
Inspecting the Solution Space of Genome-Scale Metabolic Models

 and
and
Abstract
:1. Introduction
2. Results
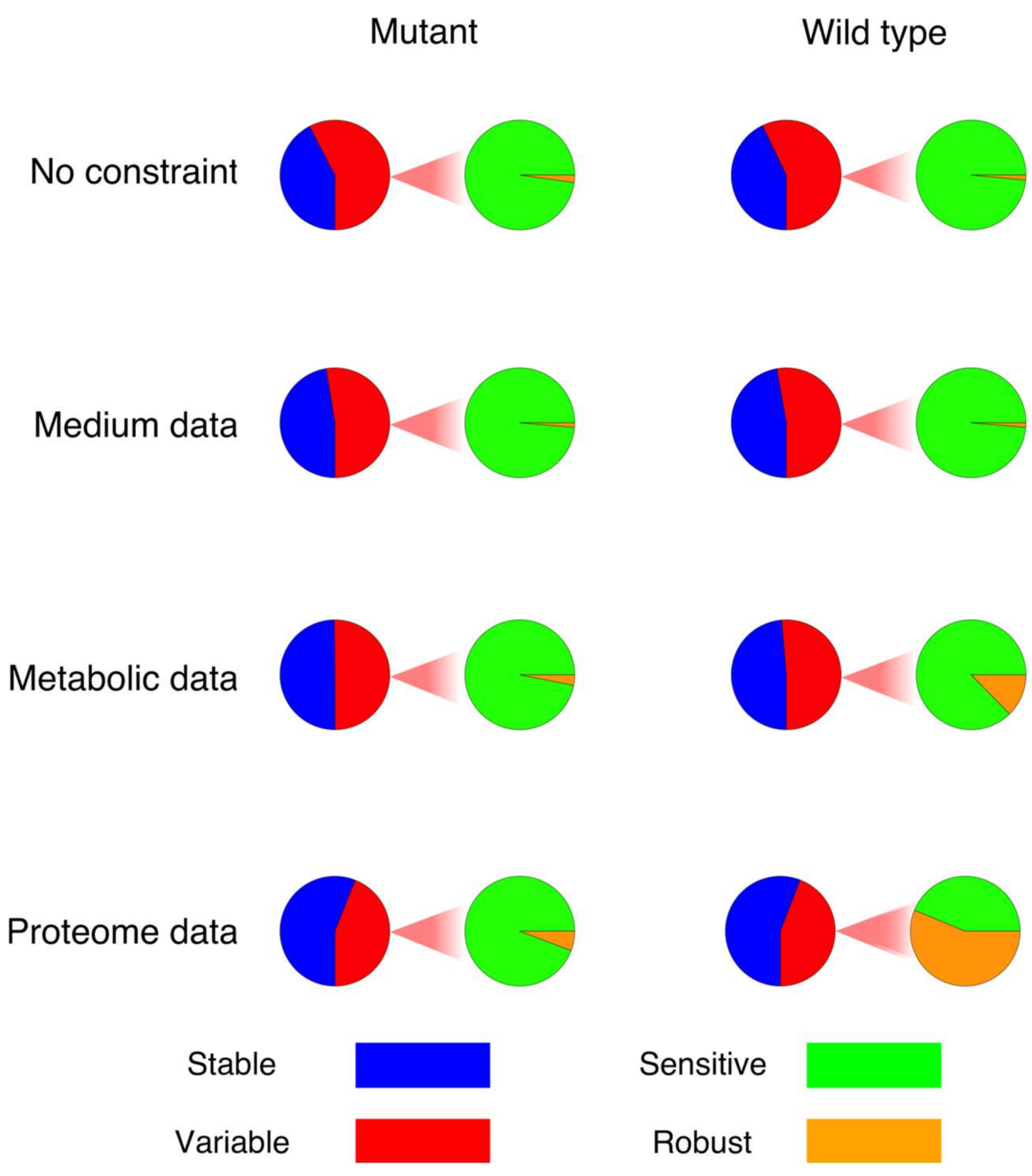
2.1. Effect of Constraints on the Solution Space in the Network
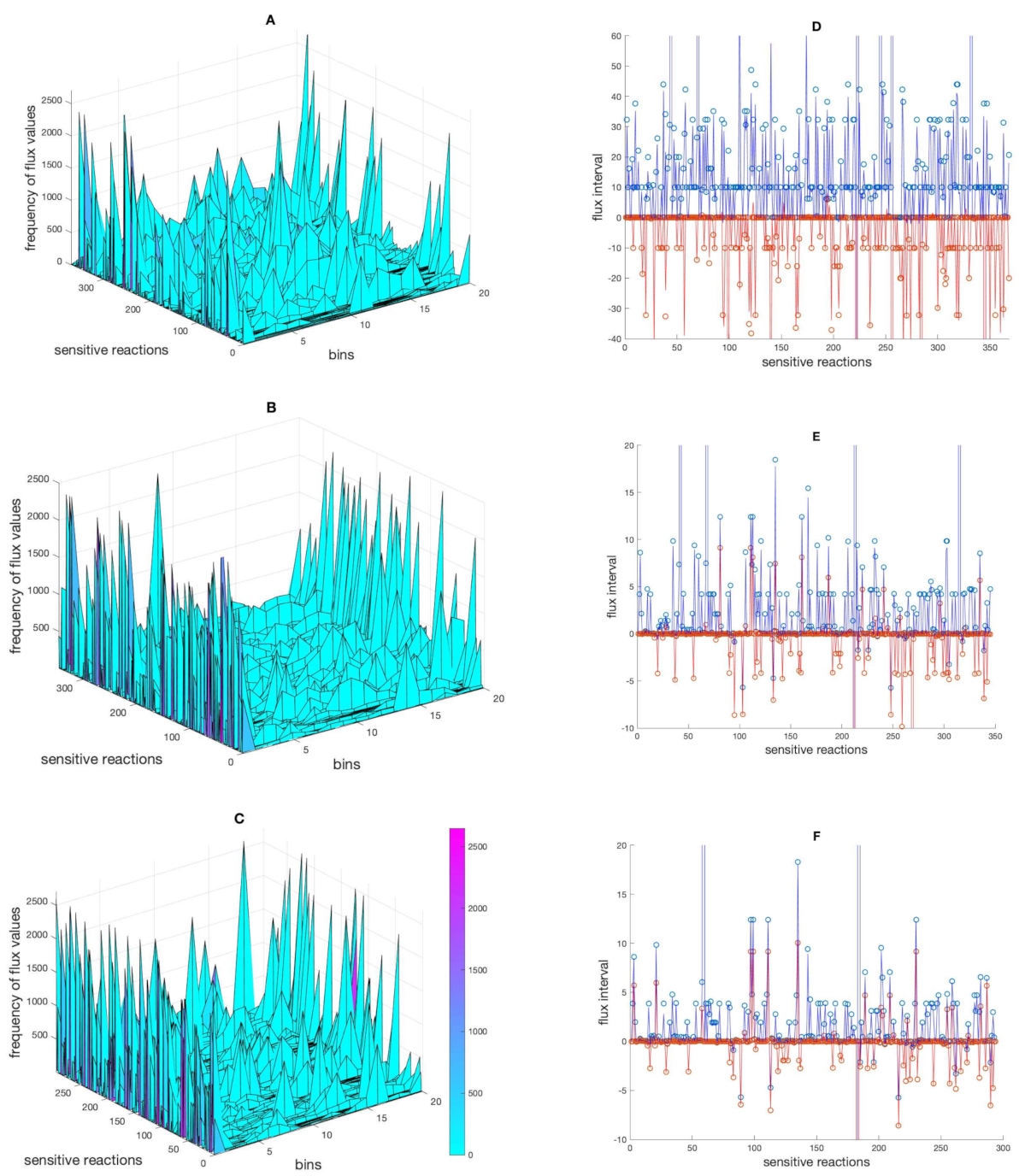
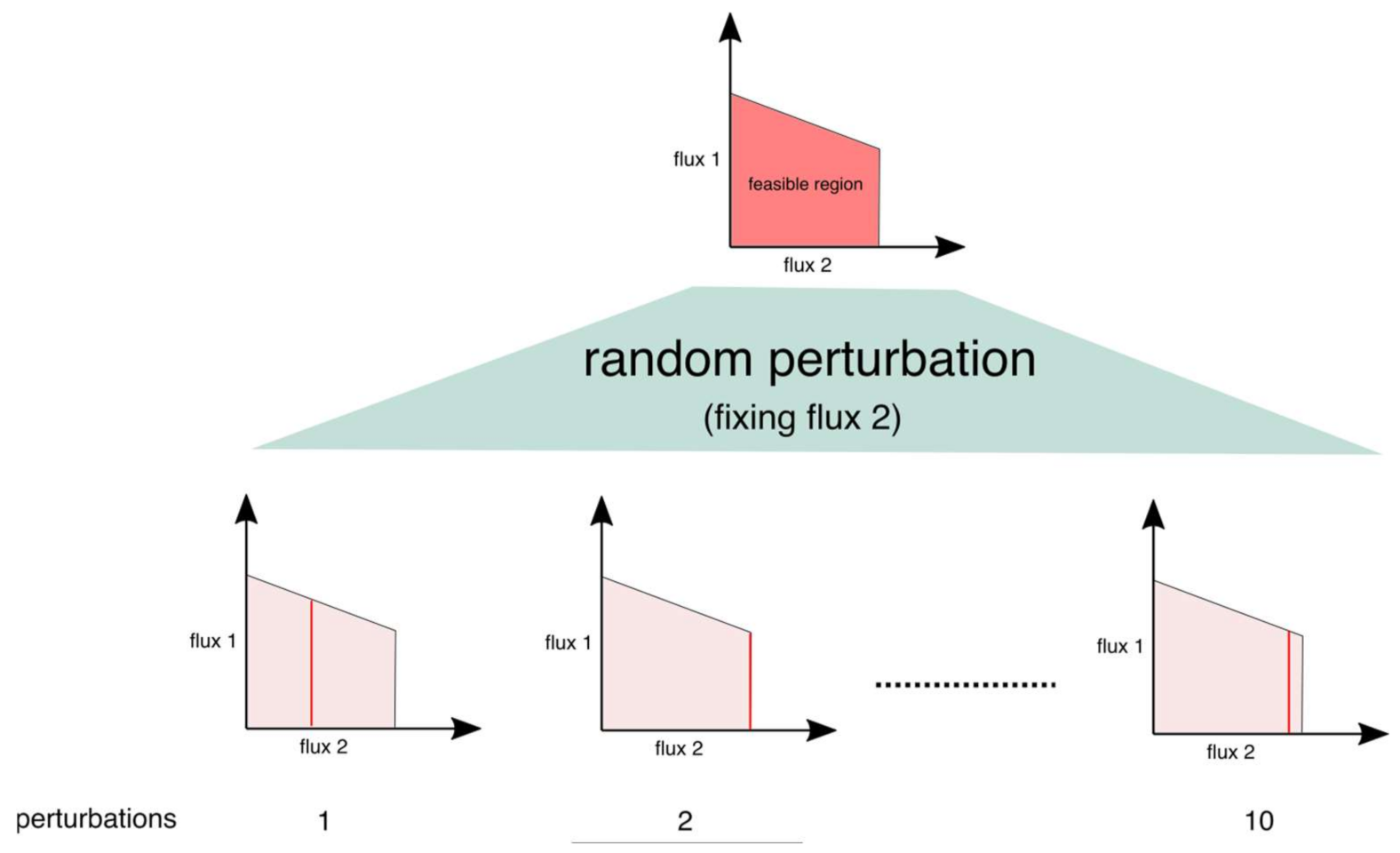
