The Role of Autophagy in White Adipose Tissue Function: Implications for Metabolic Health

Abstract
1. Introduction
2. The Role of WAT Dysfunction in Metabolic Homeostasis
3. Autophagy
Macroautophagy Pathway
4. Autophagy Involvement in WAT Expandability: Clues from Experimental Data
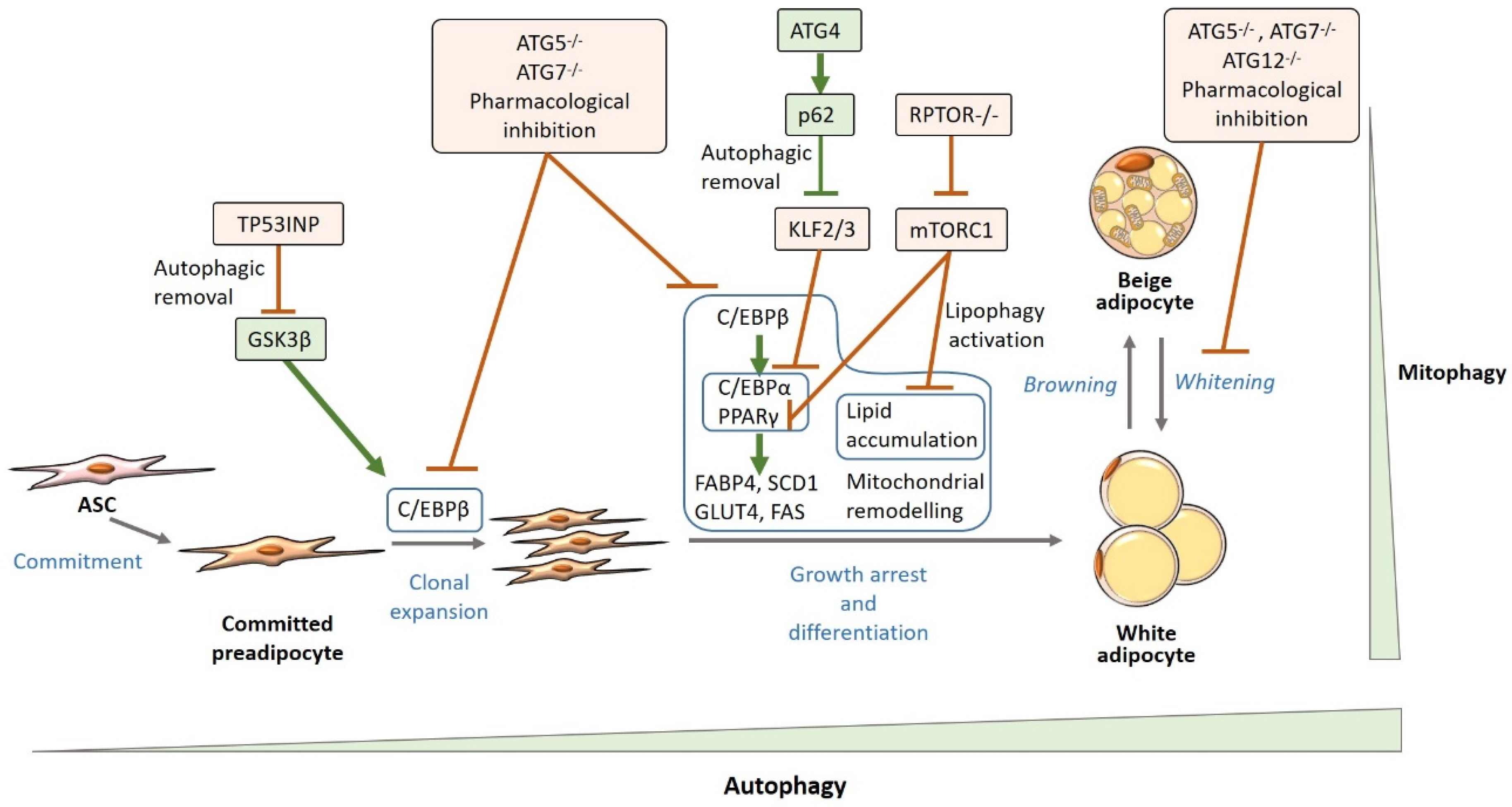
4.1. Autophagy and White Adipogenesis
Mechanisms Underlying the Relationship Between Autophagy and White Adipocyte Biology
Autophagic Regulation of Adipogenic Factors
Mitophagy
Lipophagy
4.2. Autophagy and Extracellular Matrix Regulation: Implications for WAT Remodeling
4.3. Autophagy and WAT Inflammation
5. WAT Autophagy Status in Obesity and Diabetes: Evidence from Animal and Human Studies
6. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; Naghavi, M.; et al. Health effects of overweight and obesity in 195 countries over 25 years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, S.T.; Batty, G.D.; Pentti, J.; Virtanen, M.; Alfredsson, L.; Fransson, E.I.; Goldberg, M.; Heikkilä, K.; Jokela, M.; Knutsson, A.; et al. Obesity and loss of disease-free years owing to major non-communicable diseases: A multicohort study. Lancet Public Health 2018, 3, e490–e497. [Google Scholar] [CrossRef]
- Castellano-Castillo, D.; Morcillo, S.; Clemente-Postigo, M.; Crujeiras, A.B.; Fernandez-García, J.C.; Torres, E.; Tinahones, F.J.; Macias-Gonzalez, M. Adipose tissue inflammation and VDR expression and methylation in colorectal cancer. Clin. Epigenet. 2018, 10, 1–10. [Google Scholar] [CrossRef]
- Pellegrinelli, V.; Carobbio, S.; Vidal-Puig, A. Adipose tissue plasticity: How fat depots respond differently to pathophysiological cues. Diabetologia 2016, 59, 1075–1088. [Google Scholar] [CrossRef]
- Carobbio, S.; Pellegrinelli, V.; Vidal-Puig, A. Adipose Tissue Function and Expandability as Determinants of Lipotoxicity and the Metabolic Syndrome. In Advances in Experimental Medicine and Biology; Springer New York LLC: New York, NY, USA, 2017; Volume 960, pp. 161–196. [Google Scholar]
- Stinkens, R.; Goossens, G.H.; Jocken, J.W.E.; Blaak, E.E. Targeting fatty acid metabolism to improve glucose metabolism. Obes. Rev. 2015, 16, 715–757. [Google Scholar] [CrossRef]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef]
- Lee, M.-W.; Lee, M.; Oh, K.-J. Adipose Tissue-Derived Signatures for Obesity and Type 2 Diabetes: Adipokines, Batokines and MicroRNAs. J. Clin. Med. 2019, 8, 854. [Google Scholar] [CrossRef]
- Scherer, P.E. The many secret lives of adipocytes: Implications for diabetes. Diabetologia 2019, 62, 223–232. [Google Scholar] [CrossRef]
- Villarroya, F.; Cereijo, R.; Villarroya, J.; Giralt, M. Brown adipose tissue as a secretory organ. Nat. Rev. Endocrinol. 2017, 13, 26–35. [Google Scholar] [CrossRef]
- Ferhat, M.; Funai, K.; Boudina, S. Autophagy in Adipose Tissue Physiology and Pathophysiology. Antioxid. Redox Signal. 2019, 31, 487–501. [Google Scholar] [CrossRef]
- Romero, M.; Zorzano, A. Role of autophagy in the regulation of adipose tissue biology. Cell Cycle 2019, 18, 1435–1445. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, V.; Hawkins, W.D.; Klionsky, D.J. Watch What You (Self-) Eat: Autophagic Mechanisms that Modulate Metabolism. Cell Metab. 2019, 29, 803–826. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Why do we need autophagy? A cartoon depiction. Autophagy 2018, 14, 739–742. [Google Scholar] [CrossRef]
- Karsli-Uzunbas, G.; Guo, J.Y.; Price, S.; Teng, X.; Laddha, S.V.; Khor, S.; Kalaany, N.Y.; Jacks, T.; Chan, C.S.; Rabinowitz, J.D.; et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014, 4, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Moretti, J.; Roy, S.; Bozec, D.; Martinez, J.; Chapman, J.R.; Ueberheide, B.; Lamming, D.W.; Chen, Z.J.; Horng, T.; Yeretssian, G.; et al. STING Senses Microbial Viability to Orchestrate Stress-Mediated Autophagy of the Endoplasmic Reticulum. Cell 2017, 171, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Bel, S.; Pendse, M.; Wang, Y.; Li, Y.; Ruhn, K.A.; Hassell, B.; Leal, T.; Winter, S.E.; Xavier, R.J.; Hooper, L.V. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science (80-) 2017, 357, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.J.; Codogno, P. Autophagy: A Sweet Process in Diabetes. Cell Metab. 2008, 8, 275–276. [Google Scholar] [CrossRef]
- Ezaki, J.; Matsumoto, N.; Takeda-Ezaki, M.; Komatsu, M.; Takahashi, K.; Hiraoka, Y.; Taka, H.; Fujimura, T.; Takehana, K.; Yoshida, M.; et al. Liver autophagy contributes to the maintenance of blood glucose and amino acid levels. Autophagy 2011, 7, 727–736. [Google Scholar] [CrossRef]
