
A Robust Method for Sample Preparation of Gastrointestinal Stromal Tumour for LC/MS Untargeted Metabolomics

 and
and
Abstract
1. Introduction
2. Results
2.1. Extraction Method Development
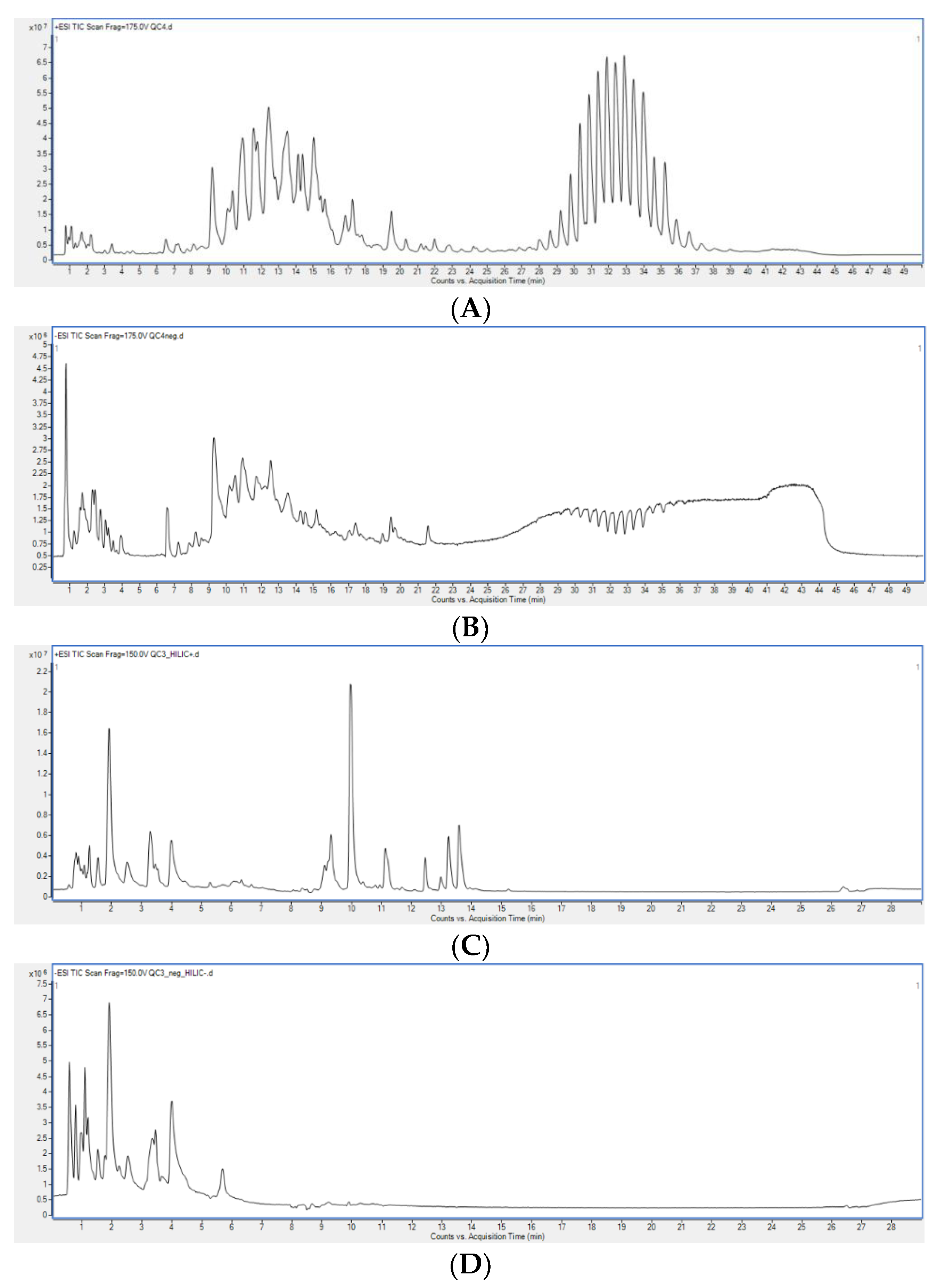
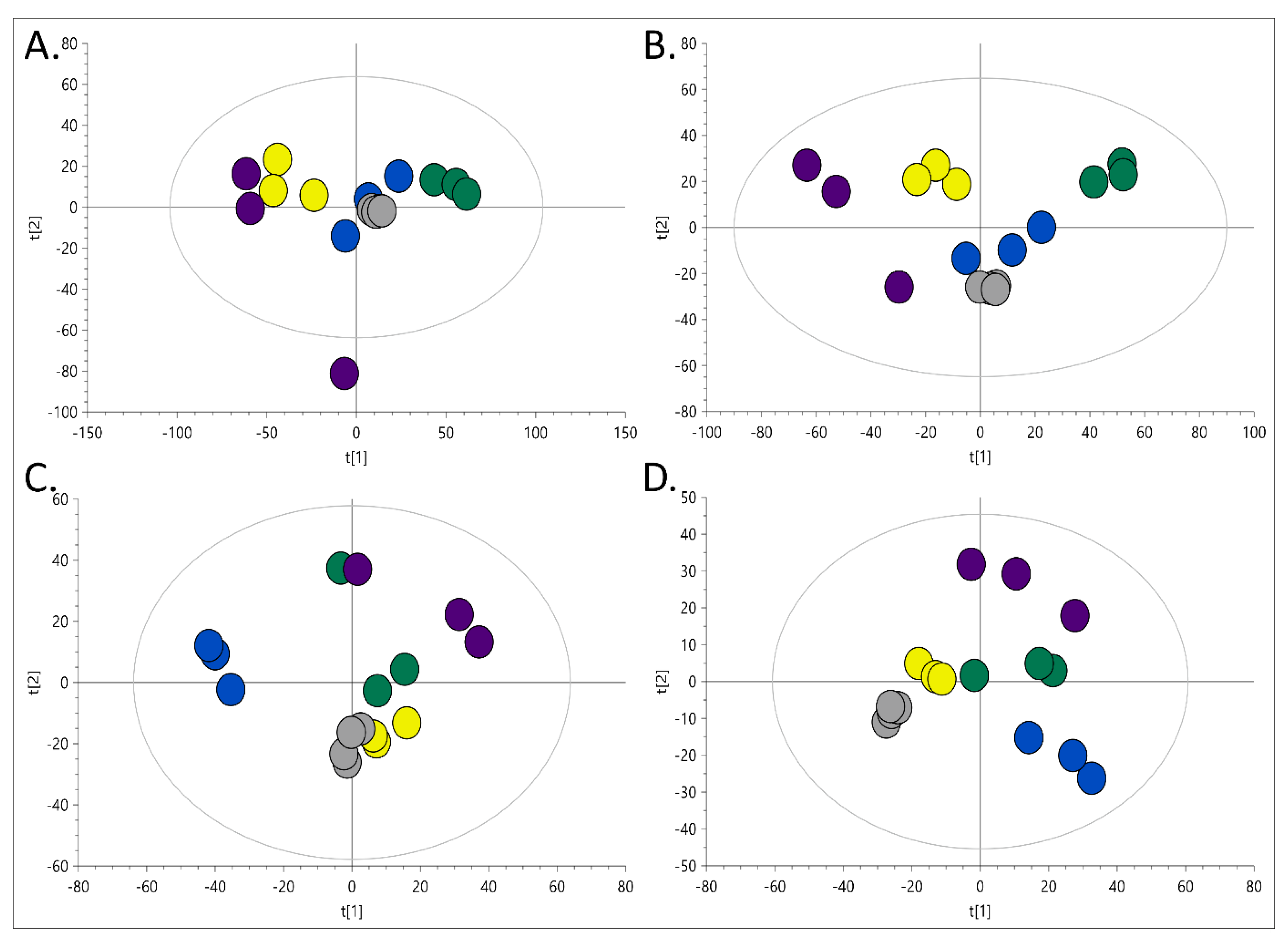
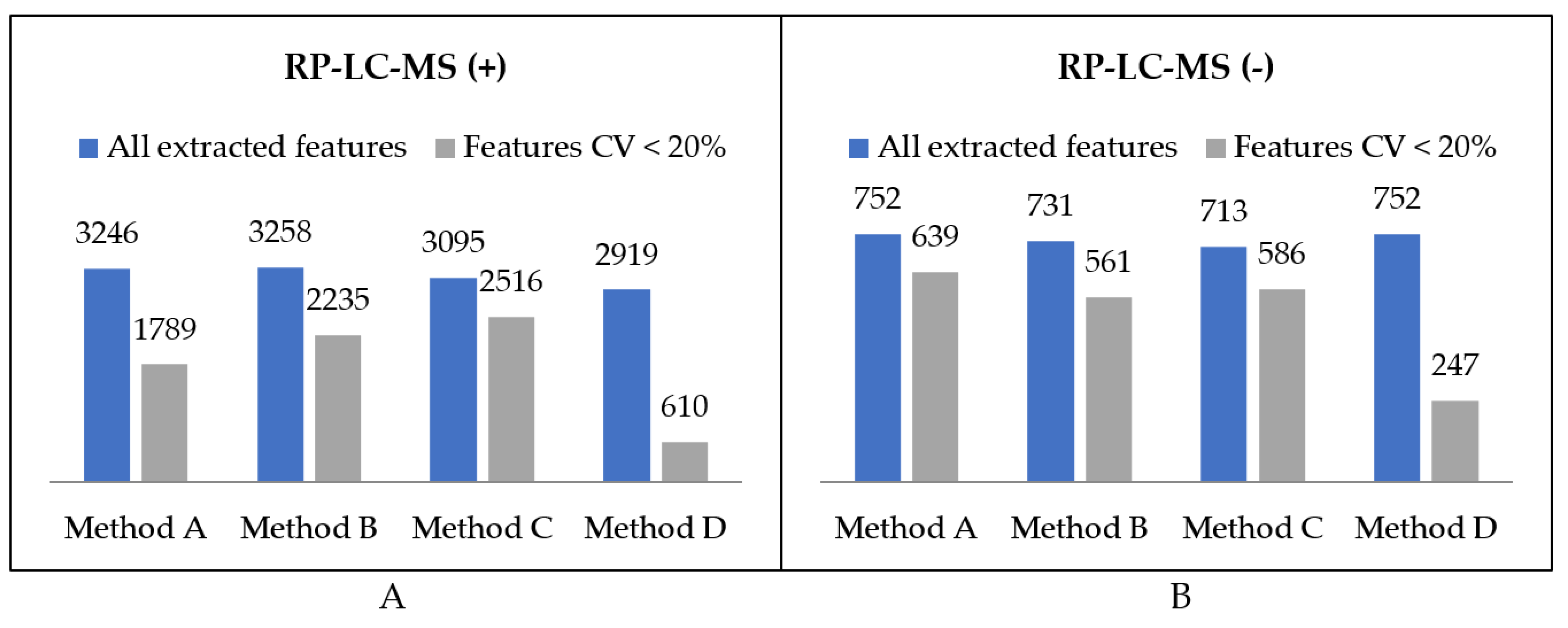
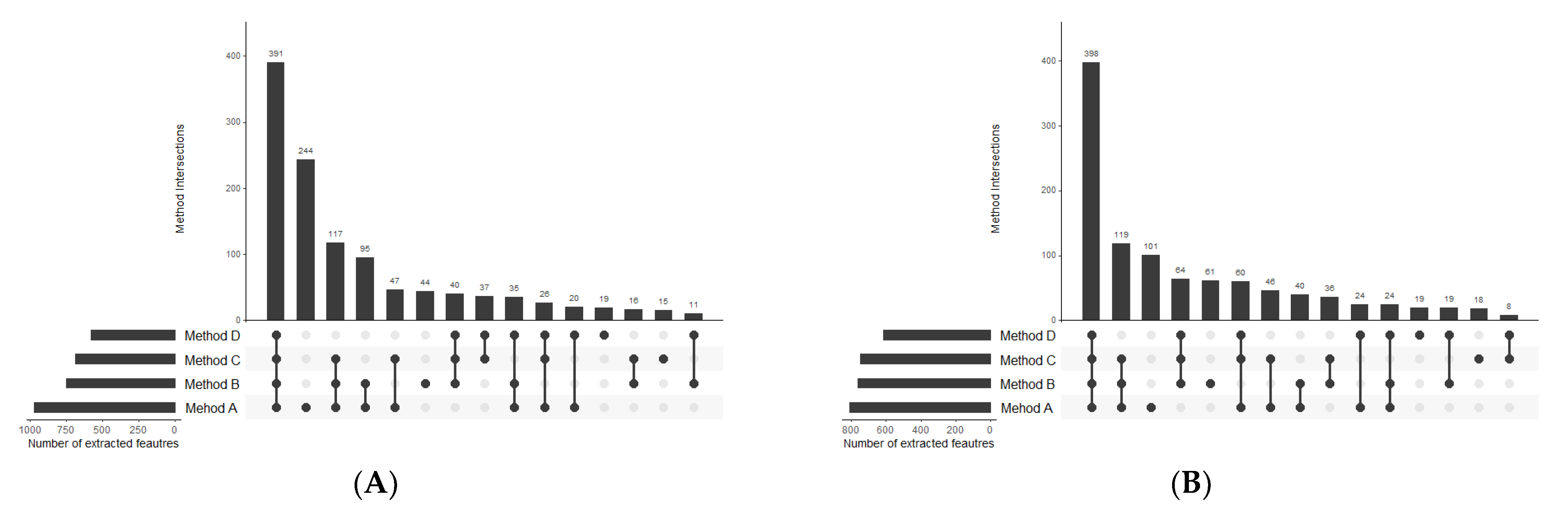
2.1.1. Organic Extracts Comparison
2.1.2. Polar Extracts Comparison
2.1.3. Metabolite Composition of Organic and Polar Fractions
2.2. Robustness Testing of the Extraction Procedure
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Sample Collection
4.3. Tissue Sample Homogenization
4.4. Methods for Metabolite Extraction
4.5. LC-MS Analysis
4.6. Data Processing
4.7. Robustness Testing of the Selected Extraction Method
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Puchades-Carrasco, L. Metabolomics Applications in Precision Medicine: An Oncological Perspective. Curr. Top. Med. Chem. 2017, 17, 2740–2751. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov. 2016, 15, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Metabolomics for Investigating Physiological and Pathophysiological Processes. Physiol. Rev. 2019, 99, 1819–1875. [Google Scholar] [CrossRef]
- Roca, M.; Alcoriza, M.I.; Garcia-Cañaveras, J.C.; Lahoz, A. Reviewing the metabolome coverage provided by LC-MS: Focus on sample preparation and chromatography-A tutorial. Anal. Chim. Acta 2021, 1147, 38–55. [Google Scholar] [CrossRef]
- Yanes, O.; Tautenhahn, R.; Patti, G.J.; Siuzdak, G. Expanding Coverage of the Metabolome for Global Metabolite Profiling. Anal. Chem. 2011, 83, 2152–2161. [Google Scholar] [CrossRef]
- Miggiels, P.; Wouters, B.; van Westen, G.J.; Dubbelman, A.-C.; Hankemeier, T. Novel technologies for metabolomics: More for less. TrAC Trends Anal. Chem. 2019, 120, 115323. [Google Scholar] [CrossRef]
