
Diethyl Succinate Modulates Microglial Polarization and Activation by Reducing Mitochondrial Fission and Cellular ROS

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
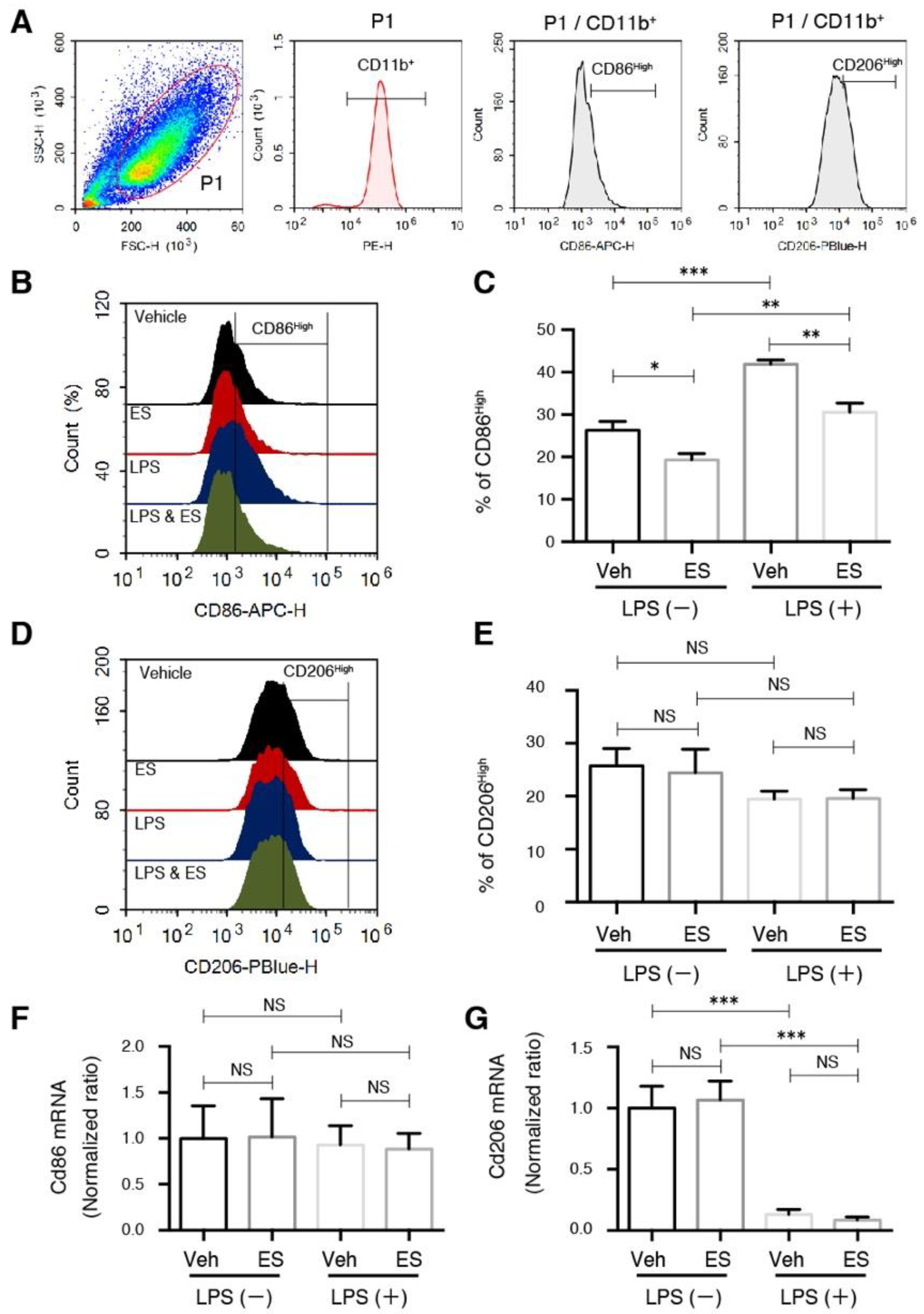
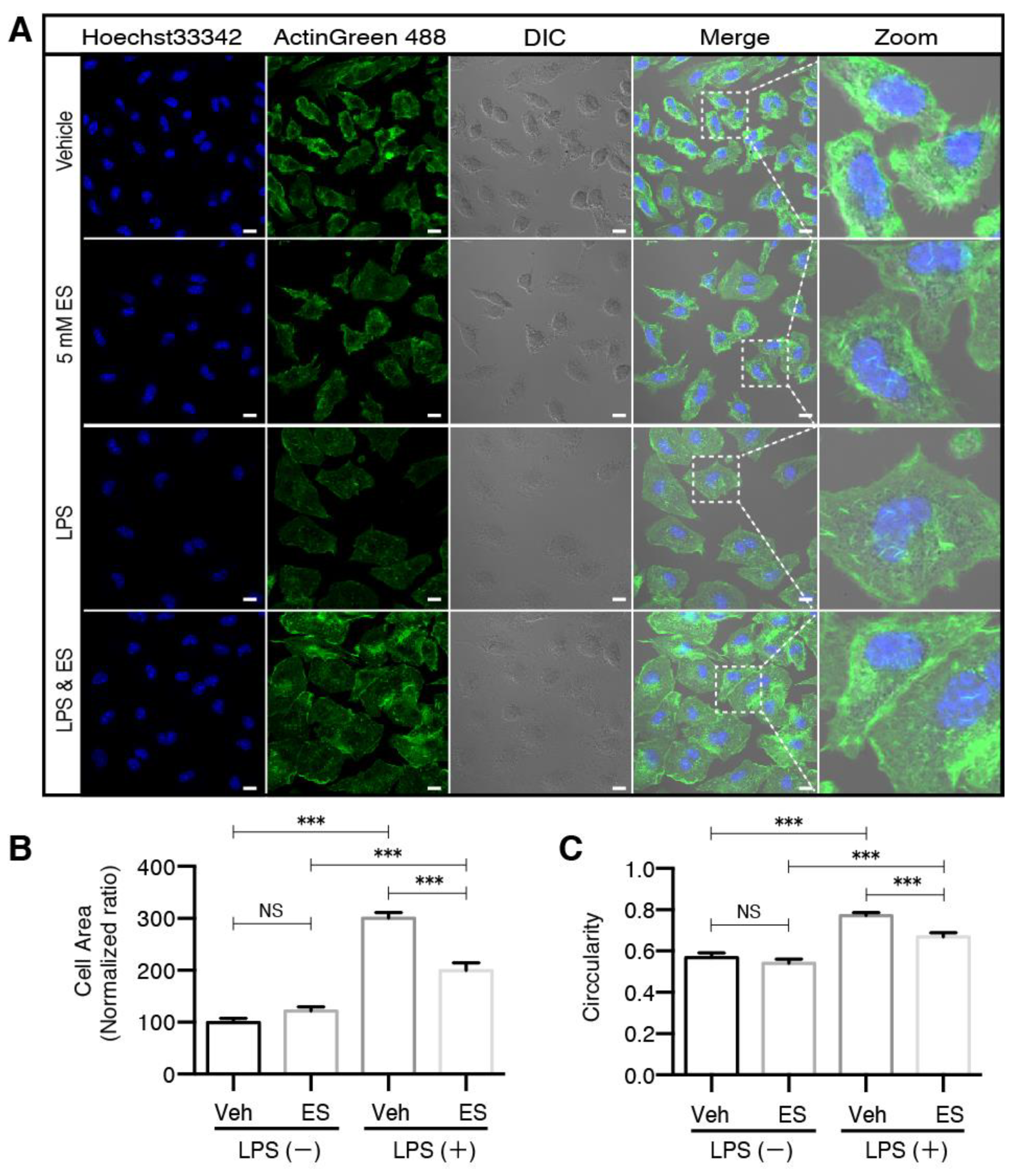
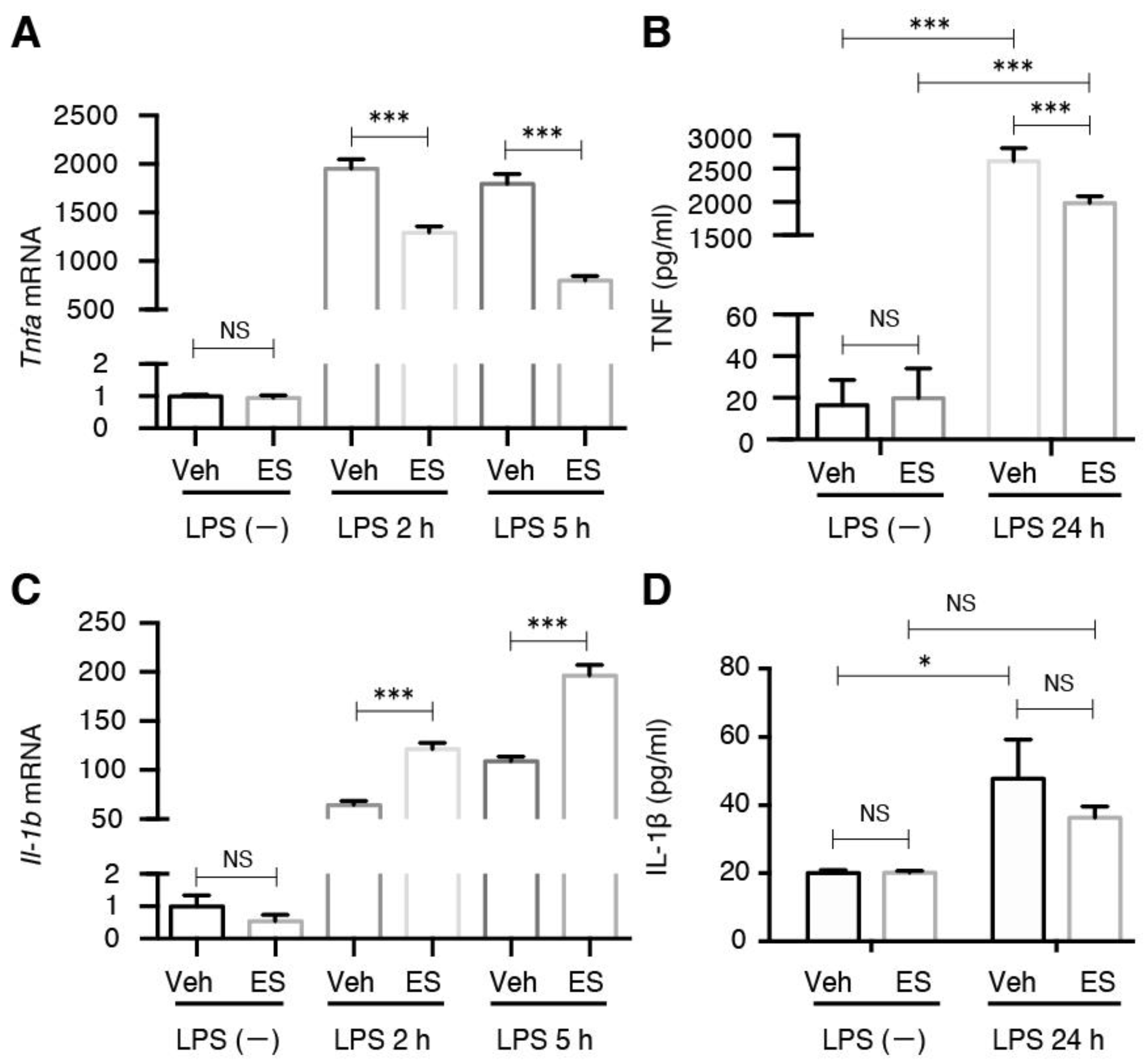
2.1. Diethyl Succinate Decreased the M1 Population in the Primary Microglial Cells
2.2. Diethyl Succinate Reduced ROS Production in the Primary Microglial Cells
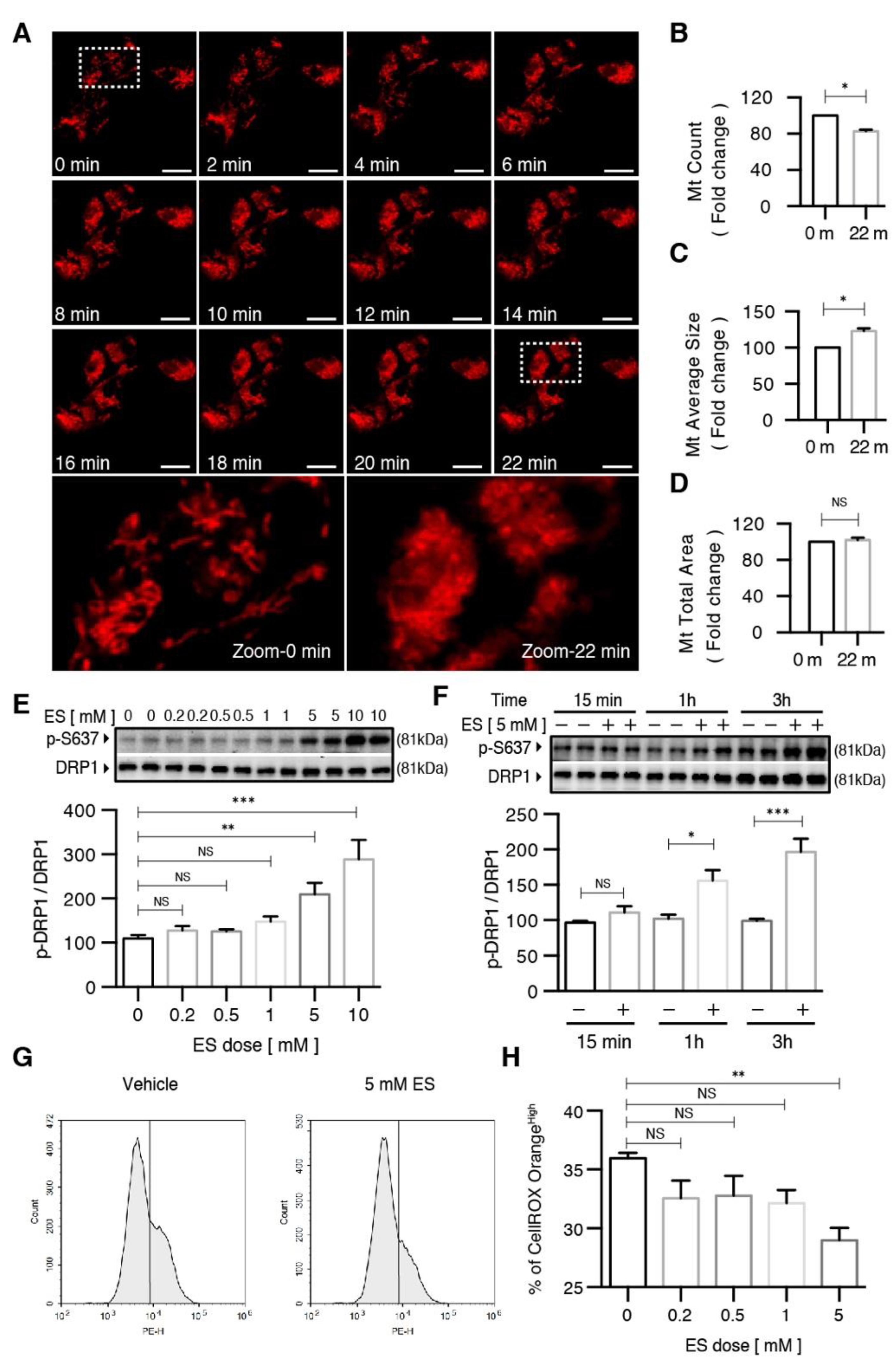
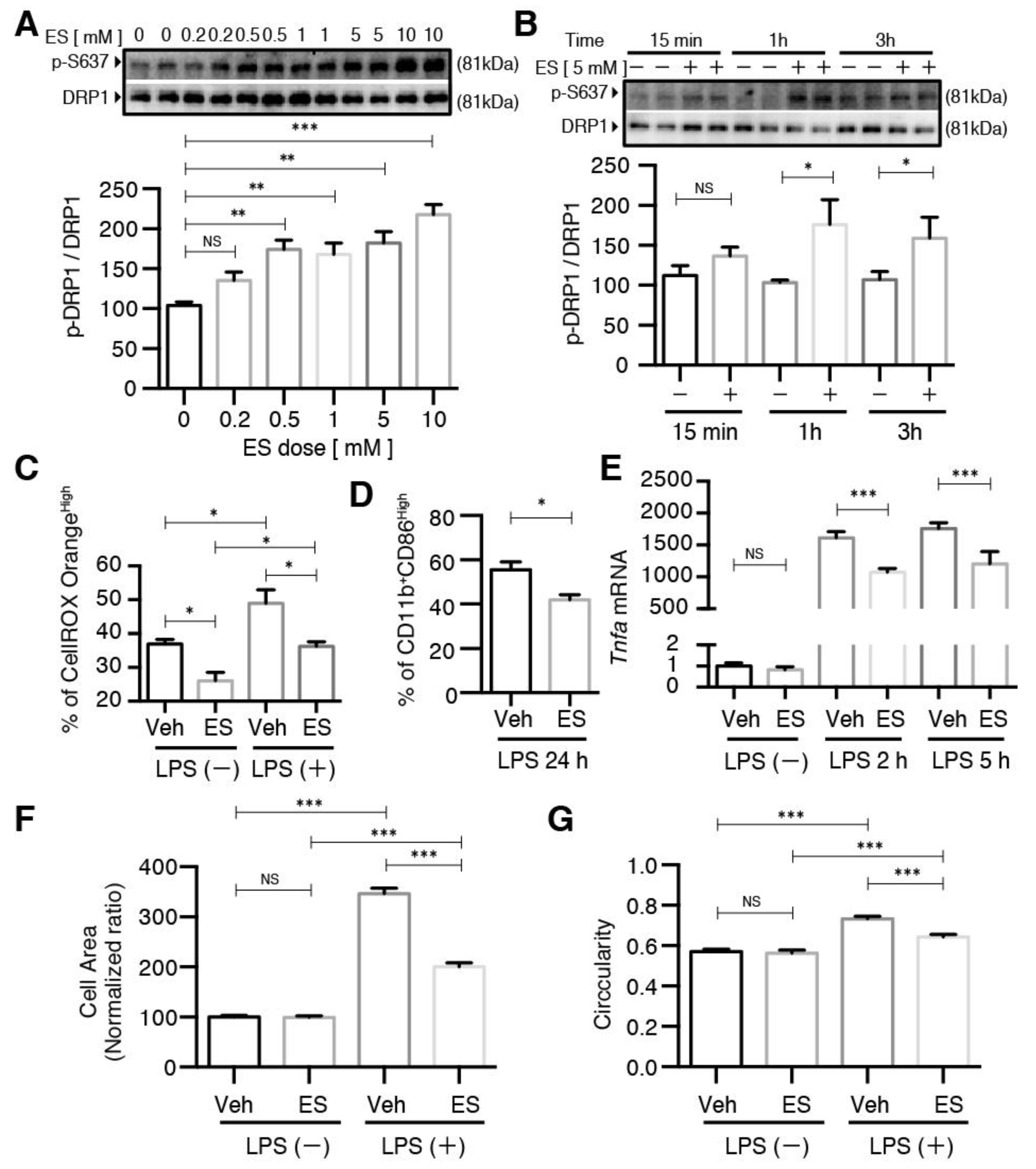
2.3. Diethyl Succinate Regulated Mitochondrial Fission
2.4. The Effects of Diethyl Succinate on Mitochondrial Fission and ROS Production Are Receptor-Independent
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fenselau, A.; Wallis, K. Substrate specificity and mechanism of action of acetoacetate coenzyme A transferase from rat heart. Biochemistry 1974, 13, 3884–3888. [Google Scholar] [CrossRef]
- Furuyama, K.; Sassa, S. Interaction between succinyl CoA synthetase and the heme-biosynthetic enzyme ALAS-E is disrupted in sideroblastic anemia. J. Clin. Investig. 2000, 105, 757–764. [Google Scholar] [CrossRef] [Green Version]
- Ostergaard, E. Disorders caused by deficiency of succinate-CoA ligase. J. Inherit. Metab. Dis. 2008, 31, 226–229. [Google Scholar] [CrossRef]