2.2. Inspecting the Solution Space Using Random Perturbations
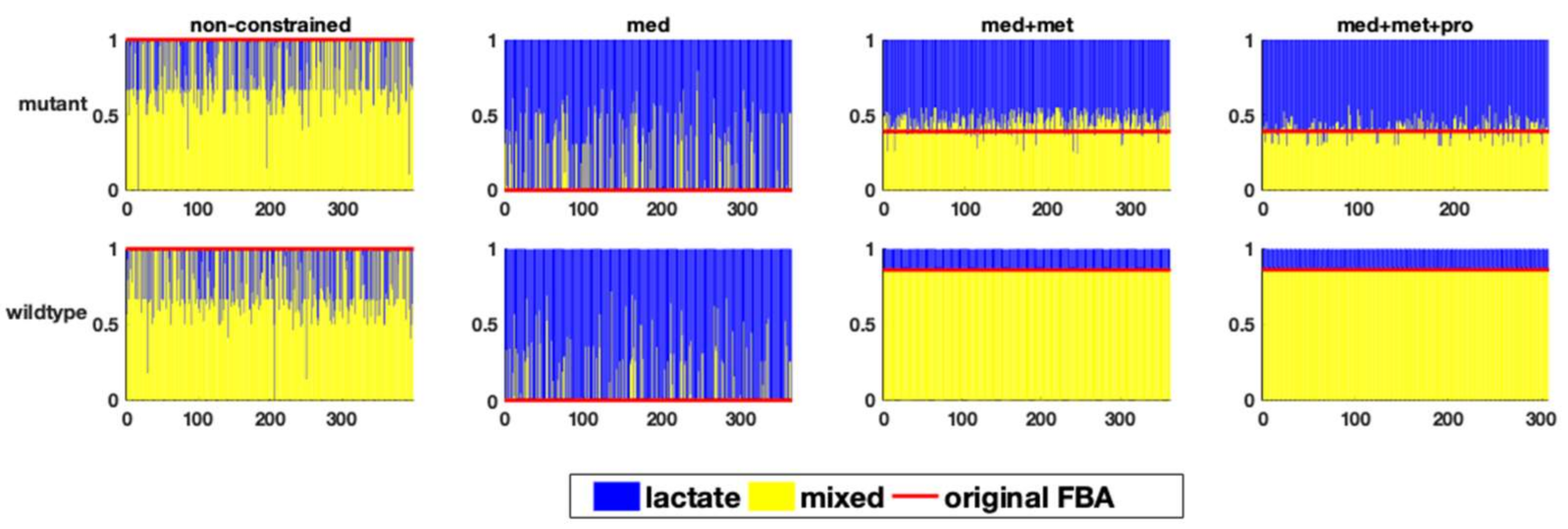
2.3. Investigating Biological Phenotypes in FBA Results
2.4. The Influence of Specific Quantitative Constraints on the Solution Space
2.5. Analysing the Solution Space Using CoPE-FBA