- Kumar, A.; Misra, B.B. Challenges and Opportunities in Cancer Metabolomics. Proteomics 2019, 19, e1900042. [Google Scholar] [CrossRef] [PubMed]
- Pezzatti, J.; Boccard, J.; Codesido, S.; Gagnebin, Y.; Joshi, A.; Picard, D.; González-Ruiz, V.; Rudaz, S. Implementation of liquid chromatography–high resolution mass spectrometry methods for untargeted metabolomic analyses of biological samples: A tutorial. Anal. Chim. Acta 2020, 1105, 28–44. [Google Scholar] [CrossRef]
- Naz, S.; Dos Santos, D.C.M.; Garcia, A.; Barbas, C. Analytical protocols based on LC–MS, GC–MS and CE–MS for nontargeted metabolomics of biological tissues. Bioanalysis 2014, 6, 1657–1677. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A Simple Method for the Isolation and Purification of Total Lipides from Animal Tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A Rapid Method of Total Lipid Extraction and Purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef]
- Sitnikov, D.G.; Monnin, C.; Vuckovic, D. Systematic Assessment of Seven Solvent and Solid-Phase Extraction Methods for Metabolomics Analysis of Human Plasma by LC-MS. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Patterson, R.E.; Ducrocq, A.J.; McDougall, D.J.; Garrett, T.J.; Yost, R.A. Comparison of blood plasma sample preparation methods for combined LC–MS lipidomics and metabolomics. J. Chromatogr. B 2015, 1002, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Gil, A.; Zhang, W.; Wolters, J.C.; Permentier, H.; Boer, T.; Horvatovich, P.; Heiner-Fokkema, M.R.; Reijngoud, D.-J.; Bischoff, R. One- vs two-phase extraction: Re-evaluation of sample preparation procedures for untargeted lipidomics in plasma samples. Anal. Bioanal. Chem. 2018, 410, 5859–5870. [Google Scholar] [CrossRef] [PubMed]