- Tretter, L.; Patocs, A.; Chinopoulos, C. Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochim. Biophys. Acta Bioenergy 2016, 1857, 1086–1101. [Google Scholar] [CrossRef]
- He, W.; Miao, F.J.-P.; Lin, D.C.-H.; Schwandner, R.T.; Wang, Z.; Gao, J.; Chen, J.-L.; Tian, H.; Ling, L. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 2004, 429, 188–193. [Google Scholar] [CrossRef]
- Regard, J.B.; Sato, I.T.; Coughlin, S.R. Anatomical Profiling of G Protein-Coupled Receptor Expression. Cell 2008, 135, 561–571. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Xie, C.; Li, X.; Yang, J.; Yu, T.; Zhang, R.; Zhang, T.; Saxena, D.; Snyder, M.; Wu, Y.; et al. Succinate and its G-protein-coupled receptor stimulates osteoclastogenesis. Nat. Commun. 2017, 8, 15621. [Google Scholar] [CrossRef]
- Connors, J.; Dawe, N.; Van Limbergen, J. The Role of Succinate in the Regulation of Intestinal Inflammation. Nutrients 2018, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Deen, P.M.; Robben, J.H. Succinate Receptors in the Kidney. J. Am. Soc. Nephrol. 2011, 22, 1416–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, R.; Cucchi, D.; Smith, J.; Pucino, V.; Macdougall, C.E.; Mauro, C. Intermediates of Metabolism: From Bystanders to Signalling Molecules. Trends Biochem. Sci. 2016, 41, 460–471. [Google Scholar] [CrossRef]