- Kaushik, S.; Rodriguez-Navarro, J.A.; Arias, E.; Kiffin, R.; Sahu, S.; Schwartz, G.J.; Cuervo, A.M.; Singh, R. Autophagy in hypothalamic agrp neurons regulates food intake and energy balance. Cell Metab. 2011, 14, 173–183. [Google Scholar] [CrossRef]
- Kahn, C.R.; Wang, G.; Lee, K.Y. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J. Clin. Investig. 2019, 129, 3990–4000. [Google Scholar] [CrossRef]
- Rodríguez, A.; Ezquerro, S.; Méndez-Giménez, L.; Becerril, S.; Frühbeck, G. Revisiting the adipocyte: A model for integration of cytokine signaling in the regulation of energy metabolism. Am. J. Physiol. Metab. 2015, 309, E691–E714. [Google Scholar] [CrossRef]
- Rosen, E.D.; Spiegelman, B.M. What We Talk About When We Talk About Fat. Cell 2014, 156, 20–44. [Google Scholar] [CrossRef] [PubMed]
- Villarroya, F.; Cereijo, R.; Gavaldà-Navarro, A.; Villarroya, J.; Giralt, M. Inflammation of brown/beige adipose tissues in obesity and metabolic disease. J. Intern. Med. 2018, 284, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Kotzbeck, P.; Giordano, A.; Mondini, E.; Murano, I.; Severi, I.; Venema, W.; Cecchini, M.P.; Kershaw, E.E.; Barbatelli, G.; Haemmerle, G.; et al. Brown adipose tissue whitening leads to brown adipocyte death and adipose tissue inflammation. J. Lipid Res. 2018, 59, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.G. Insights into an adipocyte whitening program. Adipocyte 2015, 4, 75–80. [Google Scholar] [CrossRef][Green Version]
- Hammarstedt, A.; Gogg, S.; Hedjazifar, S.; Nerstedt, A.; Smith, U. Impaired adipogenesis and dysfunctional adipose tissue in human hypertrophic obesity. Physiol. Rev. 2018, 98, 1911–1941. [Google Scholar] [CrossRef]
- Crewe, C.; An, Y.A.; Scherer, P.E. The ominous triad of adipose tissue dysfunction: Inflammation, fibrosis, and impaired angiogenesis. J. Clin. Investig. 2017, 127, 74–82. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Kajimura, S. Metabolic adaptation and maladaptation in adipose tissue. Nat. Metab. 2019, 1, 189–200. [Google Scholar] [CrossRef]
- Barbarroja, N.; López-Pedrera, R.; Mayas, M.D.; García-Fuentes, E.; Garrido-Sánchez, L.; Macías-González, M.; El Bekay, R.; Vidal-Puig, A.; Tinahones, F.J. The obese healthy paradox: Is inflammation the answer? Biochem. J. 2010, 430, 141–149. [Google Scholar] [CrossRef]
- Reaven, G. All obese individuals are not created equal: Insulin resistance is the major determinant of cardiovascular disease in overweight/obese individuals. Diabetes Vasc. Dis. Res. 2005, 2, 105–112. [Google Scholar] [CrossRef]
- Karelis, A.D.; St-Pierre, D.H.; Conus, F.; Rabasa-Lhoret, R.; Poehlman, E.T. Metabolic and body composition factors in subgroups of obesity: What do we know? J. Clin. Endocrinol. Metab. 2004, 89, 2569–2575. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Postigo, M.; Tinahones, F.J. Do metabolically healthy obese subjects exist? In Overweight and Obesity; Bellido, D., García-Almedida, J.M., López de la Torre, M., Rubio Herrera, M.Á., Eds.; Sociedad Española para el Estudio de la Obesidad (SEEDO): Madrid, Spain, 2015; pp. 175–187. ISBN 978-84-606-9324-6. [Google Scholar]
- Schulze, M.B. Metabolic health in normal-weight and obese individuals. Diabetologia 2019, 62, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Schick, F.; Häring, H.-U. Causes, Characteristics, and Consequences of Metabolically Unhealthy Normal Weight in Humans. Cell Metab. 2017, 26, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Akinci, B.; Meral, R.; Oral, E.A. Phenotypic and Genetic Characteristics of Lipodystrophy: Pathophysiology, Metabolic Abnormalities, and Comorbidities. Curr. Diab. Rep. 2018, 18, 143. [Google Scholar] [CrossRef]
- Lagathu, C.; Béréziat, V.; Gorwood, J.; Fellahi, S.; Bastard, J.-P.; Vigouroux, C.; Boccara, F.; Capeau, J. Metabolic complications affecting adipose tissue, lipid and glucose metabolism associated with HIV antiretroviral treatment. Expert Opin. Drug Saf. 2019, 18, 829–840. [Google Scholar] [CrossRef]
- Virtue, S.; Vidal-Puig, A. It’s Not How Fat You Are, It’s What You Do with It That Counts. PLoS Biol. 2008, 6, e237. [Google Scholar] [CrossRef]
- Lackey, D.E.; Olefsky, J.M. Regulation of metabolism by the innate immune system. Nat. Rev. Endocrinol. 2016, 12, 15–28. [Google Scholar] [CrossRef]
- Datta, R.; Podolsky, M.J.; Atabai, K. Fat fibrosis: Friend or foe? JCI Insight 2018, 3, e122289. [Google Scholar] [CrossRef]