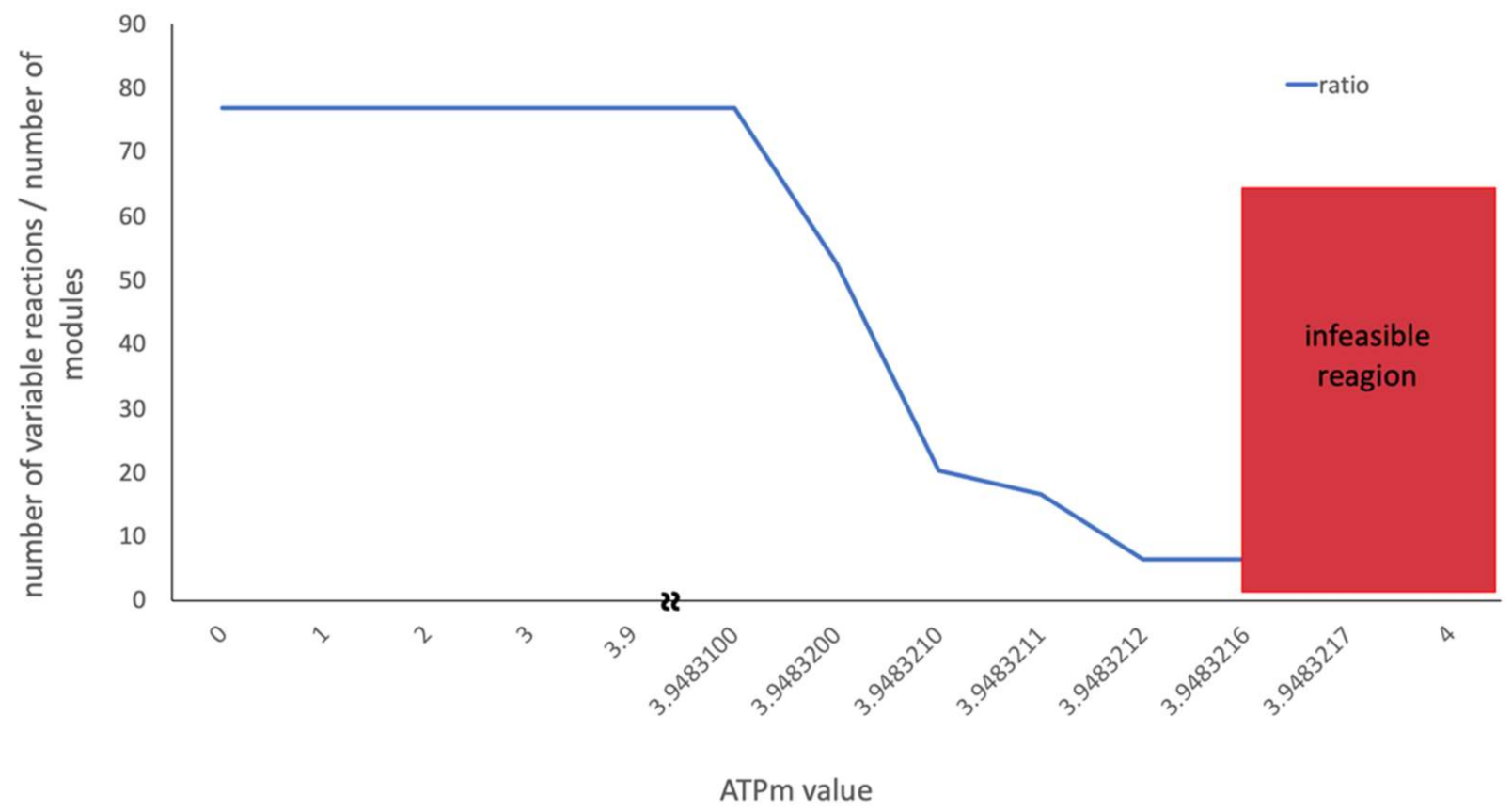
2.6. The Influence of ATP Maintenance on the Solution Space
2.7. Validating the Results Using Models of Other Species
3. Discussion
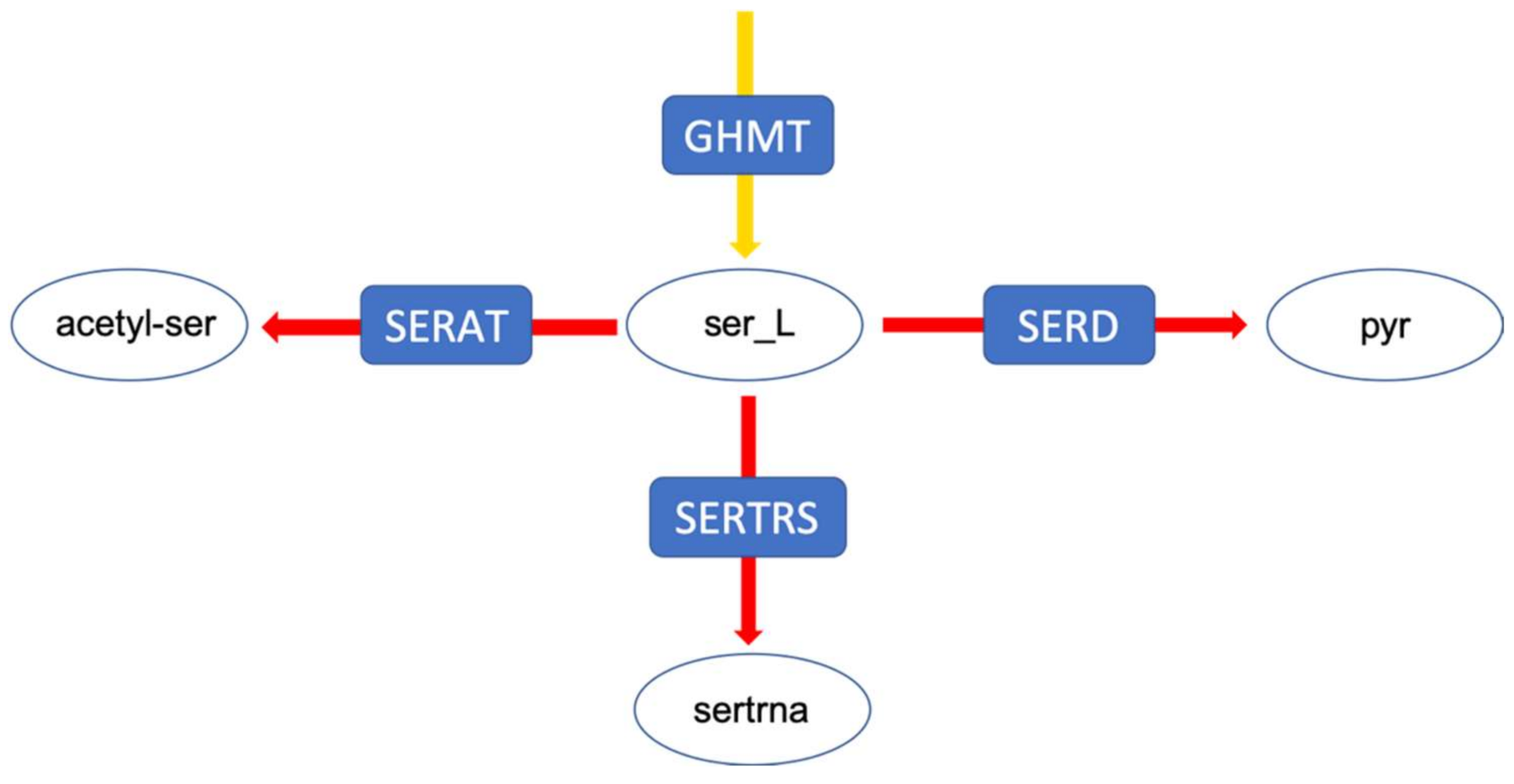
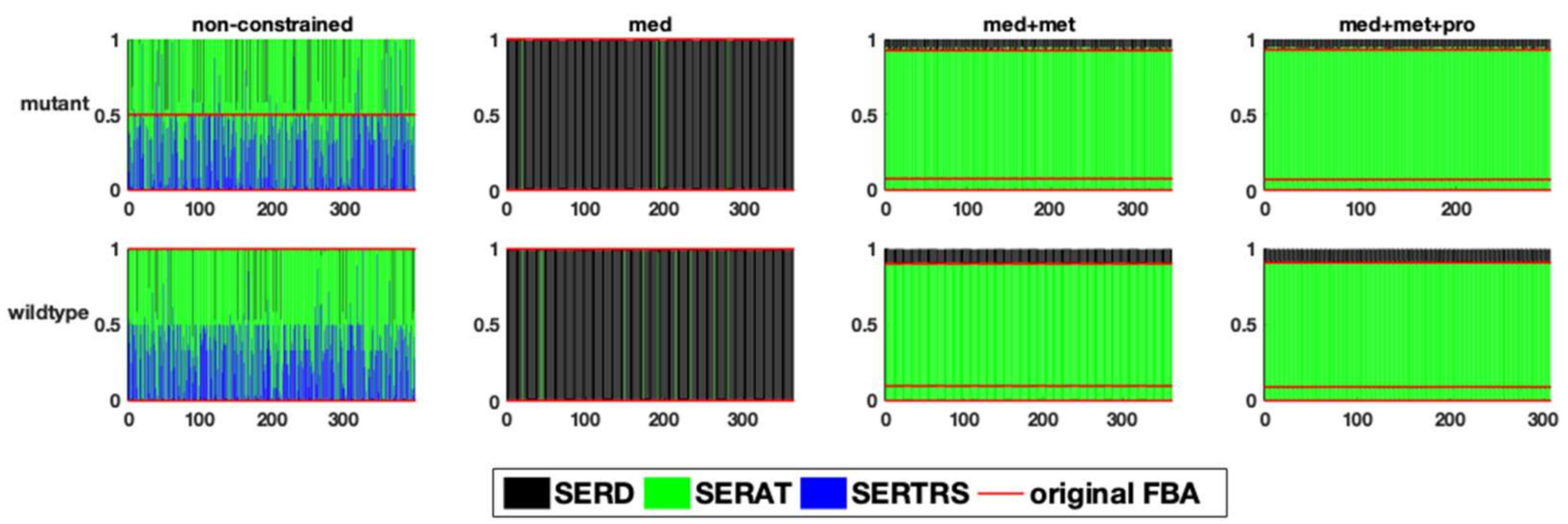
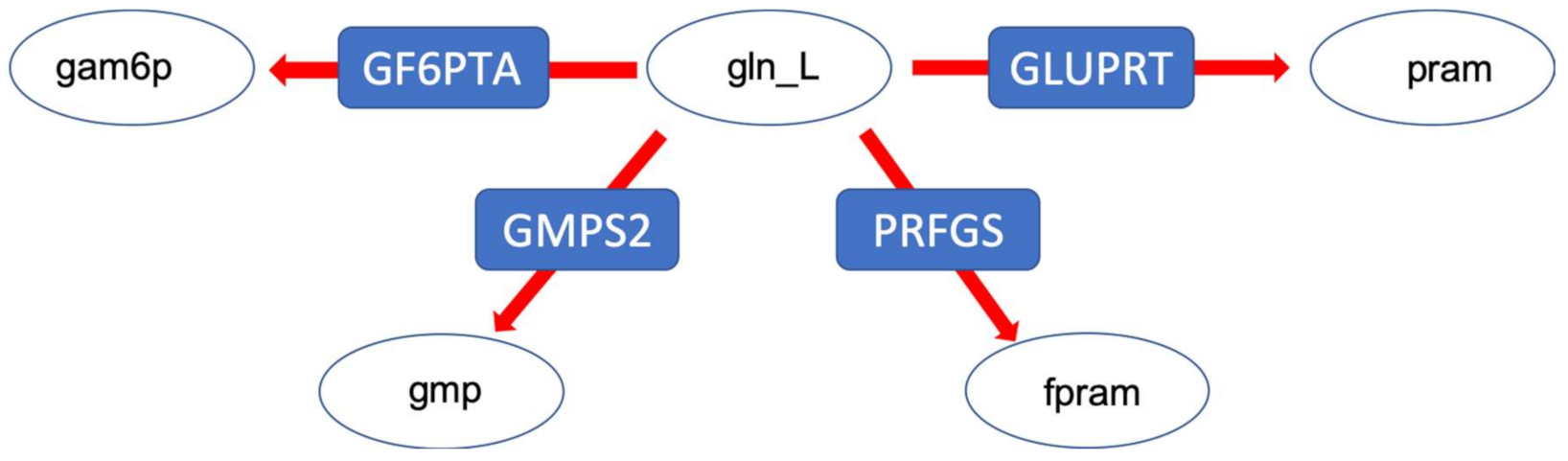