- Jiang, S.; Yan, W. Succinate in the cancer-immune cycle. Cancer Lett. 2017, 390, 45–47. [Google Scholar] [CrossRef] [PubMed]
- Littlewood-Evans, A.; Sarret, S.; Apfel, V.; Loesle, P.; Dawson, J.; Zhang, J.; Muller, A.; Tigani, B.; Kneuer, R.; Patel, S.; et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J. Exp. Med. 2016, 213, 1655–1662. [Google Scholar] [CrossRef] [PubMed]
- Macias-Ceja, D.C.; Ortiz-Masia, M.D.; Salvador, P.; Gisbert-Ferrándiz, L.; Hernandez, C.; Hausmann, M.; Rogler, G.; Esplugues, J.V.; Hinojosa, J.; Alós, R.; et al. Succinate receptor mediates intestinal inflammation and fibrosis. Mucosal Immunol. 2019, 12, 178–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, E.; O’Neill, L.A. Succinate: A metabolic signal in inflammation. Trends Cell Biol. 2014, 24, 313–320. [Google Scholar] [CrossRef] [Green Version]
- McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; Zheng, L.; Gardet, A.; Tong, Z.; Jany, S.S.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar]
- Rubic, T.; Lametschwandtner, G.; Jost, S.; Hinteregger, S.; Kund, J.; Carballido-Perrig, N.; Schwärzler, C.; Junt, T.; Voshol, H.; Meingassner, J.G.; et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat. Immunol. 2008, 9, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.H.; Choi, G.E.; Oh, J.Y.; Lee, H.J.; Kim, J.S.; Chae, C.W.; Choi, D.; Han, H.J. Succinate promotes stem cell migration through the GPR91-dependent regulation of DRP1-mediated mitochondrial fission. Sci. Rep. 2017, 7, 12582. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.-T.; Li, L.-Z.; Yang, Y.-L.; Yin, X.; Liu, Q.; Zhang, L.; Liu, K.; Liu, B.; Li, J.; Qi, L.-W. Succinate induces aberrant mitochondrial fission in cardiomyocytes through GPR91 signaling. Cell Death Dis. 2018, 9, 672. [Google Scholar] [CrossRef] [Green Version]
- Fuhrmann, D.C.; Wittig, I.; Brüne, B. TMEM126B deficiency reduces mitochondrial SDH oxidation by LPS, attenuating HIF-1α stabilization and IL-1β expression. Redox Biol. 2019, 20, 204–216. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.; Sobotka, K.S.; Joshi, P.; Gressens, P.; Fleiss, B.; Thornton, C.; Mallard, C.; Hagberg, H. Lipopolysaccharide-induced alteration of mitochondrial morphology induces a metabolic shift in microglia modulating the inflammatory response in vitro and in vivo. Glia 2019, 67, 1047–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Choi, H.; Min, J.-S.; Park, S.