- Lee, Y.S.; Wollam, J.; Olefsky, J.M. An Integrated View of Immunometabolism. Cell 2018, 172, 22–40. [Google Scholar] [CrossRef]
- Malagon, M.; Díaz-Ruiz, A.; Guzman-Ruiz, R.; Jimenez-Gomez, Y.; Moreno, N.; Garcia-Navarro, S.; Vazquez-Martinez, R.; Peinado, J. Adipobiology for Novel Therapeutic Approaches in Metabolic Syndrome. Curr. Vasc. Pharmacol. 2014, 11, 954–967. [Google Scholar] [CrossRef]
- Vishvanath, L.; Gupta, R.K.; Vishvanath, L.; Gupta, R.K. Contribution of adipogenesis to healthy adipose tissue expansion in obesity Find the latest version: Contribution of adipogenesis to healthy adipose tissue expansion in obesity. J. Clin. Investig. 2019, 129, 4022–4031. [Google Scholar] [CrossRef] [PubMed]
- Virtue, S.; Vidal-Puig, A. Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome—An allostatic perspective. Biochim. Biophys. Acta 2010, 1801, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.A.; Ludwig, R.G.; Garcia-Martin, R.; Brandão, B.B.; Kahn, C.R. Extracellular miRNAs: From Biomarkers to Mediators of Physiology and Disease. Cell Metab. 2019, 8, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.I.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORCl association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef]
- Chang, Y.Y.; Neufeld, T.P. An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol. Biol. Cell 2009, 20, 2004–2014. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, M.S. Autophagy as a crosstalk mediator of metabolic organs in regulation of energy metabolism. Rev. Endocr. Metab. Disord. 2014, 15, 11–20. [Google Scholar] [CrossRef]
- Van Niekerk, G.; Du Toit, A.; Loos, B.; Engelbrecht, A.-M. Nutrient excess and autophagic deficiency: Explaining metabolic diseases in obesity. Metabolism 2018, 82, 14–21. [Google Scholar] [CrossRef]
- Stienstra, R.; Haim, Y.; Riahi, Y.; Netea, M.; Rudich, A.; Leibowitz, G. Autophagy in adipose tissue and the beta cell: Implications for obesity and diabetes. Diabetologia 2014, 57, 1505–1516. [Google Scholar] [CrossRef]
- Marasco, M.R.; Linnemann, A.K. β-Cell Autophagy in Diabetes Pathogenesis. Endocrinology 2018, 159, 2127–2141. [Google Scholar] [CrossRef] [PubMed]
- Oami, T.; Watanabe, E.; Hatano, M.; Sunahara, S.; Fujimura, L.; Sakamoto, A.; Ito, C.; Toshimori, K.; Oda, S. Suppression of T Cell Autophagy Results in Decreased Viability and Function of T Cells Through Accelerated Apoptosis in a Murine Sepsis Model. Crit. Care Med. 2017, 45, e77–e85. [Google Scholar] [CrossRef] [PubMed]
- Fernández, Á.F.; Sebti, S.; Wei, Y.; Zou, Z.; Shi, M.; McMillan, K.L.; He, C.; Ting, T.; Liu, Y.; Chiang, W.C.; et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 2018, 558, 136–140. [Google Scholar] [CrossRef]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.-C.; Chen, C.-Y.; Lee, B.-C.; Chen, M.-F. High-fat diet induces cardiomyocyte apoptosis via the inhibition of autophagy. Eur. J. Nutr. 2016, 55, 2245–2254. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef]
- Frake, R.A.; Ricketts, T.; Menzies, F.M.; Rubinsztein, D.C. Autophagy and neurodegeneration. J. Clin. Investig. 2015, 125, 65–74. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, J.; Bao, J. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of Cytosolic Proteins by Late Endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef]
- He, C.; Levine, B. The Beclin 1 interactome. Curr. Opin. Cell Biol. 2010, 22, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef]
- Polson, H.E.J.; De Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbé, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Abada, A.; Elazar, Z. Getting ready for building: Signaling and autophagosome biogenesis. EMBO Rep. 2014, 15, 839–852. [Google Scholar] [CrossRef]
- Ktistakis, N.T.; Tooze, S.A. Digesting the Expanding Mechanisms of Autophagy. Trends Cell Biol. 2016, 26, 624–635. [Google Scholar] [CrossRef]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Cairó, M.; Villarroya, J. The role of autophagy in brown and beige adipose tissue plasticity. J. Physiol. Biochem. 2019. [Google Scholar] [CrossRef]
- Choi, J.W.; Jo, A.; Kim, M.; Park, H.S.; Chung, S.S.; Kang, S.; Park, K.S. BNIP3 is essential for mitochondrial bioenergetics during adipocyte remodelling in mice. Diabetologia 2016, 59, 571–581. [Google Scholar] [CrossRef]
- Zhang, Y.; Goldman, S.; Baerga, R.; Zhao, Y.; Komatsu, M.; Jin, S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 19860–19865. [Google Scholar] [CrossRef]
- Baerga, R.; Zhang, Y.; Chen, P.H.; Goldman, S.; Jin, S. Targeted deletion of autophagy-related 5 (atg5) impairs adipogenesis in a cellular model and in mice. Autophagy 2009, 5, 1118–1130. [Google Scholar] [CrossRef]