- Löfgren, L.; Forsberg, G.-B.; Ståhlman, M. The BUME method: A new rapid and simple chloroform-free method for total lipid extraction of animal tissue. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Masson, P.; Alves, A.C.; Ebbels, T.M.D.; Nicholson, J.K.; Want, E.J. Optimization and Evaluation of Metabolite Extraction Protocols for Untargeted Metabolic Profiling of Liver Samples by UPLC-MS. Anal. Chem. 2010, 82, 7779–7786. [Google Scholar] [CrossRef] [PubMed]
- Sostare, J.; Di Guida, R.; Kirwan, J.; Chalal, K.; Palmer, E.; Dunn, W.B.; Viant, M.R. Comparison of modified Matyash method to conventional solvent systems for polar metabolite and lipid extractions. Anal. Chim. Acta 2018, 1037, 301–315. [Google Scholar] [CrossRef]
- Ulmer, C.Z.; Jones, C.M.; Yost, R.A.; Garrett, T.J.; Bowden, J.A. Optimization of Folch, Bligh-Dyer, and Matyash sample-to-extraction solvent ratios for human plasma-based lipidomics studies. Anal. Chim. Acta 2018, 1037, 351–357. [Google Scholar] [CrossRef]
- Sarafian, M.H.; Gaudin, M.; Lewis, M.R.; Martin, F.-P.; Holmes, E.; Nicholson, J.K.; Dumas, M.-E. Objective Set of Criteria for Optimization of Sample Preparation Procedures for Ultra-High Throughput Untargeted Blood Plasma Lipid Profiling by Ultra Performance Liquid Chromatography–Mass Spectrometry. Anal. Chem. 2014, 86, 5766–5774. [Google Scholar] [CrossRef]
- Forcisi, S.; Moritz, F.; Kanawati, B.; Tziotis, D.; Lehmann, R.; Schmitt-Kopplin, P. Liquid chromatography–mass spectrometry in metabolomics research: Mass analyzers in ultra high pressure liquid chromatography coupling. J. Chromatogr. A 2013, 1292, 51–65. [Google Scholar] [CrossRef]
- Segers, K.; Declerck, S.; Mangelings, D.; Heyden, Y.V.; Eeckhaut, A.V. Analytical techniques for metabolomic studies: A review. Bioanalysis 2019, 11, 2297–2318. [Google Scholar] [CrossRef]
- Heyden, Y.V.; Nijhuis, A.; Smeyers-Verbeke, J.; Vandeginste, B.; Massart, D. Guidance for robustness/ruggedness tests in method validation. J. Pharm. Biomed. Anal. 2001, 24, 723–753. [Google Scholar] [CrossRef]
- Rutledge, J. An Introduction to Design of Experiments: A Simplified Approach. Technometrics 2000, 42, 309. [Google Scholar] [CrossRef]