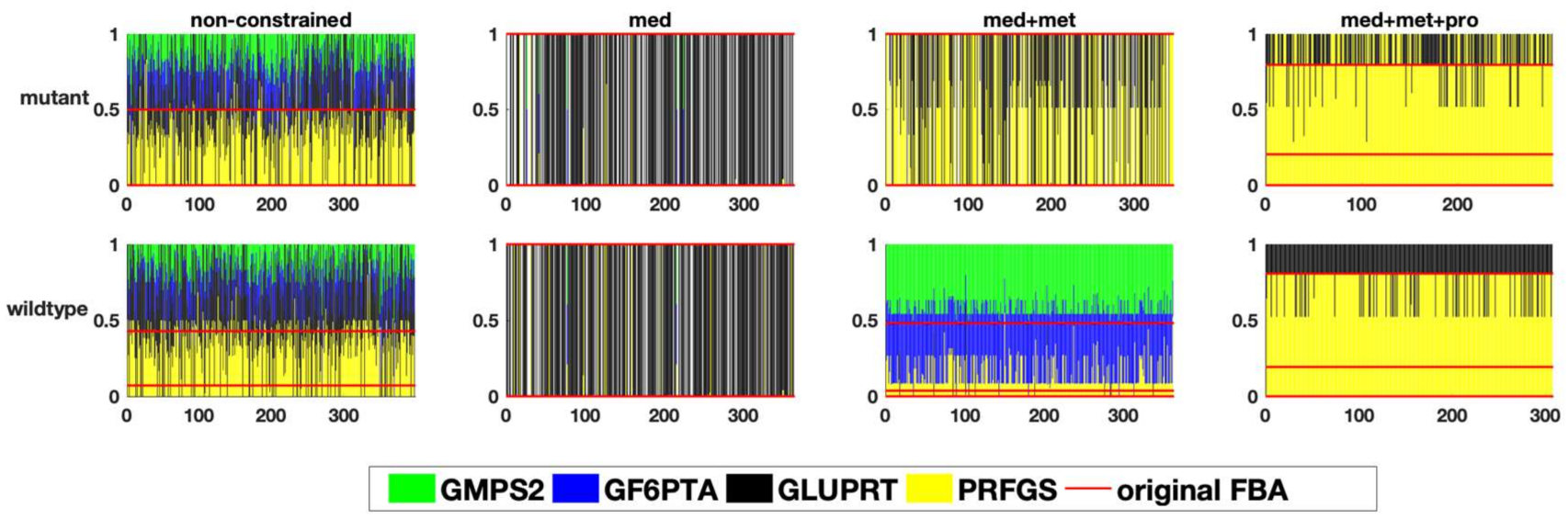
- To decrease the number of biologically inconsistent results, it is vital to integrate biological constraints. In our analysis, generally the integration of proteome data was the most effective in reducing the solution space. However, metabolic flux data on exchange reactions (metabolic uptake and production rates) can also already significantly reduce the solution space (decrease in sensitivity), e.g., at branching points. The fact that the degree of reduction is really case-specific underlines the second point [29].