- Singh, R.; Xiang, Y.; Wang, Y.; Baikati, K.; Cuervo, A.M.; Luu, Y.K.; Tang, Y.; Pessin, J.E.; Schwartz, G.J.; Czaja, M.J. Autophagy regulates adipose mass and differentiation in mice. J. Clin. Investig. 2009, 119, 3329–3339. [Google Scholar] [CrossRef]
- Goldman, S.J.; Zhang, Y.; Jin, S. Autophagic Degradation of Mitochondria in White Adipose Tissue Differentiation. Antioxid. Redox Signal. 2011, 14, 1971–1978. [Google Scholar] [CrossRef]
- Yoon, M.S.; Zhang, C.; Sun, Y.; Schoenherr, C.J.; Chen, J. Mechanistic target of rapamycin controls homeostasis of adipogenesis. J. Lipid Res. 2013, 54, 2166–2173. [Google Scholar] [CrossRef]
- Zhang, C.; He, Y.; Okutsu, M.; Ong, L.C.; Jin, Y.; Zheng, L.; Chow, P.; Yu, S.; Zhang, M.; Yan, Z. Autophagy is involved in adipogenic differentiation by repressesing proteasome-dependent PPARγ2 degradation. Am. J. Physiol. Endocrinol. Metab. 2013, 305, 530–539. [Google Scholar] [CrossRef]
- Tang, Q.Q.; Otto, T.C.; Lane, M.D. CCAAT/enhancer-binding protein β is required for mitotic clonal expansion during adipogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.W.; Klemm, D.J.; Vinson, C.; Lane, M.D. Role of CREB in Transcriptional Regulation of CCAAT/Enhancer-binding Protein β Gene during Adipogenesis. J. Biol. Chem. 2004, 279, 4471–4478. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.Q.; Grønborg, M.; Huang, H.; Kim, J.W.; Otto, T.C.; Pandey, A.; Lane, M.D. Sequential phosphorylation of CCAAT enhancer-binding protein β by MAPK and glycogen synthase kinase 3β is required for adipogenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 9766–9771. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.L.; Robinson, C.E.; Gimble, J.M. CAAT/Enhancer binding proteins directly modulate transcription from the peroxisome proliferator-activated receptor γ2 promoter. Biochem. Biophys. Res. Commun. 1997, 240, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Christy, R.J.; Kaestner, K.H.; Geiman, D.E.; Daniel Lane, M. CCAAT/enhancer binding protein gene promoter: Binding of nuclear factors during differentiation of 3T3-L1 preadipocytes. Proc. Natl. Acad. Sci. USA 1991, 88, 2593–2597. [Google Scholar] [CrossRef]
- Roldan, M.; Macias-Gonzalez, M.; Garcia, R.; Tinahones, F.J.; Martin, M. Obesity short-circuits stemness gene network in human adipose multipotent stem cells. FASEB J. 2011, 25, 4111–4126. [Google Scholar] [CrossRef]
- Pachón-Peña, G.; Serena, C.; Ejarque, M.; Petriz, J.; Duran, X.; Oliva-Olivera, W.; Simó, R.; Tinahones, F.J.; Fernández-Veledo, S.; Vendrell, J. Obesity Determines the Immunophenotypic Profile and Functional Characteristics of Human Mesenchymal Stem Cells From Adipose Tissue. Stem Cells Transl. Med. 2016, 5, 464–475. [Google Scholar] [CrossRef]
- Frazier, T.P.; Gimble, J.M.; Devay, J.W.; Tucker, H.A.; Chiu, E.S.; Rowan, B.G. Body mass index affects proliferation and osteogenic differentiation of human subcutaneous adipose tissue-derived stem cells. BMC Cell Biol. 2013, 14, 34. [Google Scholar] [CrossRef]
- Isakson, P.; Hammarstedt, A.; Gustafson, B.; Smith, U. Impaired preadipocyte differentiation in human abdominal obesity: Role of Wnt, tumor necrosis factor-α, and inflammation. Diabetes 2009, 58, 1550–1557. [Google Scholar] [CrossRef]
- Park, H.T.; Lee, E.S.; Cheon, Y.-P.; Lee, D.R.; Yang, K.-S.; Kim, Y.T.; Hur, J.Y.; Kim, S.H.; Lee, K.W.; Kim, T. The relationship between fat depot-specific preadipocyte differentiation and metabolic syndrome in obese women. Clin. Endocrinol. (Oxf.) 2012, 76, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Oliva-Olivera, W.; Coín-Aragüez, L.; Lhamyani, S.; Clemente-Postigo, M.; Torres, J.A.; Bernal-López, M.R.; El Bekay, R.; Tinahones, F.J. Adipogenic Impairment of Adipose Tissue-Derived Mesenchymal Stem Cells in Subjects With Metabolic Syndrome: Possible Protective Role of FGF2. J. Clin. Endocrinol. Metab. 2017, 102, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Skop, V.; Cahova, M.; Dankova, H.; Papackova, Z.; Palenickova, E.; Svoboda, P.; Zidkova, J.; Kazdova, L. Autophagy inhibition in early but not in later stages prevents 3T3-L1 differentiation: Effect on mitochondrial remodeling. Differentiation 2014, 87, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Guo, Y.; Yuan, F.; Chen, S.; Yin, H.; Jiang, X.; Jiao, F.; Wang, F.; Ji, H.; Hu, G.; et al. Autophagy inhibition prevents glucocorticoid-increased adiposity via suppressing BAT whitening. Autophagy 2020, 16, 451–465. [Google Scholar] [CrossRef]