- Mongomery, D.C. Montgomery: Design and Analysis of Experiments; Wiley: Hoboken, NJ, USA, 2017. [Google Scholar]
- Naik-Panvelkar, P.; Armour, C.; Saini, B. Discrete choice experiments in pharmacy: A review of the literature. Int. J. Pharm. Pr. 2013, 21, 3–19. [Google Scholar] [CrossRef]
- Ferreira, S.L.C.; Caires, A.O.; Borges, T.D.S.; Lima, A.M.; Silva, L.O.; dos Santos, W.N. Robustness evaluation in analytical methods optimized using experimental designs. Microchem. J. 2017, 131, 163–169. [Google Scholar] [CrossRef]
- Pantaleo, M.A.; Urbini, M.; Indio, V.; Ravegnini, G.; Nannini, M.; De Luca, M.; Tarantino, G.; Angelini, S.; Gronchi, A.; Vincenzi, B.; et al. Genome-Wide Analysis Identifies MEN1 and MAX Mutations and a Neuroendocrine-Like Molecular Heterogeneity in Quadruple WT GIST. Mol. Cancer Res. 2017, 15, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Astolfi, A.; Urbini, M.; Indio, V.; Nannini, M.; Genovese, C.G.; Santini, D.; Saponara, M.; Mandrioli, A.; Ercolani, G.; Brandi, G.; et al. Whole exome sequencing (WES) on formalin-fixed, paraffin-embedded (FFPE) tumor tissue in gastrointestinal stromal tumors (GIST). BMC Genom. 2015, 16, 1–11. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Indio, V.; Astolfi, A.; Tarantino, G.; Urbini, M.; Patterson, J.; Nannini, M.; Saponara, M.; Gatto, L.; Santini, D.; Valle, I.F.D.; et al. Integrated Molecular Characterization of Gastrointestinal Stromal Tumors (GIST) Harboring the Rare D842V Mutation in PDGFRA Gene. Int. J. Mol. Sci. 2018, 19, 732. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef]
- Yakoub, D.; Marks, V.; Wang, Z.; Paulus, E.; Capobianco, T.; Trent, J.C.; Walls, J.; Livingstone, A.S. Metabolomic profiling of gastrointestinal stromal tumor (GIST) in human tissue samples and xenografts. J. Clin. Oncol. 2014, 32, 10516. [Google Scholar] [CrossRef]