-J.; Kim, J.-H.; Park, H.-J.; Kim, B.; Chae, J.-I.; Yim, M.; Lee, D.-S. Mitochondrial dynamics modulate the expression of pro-inflammatory mediators in microglial cells. J. Neurochem. 2013, 127, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Lenz, K.M.; Nelson, L. Microglia and Beyond: Innate Immune Cells As Regulators of Brain Development and Behavioral Function. Front. Immunol. 2018, 9, 698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, S.; Zhou, T.; Cheng, K.; Chen, M.; Wang, Y.; Jiang, Y.; Yang, P. Carboxylic Acid Fullerene (C60) Derivatives Attenuated Neuroinflammatory Responses by Modulating Mitochondrial Dynamics. Nanoscale Res. Lett. 2015, 10, 953. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M.; Wu, B.; Nguyen, H.B.; Thai, T.Q.; Yamasaki, R.; Lu, H.; Rietsch, A.M.; Zorlu, M.M.; Shinozaki, Y.; Saitoh, Y.; et al. Polymorphic regulation of mitochondrial fission and fusion modifies phenotypes of microglia in neuroinflammation. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lively, S.; Schlichter, L.C. Microglia Responses to Pro-inflammatory Stimuli (LPS, IFNgamma+TNFalpha) and Reprogramming by Resolving Cytokines (IL-4, IL-10). Front. Cell Neurosci. 