- Shimizu, I.; Aprahamian, T.; Kikuchi, R.; Shimizu, A.; Papanicolaou, K.N.; MacLauchlan, S.; Maruyama, S.; Walsh, K. Vascular rarefaction mediates whitening of brown fat in obesity. J. Clin. Investig. 2014, 124, 2099–2112. [Google Scholar] [CrossRef]
- Green, H.; Kehinde, O. An established preadipose cell line and its differentiation in culture II. Factors affecting the adipose conversion. Cell 1975, 5, 19–27. [Google Scholar] [CrossRef]
- Müller, T.D.; Lee, S.J.; Jastroch, M.; Kabra, D.; Stemmer, K.; Aichler, M.; Abplanalp, B.; Ananthakrishnan, G.; Bhardwaj, N.; Collins, S.; et al. P62 Links β-adrenergic input to mitochondrial function and thermogenesis. J. Clin. Investig. 2013, 123, 469–478. [Google Scholar] [CrossRef]
- Frendo-Cumbo, S.; Jaldin-Fincati, J.R.; Coyaud, E.; Laurent, E.M.N.; Townsend, L.K.; Tan, J.M.J.; Xavier, R.J.; Pillon, N.J.; Raught, B.; Wright, D.C.; et al. Deficiency of the autophagy gene ATG16L1 induces insulin resistance through KLHL9/KLHL13/CUL3-mediated IRS1 degradation. J. Biol. Chem. 2019, 294, 16172–16185. [Google Scholar] [CrossRef]
- Liu, Y.; Takahashi, Y.; Desai, N.; Zhang, J.; Serfass, J.M.; Shi, Y.G.; Lynch, C.J.; Wang, H.G. Bif-1 deficiency impairs lipid homeostasis and causes obesity accompanied by insulin resistance. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Galluzzi, L.; Green, D.R. Autophagy-Independent Functions of the Autophagy Machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef]
- Guo, L.; Huang, J.-X.; Liu, Y.; Li, X.; Zhou, S.-R.; Qian, S.-W.; Liu, Y.; Zhu, H.; Huang, H.-Y.; Dang, Y.-J.; et al. Transactivation of Atg4b by C/EBPβ Promotes Autophagy To Facilitate Adipogenesis. Mol. Cell. Biol. 2013, 33, 3180–3190. [Google Scholar] [CrossRef]
- Romero, M.; Sabaté-Pérez, A.; Francis, V.A.; Castrillón-Rodriguez, I.; Díaz-Ramos, Á.; Sánchez-Feutrie, M.; Durán, X.; Palacín, M.; Moreno-Navarrete, J.M.; Gustafson, B.; et al. TP53INP2 regulates adiposity by activating β-catenin through autophagy-dependent sequestration of GSK3β. Nat. Cell Biol. 2018, 20, 443–454. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Wu, H.; Wang, Y.; Li, W.; Chen, H.; Du, L.; Liu, D.; Wang, X.; Xu, T.; Liu, L.; Chen, Q. Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy 2019, 15, 1882–1898. [Google Scholar] [CrossRef] [PubMed]
- Cummins, T.D.; Holden, C.R.; Sansbury, B.E.; Gibb, A.A.; Shah, J.; Zafar, N.; Tang, Y.; Hellmann, J.; Rai, S.N.; Spite, M.; et al. Metabolic remodeling of white adipose tissue in obesity. Am. J. Physiol. Endocrinol. Metab. 2014, 307, 262–277. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.; Gottlieb, R.A. Parkin-mediated mitophagy is downregulated in browning of white adipose tissue. Obesity 2017, 25, 704–712. [Google Scholar] [CrossRef]
- Altshuler-Keylin, S.; Shinoda, K.; Hasegawa, Y.; Ikeda, K.; Hong, H.; Kang, Q.; Yang, Y.; Perera, R.M.; Debnath, J.; Kajimura, S. Beige Adipocyte Maintenance Is Regulated by Autophagy-Induced Mitochondrial Clearance. Cell Metab. 2016, 24, 402–419. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wu, K.K.L.; Jiang, X.; Xu, A.; Cheng, K.K.Y. The role of adipose tissue senescence in obesityand ageing-related metabolic disorders. Clin. Sci. 2020, 134, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.J.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.J.; Tavaré, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef]
- Xu, M.; Palmer, A.K.; Ding, H.; Weivoda, M.M.; Pirtskhalava, T.; White, T.A.; Sepe, A.; Johnson, K.O.; Stout, M.B.; Giorgadze, N.; et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife 2015, 4. [Google Scholar] [CrossRef]
- Gustafson, B.; Nerstedt, A.; Smith, U. Reduced subcutaneous adipogenesis in human hypertrophic obesity is linked to senescent precursor cells. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Molchadsky, A.; Shats, I.; Goldfinger, N.; Pevsner-Fischer, M.; Olson, M.; Rinon, A.; Tzahor, E.; Lozano, G.; Zipori, D.; Sarig, R.; et al. P53 plays a role in mesenchymal differentiation programs, in a cell fate dependent manner. PLoS ONE 2008, 3, e3707. [Google Scholar] [CrossRef] [PubMed]
- Okita, N.; Ishikawa, N.; Mizunoe, Y.; Oku, M.; Nagai, W.; Suzuki, Y.; Matsushima, S.; Mikami, K.; Okado, H.; Sasaki, T.; et al. Inhibitory effect of p53 on mitochondrial content and function during adipogenesis. Biochem. Biophys. Res. Commun. 2014, 446, 91–97. [Google Scholar] [CrossRef]