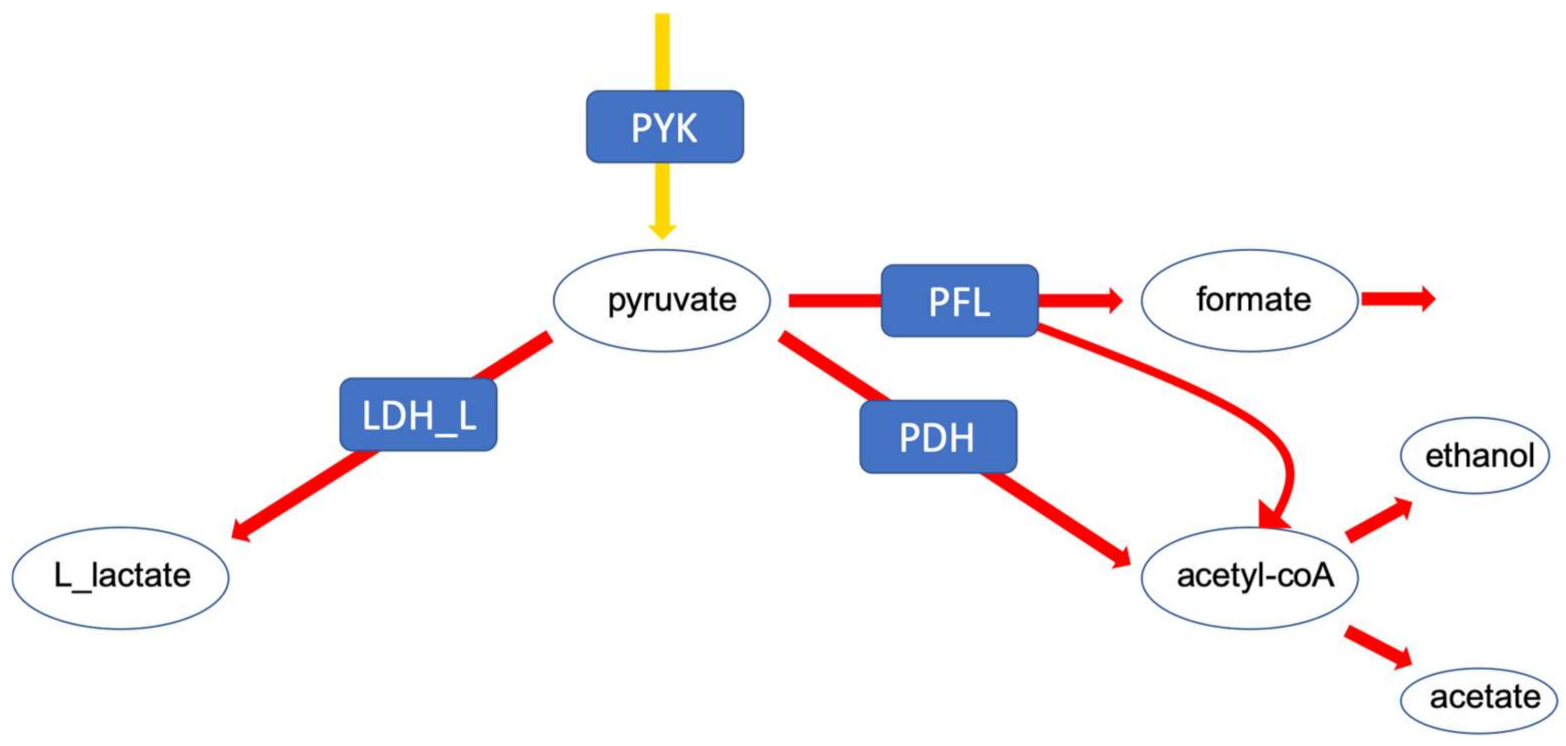
- The analysis of the solution space should be taken into account in any study using FBA. As the above cases demonstrate, constraints like metabolite exchange rates, which are arguably one of the more commonly used constraints, can effectively reduce the solution space such that biologically relevant results for certain questions (e.g., specific kind of fermentation) are achieved, but this is not the case for every model/data-set. There are different ways to investigate the solution space. In our study, sampling by perturbation was an easy and informative way to investigate the different optimal flux distributions. We suggest that the functional analysis of the solution space using our perturbation method gives an explicit account for the robustness as well as reliability of genome-scale models. This also enables us to understand which data sets and which biological phenotypes can effectively shrink the solution space and increase the predictability of models.However, there are alternative methods for the analysis of the solution space not investigated here, e.g., Monte-Carlo sampling. This approach is mostly used to calculate the probability distribution of individual fluxes as well as to determine correlated reaction sets which can be further used for experimental design [12,14]. While this aims at the probability distribution of individual fluxes, our method is focused on uncovering the uncertainty in the interplay between different metabolic fluxes. As mentioned above, Monte-Carlo sampling enables the calculation of correlated reaction sets, which can be used to select candidate reactions for flux measurements, helping to estimate the flux value of its correlated ones. Nevertheless, our method showed, while correlated, the integration of metabolic fluxes of fermentation products for which the internal reactions were reported to be correlated (e.g., LDH, PFK [12]), does not necessarily result in eliminating the physiologically inconsistent result (see analysis of the pyruvate branching point in the case of the E. faecalis mutant). Therefore, the analysis of the solution space using the perturbation procedure helped to yield more information regarding the behaviour of the network as a whole. Another difference between the method presented in this article and Monte-Carlo sampling is a far smaller sample size needed to capture the network response to different metabolic states. While Monte-Carlo sampling needs a large sample size to reveal a comprehensive overview (250,000 data points in [12]), our method uncovers different aspects of the solution space using a far smaller sample size (~10 times the number of variable reactions). However, this also implies that our method does a less complete sampling of the solution space and certain alternative solutions might be overlooked. Moreover, although it is hard to compare the computational performance since our method has a different purpose, we would like to state that our method is fast compared to the Monte-Carlo sampling methods with respect to computational time. The comparison of different Monte-Carlo sampling methods reported that the sampling time spans from 7.64 to 10.67 min, for models of comparable size to our models (especifically the model of E. faecalis) using the CHRR method (the most efficient Monte-Carlo method available right now ) on an intel Core i7 at 2.5 GHz as reported by Fallahi and colleagues [15]. In this study, a reduced version of the metabolic models was used, meaning that the reactions carrying no flux were discarded. Therefore, the number of reactions in the case of the four models, iLJ478, iSB619, iHN637 and iJN746 were reduced from 652, 743, 785 and 1054 to 380, 450, 522 and 652, respectively [15]. The perturbation process of our method took between 122 to 175 seconds depending on the model (wildtype or mutant) and how constrained a model was, in the case of the E. faecalis model on an Intel Core i5 2.3 GHz, 16 MB memory and HDD hard drive, when the flux distribution profiles were obtained using FBA (on MATLAB). Our method also allows the acquisition of flux distributions using FVA, which takes more time—in this case between 31 to 51 min for the same models on the same hardware setup. Comprehensive information regarding the run time of different models used in the above-mentioned study using CHRR and the perturbation process in this study, as well as the number of metabolites and reactions of each model used can be found in the Supplementary Tables S2a and S2b (Supplementary file 2, sheet: run time comparison). Of course; any additional statistical analysis takes further time.
- 3.
- Caution has to be taken if outcomes of FBA are close to the edge of the feasible solution space w.r.t. some parameter, e.g., ATP maintenance. This is at least true when applying methods that are based on FVA, as shown in Section 2.6, since FVA often fails under these conditions and a solution space smaller than the actual space is reported.