2018, 12, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, M.; Xu, Y.; Pearse, D.D. Cyclic AMP is a key regulator of M1 to M2a phenotypic conversion of microglia in the presence of Th2 cytokines. J. Neuroinflamm. 2016, 13, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taetzsch, T.; Levesque, S.; McGraw, C.; Brookins, S.; Luqa, R.; Bonini, M.G.; Mason, R.P.; Oh, U.; Block, M.L. Redox regulation of NF-kappaB p50 and M1 polarization in microglia. Glia 2015, 63, 423–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, E.; Foster, T.; Thomas, W. Cellular forms and functions of brain microglia. Brain Res. Bull. 1994, 34, 73–78. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Chuang, D.Y.; Simonyi, A.; Kotzbauer, P.T.; Gu, Z.; Sun, G.Y. Cytosolic phospholipase A2 plays a crucial role in ROS/NO signaling during microglial activation through the lipoxygenase pathway. J. Neuroinflamm. 2015, 12, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Burté, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef]
- Lim, H.S.; Kim, Y.J.; Kim, B.Y.; Park, G.; Jeong, S.J. The Anti-neuroinflammatory Activity of Tectorigenin Pretreatment via Downregulated NF-kappaB and ERK/JNK Pathways in BV-2 Microglial and Microglia Inactivation in Mice with Lipopolysaccharide. Front. Pharmacol. 2018, 9, 462. [Google Scholar] [CrossRef] [PubMed]