- Fu, W.; Liu, Y.; Sun, C.; Yin, H. Transient p53 inhibition sensitizes aged white adipose tissue for beige adipocyte recruitment by blocking mitophagy. FASEB J. 2019, 33, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; Miller, J.D.; LeBrasseur, N.K. Cellular senescence: Implications for metabolic disease. Mol. Cell. Endocrinol. 2017, 455, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, D.; Wang, C.; Luo, Y.; Ding, X.; Yang, X.; Silva, F.; Arenas, S.; Weaver, J.M.; Mandell, M.; et al. Sustained activation of autophagy suppresses adipocyte maturation via a lipolysis-dependent mechanism. Autophagy 2019, 1–15. [Google Scholar] [CrossRef]
- Polak, P.; Cybulski, N.; Feige, J.N.; Auwerx, J.; Rüegg, M.A.; Hall, M.N. Adipose-Specific Knockout of raptor Results in Lean Mice with Enhanced Mitochondrial Respiration. Cell Metab. 2008, 8, 399–410. [Google Scholar] [CrossRef]
- Bell, A.; Grunder, L.; Sorisky, A. Rapamycin Inhibits Human Adipocyte Differentiation in Primary Culture. Obes. Res. 2000, 8, 249–254. [Google Scholar] [CrossRef]
- Yeh, W.C.; Bierer, B.E.; McKnight, S.L. Rapamycin inhibits clonal expansion and adipogenic differentiation of 3T3-L1 cells. Proc. Natl. Acad. Sci. USA 1995, 92, 11086–11090. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Manning, B.D. mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol. 2017, 45, 72–82. [Google Scholar] [CrossRef]
- Öst, A.; Svensson, K.; Ruishalme, I.; Brännmark, C.; Franck, N.; Krook, H.; Sandström, P.; Kjolhede, P.; Strålfors, P. Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol. Med. 2010, 16, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Feil, S.; Valtcheva, N.; Feil, R. Inducible cre mice. Methods Mol. Biol. 2009, 530, 343–363. [Google Scholar] [CrossRef]
- Cai, J.; Pires, K.M.; Ferhat, M.; Chaurasia, B.; Buffolo, M.A.; Smalling, R.; Sargsyan, A.; Atkinson, D.L.; Summers, S.A.; Graham, T.E.; et al. Autophagy Ablation in Adipocytes Induces Insulin Resistance and Reveals Roles for Lipid Peroxide and Nrf2 Signaling in Adipose-Liver Crosstalk. Cell Rep. 2018, 25, 1708–1717.e5. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.D.; Manou, D.; Karamanos, N.K. The extracellular matrix as a multitasking player in disease. FEBS J. 2019, 286, 2830–2869. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O.; Naba, A. Overview of the Matrisome—An Inventory of Extracellular Matrix Constituents and Functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. 2016, 49, 10–24. [Google Scholar] [CrossRef]
- Lin, D.; Chun, T.H.; Kang, L. Adipose extracellular matrix remodelling in obesity and insulin resistance. Biochem. Pharmacol. 2016, 119, 8–16. [Google Scholar] [CrossRef]
- Shao, X.; Taha, I.N.; Clauser, K.R.; Gao, Y.; Naba, A. MatrisomeDB: The ECM-protein knowledge database. Nucleic Acids Res. 2020, 48, D1136–D1144. [Google Scholar] [CrossRef]
- McWhorter, F.; Davis, C.; Liu, W. Physical and mechanical regulation of macrophage phenotype and function. Cell. Mol. Life Sci. 2015, 72, 1303–1316. [Google Scholar] [CrossRef]
- Karamanos, N.K.; Theocharis, A.D.; Neill, T.; Iozzo, R.V. Matrix modeling and remodeling: A biological interplay regulating tissue homeostasis and diseases. Matrix Biol. 2019, 75–76, 1–11. [Google Scholar] [CrossRef]
- Sun, K.; Tordjman, J.; Clément, K.; Scherer, P.E. Fibrosis and adipose tissue dysfunction. Cell Metab. 2013, 18, 470–477. [Google Scholar] [CrossRef]
- Vila, I.K.; Badin, P.-M.; Marques, M.-A.; Monbrun, L.; Lefort, C.; Mir, L.; Louche, K.; Bourlier, V.; Roussel, B.; Gui, P.; et al. Immune cell Toll-like receptor 4 mediates the development of obesity- and endotoxemia-associated adipose tissue fibrosis. Cell Rep. 2014, 7, 1116–1129. [Google Scholar] [CrossRef]
- Lech, M.; Anders, H.J. Macrophages and fibrosis: How resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 989–997. [Google Scholar] [CrossRef]
- Xu, J.; Liu, L.-Q.; Xu, L.-L.; Xing, Y.; Ye, S. Metformin alleviates renal injury in diabetic rats by inducing Sirt1/FoxO1 autophagic signal axis. Clin. Exp. Pharmacol. Physiol. 2020, 47, 599–608. [Google Scholar] [CrossRef]
- Bao, C.; Yang, Z.; Cai, Q.; Li, Q.; Li, H.; Shu, B. Incremental load training improves renal fibrosis by regulating the TGF-β1/TAK1/MKK3/p38MAPK signaling pathway and inducing the activation of autophagy in aged mice. Int. J. Mol. Med. 2019, 44, 1677–1686. [Google Scholar] [CrossRef]