4. Materials and Methods
4.1. Models, Experimental Data and Constraints Integration
4.2. Perturbation Procedure
4.3. Analysing the Solution Space Using CoPE-FBA
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bordel, S.; Agren, R.; Nielsen, J. Sampling the Solution Space in Genome-Scale Metabolic Networks Reveals Transcriptional Regulation in Key Enzymes. PLoS Comput. Biol. 2010, 6, e1000859. [Google Scholar] [CrossRef]
- Varma, A.; Palsson, B.O. Metabolic Flux Balancing: Basic Concepts, Scientific and Practical Use. Bio/Technology 1994, 12, 994–998. [Google Scholar] [CrossRef]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What Is Flux Balance Analysis? Nat. Biotechnol. 2010, 28, 245. [Google Scholar] [CrossRef] [PubMed]
- Price, N.D.; Reed, J.L.; Palsson, B. Genome-Scale Models of Microbial Cells: Evaluating the Consequences of Constraints. Nat. Rev. Microbiol. 2004, 2, 886–897. [Google Scholar] [CrossRef]
- Becker, S.A.; Palsson, B.O. Context-Specific Metabolic Networks Are Consistent with Experiments. PLoS Comput. Biol. 2008, 4, e1000082. [Google Scholar] [CrossRef]
- Colijn, C.; Brandes, A.; Zucker, J.; Lun, D.S.; Weiner, B.; Farhat, M.R.; Cheng, T.Y.; Moody, D.B.; Murray, M.; Galagan, J.E. Interpreting Expression Data with Metabolic Flux Models: Predicting Mycobacterium Tuberculosis Mycolic Acid Production. PLoS Comput. Biol. 2009, 5, e1000489. [Google Scholar] [CrossRef]
- Großeholz, R.; Koh, C.-C.; Veith, N.; Fiedler, T.; Strauss, M.; Olivier, B.; Collins, B.C.; Schubert, O.T.; Bergmann, F.; Kreikemeyer, B.; et al. Integrating Highly Quantitative Proteomics and Genome-Scale Metabolic Modeling to Study PH Adaptation in the Human Pathogen Enterococcus Faecalis. NPJ Syst. Biol. Appl. 2016, 2, 16017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, P.A.; Papin, J.A. Functional Integration of a Metabolic Network Model and Expression Data without Arbitrary Thresholding. Bioinformatics 2011, 27, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Famili, I.; Palsson, B.O. The Convex Basis of the Left Null Space of the Stoichiometric Matrix Leads to the Definition of Metabolically Meaningful Pools. Biophys. J. 2003, 85, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Mahadevan, R.; Schilling, C.H. The Effects of Alternate Optimal Solutions in Constraint-Based Genome-Scale Metabolic Models. Metab. Eng. 2003, 5, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Kelk, S.M.; Olivier, B.G.; Stougie, L.; Bruggeman, F.J. Optimal Flux Spaces of Genome-Scale Stoichiometric Models Are Determined by a Few Subnetworks. Sci. Rep. 2012, 2, 44–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, N.D.; Schellenberger, J.; Palsson, B.O. Uniform Sampling of Steady-State Flux Spaces: Means to Design Experiments and to Interpret Enzymopathies. Biophys. J. 2004, 87, 2172–2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papin, J.A.; Reed, J.L.; Palsson, B.O. Hierarchical Thinking in Network Biology: The Unbiased Modularization of Biochemical Networks. Trends Biochem. Sci. 2004, 29, 641–647. [Google Scholar] [CrossRef]
- Thiele, I.; Price, N.D.; Vo, T.D.; Palsson, B. Candidate Metabolic Network States in Human Mitochondria. Impact of Diabetes, Ischemia, and Diet. J. Biol. Chem. 2005, 280, 11683–11695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallahi, S.; Skaug, H.J.; Alendal, G. A Comparison of Monte Carlo Sampling Methods for Metabolic Network Models. PLoS ONE 2020, 15, e0235393. [Google Scholar] [CrossRef]
- Pinhal, S.; Ropers, D.; Geiselmann, J.; de Jong, H. Acetate Metabolism and the Inhibition of Bacterial Growth by Acetate. J. Bacteriol. 2021, 201, e00147-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinh, H.V.; Sarkar, D.; Maranas, C.D. Quantifying the Propagation of Parametric Uncertainty on Flux Balance Analysis. Metab. Eng. 2022, 69, 26–39. [Google Scholar] [CrossRef]
- Maranas, C.D.; Zomorrodi, A.R. Optimization Methods in Metabolic Networks; John Wiley & Sons: New York, NY, USA, 2016; ISBN 1119189012. [Google Scholar]
- Veith, N.; Solheim, M.; van Grinsven, K.W.A.; Olivier, B.G.; Levering, J.; Grosseholz, R.; Hugenholtz, J.; Holo, H.; Nes, I.; Teusink, B.; et al. Using a Genome-Scale Metabolic Model of Enterococcus Faecalis V583 to Assess Amino Acid Uptake and Its Impact on Central Metabolism. Appl. Environ. Microbiol. 2015, 81, 1622–1633. [Google Scholar] [CrossRef] [Green Version]
- Levering, J.; Fiedler, T.; Sieg, A.; van Grinsven, K.W.A.; Hering, S.; Veith, N.; Olivier, B.G.; Klett, L.; Hugenholtz, J.; Teusink, B.; et al. Genome-Scale Reconstruction of the Streptococcus Pyogenes M49 Metabolic Network Reveals Growth Requirements and Indicates Potential Drug Targets. J. Biotechnol. 2016, 232, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Flahaut, N.A.L.; Wiersma, A.; van de Bunt, B.; Martens, D.E.; Schaap, P.J.; Sijtsma, L.; dos Santos, V.A.M.; de Vos, W.M. Genome-Scale Metabolic Model for Lactococcus Lactis MG1363 and Its Application to the Analysis of Flavor Formation. Appl. Microbiol. Biotechnol. 2013, 97, 8729–8739. [Google Scholar] [CrossRef] [PubMed]
- Olivier, B.; Gottstein, W. CBMPy Release 0.8.2. Zenodo 2021. [Google Scholar] [CrossRef]