- Sarah, K.A.; Jenner, A.M.; Mitchell, T.; Brown, S.; Halliday, G.; Andrew, M.J. An Improved High-Throughput Lipid Extraction Method for the Analysis of Human Brain Lipids. Lipids 2013, 48, 307–318. [Google Scholar] [CrossRef]
- Pellegrino, R.M.; Di Veroli, A.; Valeri, A.; Goracci, L.; Cruciani, G. LC/MS lipid profiling from human serum: A new method for global lipid extraction. Anal. Bioanal. Chem. 2014, 406, 7937–7948. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, Y.; Sun, J.; Fan, S.; Yao, X.; Ran, X.; Zheng, C.; Huang, M.; Bi, H. Optimization of lipid extraction and analytical protocols for UHPLC-ESI-HRMS-based lipidomic analysis of adherent mammalian cancer cells. Anal. Bioanal. Chem. 2017, 409, 5349–5358. [Google Scholar] [CrossRef]
- Wang, H.; Xu, J.; Chen, Y.; Zhang, R.; He, J.; Wang, Z.; Zang, Q.; Wei, J.; Song, X.; Abliz, Z. Optimization and Evaluation Strategy of Esophageal Tissue Preparation Protocols for Metabolomics by LC–MS. Anal. Chem. 2016, 88, 3459–3464. [Google Scholar] [CrossRef]
- Chen, S.; Hoene, M.; Li, J.; Li, Y.; Zhao, X.; Häring, H.-U.; Schleicher, E.D.; Weigert, C.; Xu, G.; Lehmann, R. Simultaneous extraction of metabolome and lipidome with methyl tert-butyl ether from a single small tissue sample for ultra-high performance liquid chromatography/mass spectrometry. J. Chromatogr. A 2013, 1298, 9–16. [Google Scholar] [CrossRef]
- Anwar, M.A.; Vorkas, P.; Li, J.; Shalhoub, J.; Want, E.J.; Davies, A.H.; Holmes, E. Optimization of metabolite extraction of human vein tissue for ultra performance liquid chromatography-mass spectrometry and nuclear magnetic resonance-based untargeted metabolic profiling. Analyst 2015, 140, 7586–7597. [Google Scholar] [CrossRef]
- Dudzik, D.; Barbas-Bernardos, C.; García, A.; Barbas, C. Quality assurance procedures for mass spectrometry untargeted metabolomics. a review. J. Pharm. Biomed. Anal. 2018, 147, 149–173. [Google Scholar] [CrossRef] [PubMed]