- Jia, D.; Wang, Y.Y.; Wang, P.; Huang, Y.; Liang, D.Y.; Wang, D.; Cheng, C.; Zhang, C.; Guo, L.; Liang, P.; et al. SVIP alleviates CCl4-induced liver fibrosis via activating autophagy and protecting hepatocytes. Cell Death Dis. 2019, 10, 71. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-X.; Cui, L.-H.; Zhuo, Y.-Z.; Hu, J.-G.; Cui, N.-Q.; Zhang, S.-K. Inhibiting autophagy promotes collagen degradation by regulating matrix metalloproteinases in pancreatic stellate cells. Life Sci. 2018, 208, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Gubbiotti, M.A.; Seifert, E.; Rodeck, U.; Hoek, J.B.; Iozzo, R.V. Metabolic reprogramming of murine cardiomyocytes during autophagy requires the extracellular nutrient sensor decorin. J. Biol. Chem. 2018, 293, 16940–16950. [Google Scholar] [CrossRef] [PubMed]
- Bernard, M.; Yang, B.; Migneault, F.; Turgeon, J.; Dieudé, M.; Olivier, M.-A.; Cardin, G.B.; El-Diwany, M.; Underwood, K.; Rodier, F.; et al. Autophagy drives fibroblast senescence through MTORC2 regulation. Autophagy 2020, 1–13. [Google Scholar] [CrossRef]
- Manou, D.; Caon, I.; Bouris, P.; Triantaphyllidou, I.-E.; Giaroni, C.; Passi, A.; Karamanos, N.K.; Vigetti, D.; Theocharis, A.D. The Complex Interplay Between Extracellular Matrix and Cells in Tissues. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2019; Volume 1952, pp. 1–20. [Google Scholar]
- Roedig, H.; Nastase, M.V.; Wygrecka, M.; Schaefer, L. Breaking down chronic inflammatory diseases: The role of biglycan in promoting a switch between inflammation and autophagy. FEBS J. 2019, 286, 2965–2979. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Yuan, Q.; Chen, Y.; Huang, Z.; Fang, X.; Zhang, H.; Peng, L.; Xiao, P. LncRNA ENST00000453774.1 contributes to oxidative stress defense dependent on autophagy mediation to reduce extracellular matrix and alleviate renal fibrosis. J. Cell. Physiol. 2019, 234, 9130–9143. [Google Scholar] [CrossRef] [PubMed]
- Castagnaro, S.; Pellegrini, C.; Pellegrini, M.; Chrisam, M.; Sabatelli, P.; Toni, S.; Grumati, P.; Ripamonti, C.; Pratelli, L.; Maraldi, N.M.; et al. Autophagy activation in COL6 myopathic patients by a low-protein-diet pilot trial. Autophagy 2016, 12, 2484–2495. [Google Scholar] [CrossRef] [PubMed]
- Pavel, M.; Renna, M.; Park, S.J.; Menzies, F.M.; Ricketts, T.; Füllgrabe, J.; Ashkenazi, A.; Frake, R.A.; Lombarte, A.C.; Bento, C.F.; et al. Contact inhibition controls cell survival and proliferation via YAP/TAZ-autophagy axis. Nat. Commun. 2018, 9, 2961. [Google Scholar] [CrossRef]
- Marcelin, G.; Da Cunha, C.; Gamblin, C.; Suffee, N.; Rouault, C.; Leclerc, A.; Lacombe, A.; Sokolovska, N.; Gautier, E.L.; Clément, K.; et al. Autophagy inhibition blunts PDGFRA adipose progenitors’ cell-autonomous fibrogenic response to high-fat diet. Autophagy 2020, 1–11. [Google Scholar] [CrossRef]
- Iwayama, T.; Steele, C.; Yao, L.; Dozmorov, M.G.; Karamichos, D.; Wren, J.D.; Olson, L.E. PDGFRα signaling drives adipose tissue fibrosis by targeting progenitor cell plasticity. Genes Dev. 2015, 29, 1106–1119. [Google Scholar] [CrossRef]
- Marcelin, G.; Ferreira, A.; Liu, Y.; Atlan, M.; Aron-Wisnewsky, J.; Pelloux, V.; Botbol, Y.; Ambrosini, M.; Fradet, M.; Rouault, C.; et al. A PDGFRα-Mediated Switch toward CD9high Adipocyte Progenitors Controls Obesity-Induced Adipose Tissue Fibrosis. Cell Metab. 2017, 25, 673–685. [Google Scholar] [CrossRef]
- Lee, Y.H.; Petkova, A.P.; Mottillo, E.P.; Granneman, J.G. In vivo identification of bipotential adipocyte progenitors recruited by β3-adrenoceptor activation and high-fat feeding. Cell Metab. 2012, 15, 480–491. [Google Scholar] [CrossRef]
- Berry, R.; Rodeheffer, M.S. Characterization of the adipocyte cellular lineage in vivo. Nat. Cell Biol. 2013, 15, 302–308. [Google Scholar] [CrossRef]
- Qian, M.; Fang, X.; Wang, X. Autophagy and inflammation. Clin. Transl. Med. 2017, 6, 1–11. [Google Scholar] [CrossRef]
- Jang, Y.J.; Kim, J.H.; Byun, S. Modulation of Autophagy for Controlling Immunity. Cells 2019, 8, 138. [Google Scholar] [CrossRef] [PubMed]
- Joven, J.; Guirro, M.; Mariné-Casadó, R.; Rodríguez-Gallego, E.; Menéndez, J.A. Autophagy Is an Inflammation-Related Defensive Mechanism Against Disease. In Advances in Experimental Medicine and Biology; Springer New York LLC: New York, NY, USA, 2014; Volume 824, pp. 43–59. ISBN 9783319073194. [Google Scholar]