- Sevenich, L. Brain-Resident Microglia and Blood-Borne Macrophages Orchestrate Central Nervous System Inflammation in Neurodegenerative Disorders and Brain Cancer. Front. Immunol. 2018, 9, 697. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Park, J.; Choi, H.; Kim, B.; Chae, U.; Lee, D.G.; Lee, S.R.; Lee, S.; Lee, H.S.; Lee, D.S. Peroxiredoxin 5 (Prx5) decreases LPS-induced microglial activation through regulation of Ca(2+)/calcineurin-Drp1-dependent mitochondrial fission. Free Radic. Biol. Med. 2016, 99, 392–404. [Google Scholar] [CrossRef]
- Kalia, R.; Wang, R.Y.-R.; Yusuf, A.; Thomas, P.V.; Agard, D.A.; Shaw, J.M.; Frost, A. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 2018, 558, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, T.B.; Sánchez-Guerrero, Á.; Milosevic, I.; Raimundo, N. Mitochondrial fission requires DRP1 but not dynamins. Nature 2019, 570, E34–E42. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Jiang, H.; Chen, S.; Du, F.; Wang, X. The Mitochondrial Phosphatase PGAM5 Functions at the Convergence Point of Multiple Necrotic Death Pathways. Cell 2012, 148, 228–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Zhang, Y.; Liu, S.; Liu, Y.; Yang, X.; Liu, G.; Shimizu, T.; Ikenaka, K.; Fan, K.; Ma, J. Cathepsin C promotes microglia M1 polarization and aggravates neuroinflammation via activation of Ca(2+)-dependent PKC/p38MAPK/NF-kappaB pathway. J. Neuroinflamm. 