- Godzien, J.; Alonso-Herranz, V.; Barbas, C.; Armitage, E.G. Controlling the quality of metabolomics data: New strategies to get the best out of the QC sample. Metabolomics 2014, 11, 518–528. [Google Scholar] [CrossRef]
- Dunn, W.B.; Wilson, I.D.; Nicholls, A.W.; Broadhurst, D. The importance of experimental design and QC samples in large-scale and MS-driven untargeted metabolomic studies of humans. Bioanalysis 2012, 4, 2249–2264. [Google Scholar] [CrossRef] [PubMed]
- Bokor, I.; Sdraulig, S.; Sanagou, M. Robustness testing of an In-Situ caesium extraction unit. J. Environ. Radioact. 2017, 178–179, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Omena, E.; Oenning, A.; Merib, J.; Richter, P.; Rosero-Moreano, M.; Carasek, E. A green and simple sample preparation method to determine pesticides in rice using a combination of SPME and rotating disk sorption devices. Anal. Chim. Acta 2019, 1069, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Gil-De-La-Fuente, A.; Godzien, J.; Saugar, S.; Garcia-Carmona, R.; Badran, H.; Wishart, D.S.; Barbas, C.; Otero, A. CEU Mass Mediator 3.0: A Metabolite Annotation Tool. J. Proteome Res. 2019, 18, 797–802. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pattern | Metanol [µL] | Mtbe1 [µL] | vortex1 [min] | MTBE2 [µL] | H20 [µL] | Vortex2 [min] | Centrifugation [min] | Volume for Evaporation [µL] | Evaporation Temperature [°C] | Solvent Volume for Reconstitution [µL] | Dummy Factor | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | +−−−+−−+−++ | 102 | 196 | 165 | 315 | 234 | 50 | 9 | 204 | 33 | 204 | 1 |
| 2 | +++−−−+−−+− | 102 | 204 | 195 | 315 | 226 | 50 | 11 | 196 | 33 | 204 | −1 |
| 3 | −−+−+++−−−+ | 98 | 196 | 195 | 315 | 234 | 70 | 11 | 196 | 33 | 196 | 1 |
| 4 | −+−−+−+++−− | 98 | 204 | 165 | 315 | 234 | 50 | 11 | 204 | 37 | 196 | −1 |
| 5 | −−−+−−+−+++ | 98 | 196 | 165 | 325 | 226 | 50 | 11 | 196 | 37 | 204 | 1 |