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Münz, C.; Yoshimori, T. Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 2015, 16, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa-Ishimoto, Y.; Hwang, S.; Cadwell, K. Autophagy and Inflammation. Annu. Rev. Immunol. 2018, 36, 73–101. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Jounai, N.; Kobiyama, K.; Shiina, M.; Ogata, K.; Ishii, K.J.; Takeshita, F. NLRP4 Negatively Regulates Autophagic Processes through an Association with Beclin1. J. Immunol. 2011, 186, 1646–1655. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.H.; Cho, M.H.; Kim, J.Y.; Kwon, M.S.; Peak, J.J.; Kang, S.W.; Yoon, S.Y.; Song, Y. Impaired macrophage autophagy induces systemic insulin resistance in obesity. Oncotarget 2016, 7, 35577–35591. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Sanchez-Lopez, E.; Karin, M. Autophagy, Inflammation, and Immunity: A Troika Governing Cancer and Its Treatment. Cell 2016, 166, 288–298. [Google Scholar] [CrossRef]
- Zmora, N.; Bashiardes, S.; Levy, M.; Elinav, E. The Role of the Immune System in Metabolic Health and Disease. Cell Metab. 2017, 25, 506–521. [Google Scholar] [CrossRef]
- Kratz, M.; Coats, B.R.; Hisert, K.B.; Hagman, D.; Mutskov, V.; Peris, E.; Schoenfelt, K.Q.; Kuzma, J.N.; Larson, I.; Billing, P.S.; et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014, 20, 614–625. [Google Scholar] [CrossRef]
- Madsen, D.H.; Leonard, D.; Masedunskas, A.; Moyer, A.; Jürgensen, H.J.; Peters, D.E.; Amornphimoltham, P.; Selvaraj, A.; Yamada, S.S.; Brenner, D.A.; et al. M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J. Cell Biol. 2013, 202, 951–966. [Google Scholar] [CrossRef]
- Liu, K.; Zhao, E.; Ilyas, G.; Lalazar, G.; Lin, Y.; Haseeb, M.; Tanaka, K.E.; Czaja, M.J. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 2015, 11, 271–284. [Google Scholar] [CrossRef]
- Litwinoff, E.M.S.; Gold, M.Y.; Singh, K.; Hu, J.; Li, H.; Cadwell, K.; Schmidt, A.M. Myeloid ATG16L1 does not affect adipose tissue inflammation or body mass in mice fed high fat diet. Obes. Res. Clin. Pract. 2018, 12, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Grijalva, A.; Xu, X.; Ferrante, A.W. Autophagy Is Dispensable for Macrophage-Mediated Lipid Homeostasis in Adipose Tissue. Diabetes 2016, 65, 967–980. [Google Scholar] [CrossRef]
- Thomas, D.; Apovian, C. Macrophage functions in lean and obese adipose tissue. Metabolism. 2017, 72, 120–143. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic Endotoxemia Initiates Obesity and Insulin Resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Postigo, M.; Queipo-Ortuño, M.I.; Murri, M.; Boto-Ordoñez, M.; Perez-Martinez, P.; Andres-Lacueva, C.; Cardona, F.; Tinahones, F.J. Endotoxin increase after fat overload is related to postprandial hypertriglyceridemia in morbidly obese patients. J. Lipid Res. 2012, 53, 973–978. [Google Scholar] [CrossRef]
- Cani, P.D.; Possemiers, S.; Van de Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; Geurts, L.; Naslain, D.; Neyrinck, A.; Lambert, D.M.; et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009, 58, 1091–1103. [Google Scholar] [CrossRef]
- Clemente-Postigo, M.; Oliva-Olivera, W.; Coin-Aragüez, L.; Ramos-Molina, B.; Giraldez-Perez, R.M.; Lhamyani, S.; Alcaide-Torres, J.; Perez-Martinez, P.; El Bekay, R.; Cardona, F.; et al. Metabolic endotoxemia promotes adipose dysfunction and inflammation in human obesity. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E319–E332. [Google Scholar] [CrossRef]
- Jansen, H.J.; Van Essen, P.; Koenen, T.; Joosten, L.A.B.; Netea, M.G.; Tack, C.J.; Stienstra, R. Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology 2012, 153, 5866–5874. [Google Scholar] [CrossRef]
- Kovsan, J.; Blüher, M.; Tarnovscki, T.; Klöting, N.; Kirshtein, B.; Madar, L.; Shai, I.; Golan, R.; Harman-Boehm, I.; Schön, M.R.; et al. Altered autophagy in human adipose tissues in obesity. J. Clin. Endocrinol. Metab. 2011, 96, 268–277. [Google Scholar] [CrossRef]
- Kosacka, J.; Kern, M.; Klöting, N.; Paeschke, S.; Rudich, A.; Haim, Y.; Gericke, M.; Serke, H.; Stumvoll, M.; Bechmann, I.; et al. Autophagy in adipose tissue of patients with obesity and type 2 diabetes. Mol. Cell. Endocrinol. 2015, 409, 21–32. [Google Scholar] [CrossRef]
- Xu, Q.; Mariman, E.C.M.; Roumans, N.J.T.; Vink, R.G.; Goossens, G.H.; Blaak, E.E.; Jocken, J.W.E. Adipose tissue autophagy related gene expression is associated with glucometabolic status in human obesity. Adipocyte 2018, 7, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, T.; Kusunoki, C.; Kondo, M.; Yasuda, M.; Kume, S.; Morino, K.; Sekine, O.; Ugi, S.; Uzu, T.; Nishio, Y.; et al. Autophagy regulates inflammation in adipocytes. Biochem. Biophys. Res. Commun. 2012, 417, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Mizunoe, Y.; Sudo, Y.; Okita, N.; Hiraoka, H.; Mikami, K.; Narahara, T.; Negishi, A.; Yoshida, M.; Higashibata, R.; Watanabe, S.; et al. