffner, K.; Barton, P.I. DFBAlab: A fast and reliable MATLAB code for dynamic flux balance analysis. BMC Bioinform. 2014, 15, 409. [Google Scholar] [CrossRef] [PubMed]
- Westermark, S.; Steuer, R. Toward multiscale models of cyanobacterial growth: A modular approach. Front. Bioeng. Biotechnol. 2016, 4, 95. [Google Scholar] [CrossRef]
- Henson, M.A. Genome-scale modeling of microbial metabolism with temporal and spatial resolution. Biochem. Soc. Trans. 2017, 43, 1164–1171. [Google Scholar] [CrossRef]
- Kuriya, Y.; Araki, M. Dynamic flux balance analysis to evaluate the strain production performance on shikimic acid production in Escherichia coli. Metabolites 2020, 10, 198. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, B.J.; Zhang, C.; Nilsson, A.; Lahtvee, P.J.; Kerkhoven, E.J.; Nielsen, J. Improving the phenotype predictions of a yeast genome-scale metabolic model by incorporating enzymatic constraints. Mol. Syst. Biol. 2017, 13, 935. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, F.; Mao, J.; Chen, Y.; Nielsen, J. Yeast optimizes metal utilization based on metabolic network and enzyme kinetics. Proc. Natl. Acad. Sci. USA 2021, 118, e2020154118. [Google Scholar] [CrossRef]
- Broddrick, J.T.; Du, N.; Smith, S.R.; Tsuji, Y.; Jallet, D.; Ware, M.A.; Peers, G.; Matsuda, Y.; Dupont, C.L.; Mitchell, B.G.; et al. Cross-compartment metabolic coupling enables flexible photoprotective mechanisms in the diatom Phaeodactylum tricornutum. New Phytol. 2019, 222, 1364–1379. [Google Scholar] [CrossRef]
- van Tol, H.M.; Armbrust, E.V. Genome-scale metabolic model of the diatom Thalassiosira pseudonana highlights the importance of nitrogen and sulfur metabolism in redox balance. PLoS ONE 2021, 16, e0241960. [Google Scholar] [CrossRef] [PubMed]
- Antoniewicz, M.R. A guide to metabolic flux analysis in metabolic engineering: Methods, tools and applications. Metab. Eng. 2021, 63, 2–12. [Google Scholar] [CrossRef]
- Niklas, J.; Schneider, K.; Heinzle, E. Metabolic flux analysis in eukaryotes. Curr. Opin. Biotechnol. 2010, 21, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, R.; Schilling, C.H. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab. Eng. 2003, 5, 264–276. [Google Scholar] [CrossRef]
- Burgard, A.P.; Nikolaev, E.V.; Schilling, C.H.; Maranas, C.D. Flux coupling analysis of genome-scale metabolic network reconstructions. Genome Res. 2004, 14, 301–312. [Google Scholar] [CrossRef]
- Kelk, S.M.; Olivier, B.G.; Stougie, L.; Bruggeman, F.J. Optimal flux spaces of genome-scale stoichiometric models are determined by a few subnetworks. Sci. Rep. 2012, 2, 580. [Google Scholar] [CrossRef]
- Gomes de Oliveira Dal’Molin, C.; Quek, L.-E.; Saa, P.A.; Nielsen, L.K. A multi-tissue genome-scale metabolic modeling framework for the analysis of whole plant systems. Front. Plant Sci. 2015, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Hanna, E.M.; Zhang, X.; Eide, M.; Fallahi, S.; Furmanek, T.; Yadetie, F.; Zielinski, D.C.; Goksøyr, A.; Jonassen, I. ReCodLiver0.9: Overcoming Challenges in Genome-Scale Metabolic Reconstruction of a Non-model Species. Front. Mol. Biosci. 2020, 7, 345. [Google Scholar] [CrossRef]
- Bernstein, D.B.; Sulheim, S.; Almaas, E.; Segrè, D. Addressing uncertainty in genome-scale metabolic model reconstruction and analysis. Genome Biol. 2021, 22, 64. [Google Scholar] [CrossRef]