| 6 | 0 | 100 | 200 | 180 | 320 | 230 | 60 | 10 | 200 | 35 | 200 | 0 |
| 7 | +−+++−−−+−− | 102 | 196 | 195 | 325 | 234 | 50 | 9 | 196 | 37 | 196 | −1 |
| 8 | −+++−−−+−−+ | 98 | 204 | 195 | 325 | 226 | 50 | 9 | 204 | 33 | 196 | 1 |
| 9 | −−+−−+−+++− | 98 | 196 | 195 | 315 | 226 | 70 | 9 | 204 | 37 | 204 | −1 |
| 10 | 0 | 100 | 200 | 180 | 320 | 230 | 60 | 10 | 200 | 35 | 200 | 0 |
| 11 | ++−−−+−−+−+ | 102 | 204 | 165 | 315 | 226 | 70 | 9 | 196 | 37 | 196 | 1 |
| 12 | +++++++++++ | 102 | 204 | 195 | 325 | 234 | 70 | 11 | 204 | 37 | 204 | 1 |
| 13 | +−−+−+++−−− | 102 | 196 | 165 | 325 | 226 | 70 | 11 | 204 | 33 | 196 | −1 |
| 14 | −+−+++−−−+− | 98 | 204 | 165 | 325 | 234 | 70 | 9 | 196 | 33 | 204 | −1 |
| 15 | 0 | 100 | 200 | 180 | 320 | 230 | 60 | 10 | 200 | 35 | 200 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macioszek, S.; Dudzik, D.; Jacyna, J.; Wozniak, A.; Schöffski, P.; Markuszewski, M.J. A Robust Method for Sample Preparation of Gastrointestinal Stromal Tumour for LC/MS Untargeted Metabolomics. Metabolites 2021, 11, 554. https://doi.org/10.3390/metabo11080554
Macioszek S, Dudzik D, Jacyna J, Wozniak A, Schöffski P, Markuszewski MJ. A Robust Method for Sample Preparation of Gastrointestinal Stromal Tumour for LC/MS Untargeted Metabolomics. Metabolites. 2021; 11(8):554. https://doi.org/10.3390/metabo11080554
Chicago/Turabian StyleMacioszek, Szymon, Danuta Dudzik, Julia Jacyna, Agnieszka Wozniak, Patrick Schöffski, and Michał J. Markuszewski. 2021. "A Robust Method for Sample Preparation of Gastrointestinal Stromal Tumour for LC/MS Untargeted Metabolomics" Metabolites 11, no. 8: 554. https://doi.org/10.3390/metabo11080554
APA StyleMacioszek, S., Dudzik, D., Jacyna, J., Wozniak, A., Schöffski, P., & Markuszewski, M. J. (2021). A Robust Method for Sample Preparation of Gastrointestinal Stromal Tumour for LC/MS Untargeted Metabolomics. Metabolites, 11(8), 554. https://doi.org/10.3390/metabo11080554