- Gu, C.; Kim, G.B.; Kim, W.J.; Kim, H.U.; Lee, S.Y. Current Status and Applications of Genome-Scale Metabolic Models. Genome Biol. 2019, 20, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oftadeh, O.; Salvy, P.; Masid, M.; Curvat, M.; Miskovic, L.; Hatzimanikatis, V. A Genome-Scale Metabolic Model of Saccharomyces Cerevisiae That Integrates Expression Constraints and Reaction Thermodynamics. Nat. Commun. 2021, 12, 4790. [Google Scholar] [CrossRef] [PubMed]
- Salvy, P.; Hatzimanikatis, V. The ETFL Formulation Allows Multi-Omics Integration in Thermodynamics-Compliant Metabolism and Expression Models. Nat. Commun. 2020, 11, 30. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, C.J.; Ebrahim, A.; Yang, L.; King, Z.A.; Catoiu, E.; O’Brien, E.J.; Liu, J.K.; Palsson, B.O. COBRAme: A Computational Framework for Genome-Scale Models of Metabolism and Gene Expression. PLoS Comput. Biol. 2018, 14, e1006302. [Google Scholar] [CrossRef] [PubMed]
- Garcia, S.; Thompson, R.A.; Giannone, R.J.; Dash, S.; Maranas, C.D.; Trinh, C.T. Development of a Genome-Scale Metabolic Model of Clostridium Thermocellum and Its Applications for Integration of Multi-Omics Datasets and Computational Strain Design. Front. Bioeng. Biotechnol. 2020, 8, 772. [Google Scholar] [CrossRef]
- Teusink, B.; Wiersma, A.; Molenaar, D.; Francke, C.; de Vos, W.M.; Siezen, R.J.; Smid, E.J. Analysis of Growth of Lactobacillus Plantarum WCFS1 on a Complex Medium Using a Genome-Scale Metabolic Model*. J. Biol. Chem. 2006, 281, 40041–40048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razmilic, V.; Castro, J.F.; Andrews, B.; Asenjo, J.A. Analysis of Metabolic Networks of Streptomyces Leeuwenhoekii C34 by Means of a Genome Scale Model: Prediction of Modifications That Enhance the Production of Specialized Metabolites. Biotechnol. Bioeng. 2018, 115, 1815–1828. [Google Scholar] [CrossRef]
- Loghmani, S.B.; Zitzow, E.; Koh, G.C.-C.; Ulmer, A.; Veith, N.; Grosszligeholz, R.; Rossnagel, M.; Loesch, M.; Aebersold, R.; Kreikemeyer, B.; et al. All Driven by Energy Demand? Integrative Comparison of Metabolism of Enterococcus Faecalis Wildtype and a Glutamine Synthase Mutant. bioRxiv 2021. [Google Scholar] [CrossRef]
- Loghmani, S.B. FBA_perturbation. Available online: https://Github.Com/Babakml/FBA_perturbation.git (accessed on 2 November 2021).
- Vlassis, N.; Pacheco, M.P.; Sauter, T. Fast Reconstruction of Compact Context-Specific Metabolic Network Models. PLoS Comput. Biol. 2014, 10, e1003424. [Google Scholar] [CrossRef]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and Analysis of Biochemical Constraint-Based Models Using the COBRA Toolbox v.3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MATLAB. MATLAB Version 9.4.0.813654 (R2018a); MathWorks Inc.: Natick, MA, USA, 2018. [Google Scholar]
- IBM. ILOG CPLEX User’s Manual. 2017, 596. Available online: https://www.ibm.com/docs/en/icos/12.8.0.0?topic=cplex-users-manual (accessed on 27 February 2020).
- Makhorin, A. GLPK (GNU Linear Programming Kit). 2008. Available online: http//www.gnu.org/s/glpk/glpk.html (accessed on 22 August 2017).
- Van Rossum, G.; Drake, F.L., Jr. Python Reference Manual; Centrum voor Wiskunde en Informatica: Amsterdam, The Netherlands, 1995. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Name | Number of Variable Reactions Variability > 10−6 | Number of Variable Reactions Variability > 10−3 | No of Reactions |
|---|---|---|---|
| mt + nc | 397 | 397 | 708 |
| mt + med | 362 | 340 | 708 |
| mt + med + met | 347 | 315 | 708 |
| mt + med + met + pro | 298 | 289 | 708 |
| wt + nc | 398 | 398 | 709 |
| wt + med | 363 | 341 | 709 |
| wt + med + met | 362 | 340 | 709 |
| wt + med + met + pro | 307 | 85 | 709 |
| Model Name | Number of Reactions in Each Module |
|---|---|
| mt + nc | 399 |
| mt + med | 360, 4 |
| mt + med + met | 345, 4 |
| mt + med + met + pro | 286, 5, 4, 4 |
| wt + nc | 400 |
| wt + med | 361, 4 |
| wt + med + met | 360, 4 |
| wt + med + met + pro | 295, 5, 4, 4 |
| Wt + Med + Met + Pro-Edge | Wt + Med + Met + Pro | |||
|---|---|---|---|---|
| optimality tolerance | 100 | 99.9 | 100 | 99.9 |
| FVA | 209 | 387 | 307 | 387 |
| #reactions, sensitive to perturbation | 94 | 137 | 137 | 151 |
| #modules according to CoPE-FBA | 4, 13, 7, 5, 4, 4, 12, 3 | 295, 5, 4, 4 | 295, 5, 4, 4 | 295, 5, 4, 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loghmani, S.B.; Veith, N.; Sahle, S.; Bergmann, F.T.; Olivier, B.G.; Kummer, U. Inspecting the Solution Space of Genome-Scale Metabolic Models. Metabolites 2022, 12, 43. https://doi.org/10.3390/metabo12010043
Loghmani SB, Veith N, Sahle S, Bergmann FT, Olivier BG, Kummer U. Inspecting the Solution Space of Genome-Scale Metabolic Models. Metabolites. 2022; 12(1):43. https://doi.org/10.3390/metabo12010043
Chicago/Turabian StyleLoghmani, Seyed Babak, Nadine Veith, Sven Sahle, Frank T. Bergmann, Brett G. Olivier, and Ursula Kummer. 2022. "Inspecting the Solution Space of Genome-Scale Metabolic Models" Metabolites 12, no. 1: 43. https://doi.org/10.3390/metabo12010043
APA StyleLoghmani, S. B., Veith, N., Sahle, S., Bergmann, F. T., Olivier, B. G., & Kummer, U. (2022). Inspecting the Solution Space of Genome-Scale Metabolic Models. Metabolites, 12(1), 43. https://doi.org/10.3390/metabo12010043