- Mendoza, S.N.; Olivier, B.G.; Molenaar, D.; Teusink, B. A systematic assessment of current genome-scale metabolic reconstruction tools. Genome Biol. 2019, 20, 158. [Google Scholar] [CrossRef]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.-W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef]
- Lieven, C.; Beber, M.E.; Olivier, B.G.; Bergmann, F.T.; Ataman, M.; Babaei, P.; Bartell, J.A.; Blank, L.M.; Chauhan, S.; Correia, K.; et al. MEMOTE for standardized genome-scale metabolic model testing. Nat. Biotechnol. 2020, 38, 272–276. [Google Scholar] [CrossRef] [PubMed]
- van Rosmalen, R.P.; Smith, R.W.; Martins dos Santos, V.A.P.; Fleck, C.; Suarez-Diez, M. Model reduction of genome-scale metabolic models as a basis for targeted kinetic models. Metab. Eng. 2021, 64, 74–84. [Google Scholar] [CrossRef]
- St. John, P.C.; Bomble, Y.J. Approaches to Computational Strain Design in the Multiomics Era. Front. Microbiol. 2019, 10, 597. [Google Scholar] [CrossRef]
- Tarazona, S.; Arzalluz-Luque, A.; Conesa, A. Undisclosed, unmet and neglected challenges in multi-omics studies. Nat. Comput. Sci. 2021, 1, 395–402. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef] [PubMed]
- Pinu, F.R.; Beale, D.J.; Paten, A.M.; Kouremenos, K.; Swarup, S.; Schirra, H.J.; Wishart, D. Systems Biology and Multi-Omics Integration: Viewpoints from the Metabolomics Research Community. Metabolites 2019, 9, 76. [Google Scholar] [CrossRef] [PubMed]
- Fondi, M.; Liò, P. Multi -omics and metabolic modelling pipelines: Challenges and tools for systems microbiology. Microbiol. Res. 2015, 171, 52–64. [Google Scholar] [CrossRef]
- Pal, S.; Mondal, S.; Das, G.; Khatua, S.; Ghosh, Z. Big data in biology: The hope and present-day challenges in it. Gene Rep. 2020, 21, 100869. [Google Scholar] [CrossRef]
- Choi, Y.-M.; Lee, Y.Q.; Song, H.-S.; Lee, D.-Y. Genome scale metabolic models and analysis for evaluating probiotic potentials. Biochem. Soc. Trans. 2020, 48, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Berding, K.; Donovan, S.M. Diet Can Impact Microbiota Composition in Children With Autism Spectrum Disorder. Front. Neurosci. 2018, 12, 515. [Google Scholar] [CrossRef] [PubMed]
- Devika, N.T.; Raman, K. Deciphering the metabolic capabilities of Bifidobacteria using genome-scale metabolic models. Sci. Rep. 2019, 9, 18222. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Passi, A.; Tibocha-Bonilla, J.D.; Kumar, M.; Tec-Campos, D.; Zengler, K.; Zuniga, C. Genome-Scale Metabolic Modeling Enables In-Depth Understanding of Big Data. Metabolites 2022, 12, 14. https://doi.org/10.3390/metabo12010014
Passi A, Tibocha-Bonilla JD, Kumar M, Tec-Campos D, Zengler K, Zuniga C. Genome-Scale Metabolic Modeling Enables In-Depth Understanding of Big Data. Metabolites. 2022; 12(1):14. https://doi.org/10.3390/metabo12010014
Chicago/Turabian StylePassi, Anurag, Juan D. Tibocha-Bonilla, Manish Kumar, Diego Tec-Campos, Karsten Zengler, and Cristal Zuniga. 2022. "Genome-Scale Metabolic Modeling Enables In-Depth Understanding of Big Data" Metabolites 12, no. 1: 14. https://doi.org/10.3390/metabo12010014
APA StylePassi, A., Tibocha-Bonilla, J. D., Kumar, M., Tec-Campos, D., Zengler, K., & Zuniga, C. (2022). Genome-Scale Metabolic Modeling Enables In-Depth Understanding of Big Data. Metabolites, 12(1), 14. https://doi.org/10.3390/metabo12010014