2019, 16, 10. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Zhang, Y.; Kiprowska, M.; Guo, Y.; Yamamoto, K.; Li, X. Diethyl Succinate Modulates Microglial Polarization and Activation by Reducing Mitochondrial Fission and Cellular ROS. Metabolites 2021, 11, 854. https://doi.org/10.3390/metabo11120854
Wang L, Zhang Y, Kiprowska M, Guo Y, Yamamoto K, Li X. Diethyl Succinate Modulates Microglial Polarization and Activation by Reducing Mitochondrial Fission and Cellular ROS. Metabolites. 2021; 11(12):854. https://doi.org/10.3390/metabo11120854
Chicago/Turabian StyleWang, Lixiang, Yanli Zhang, Magdalena Kiprowska, Yuqi Guo, Ken Yamamoto, and Xin Li. 2021. "Diethyl Succinate Modulates Microglial Polarization and Activation by Reducing Mitochondrial Fission and Cellular ROS" Metabolites 11, no. 12: 854. https://doi.org/10.3390/metabo11120854
APA StyleWang, L., Zhang, Y., Kiprowska, M., Guo, Y., Yamamoto, K., & Li, X. (2021). Diethyl Succinate Modulates Microglial Polarization and Activation by Reducing Mitochondrial Fission and Cellular ROS. Metabolites, 11(12), 854. https://doi.org/10.3390/metabo11120854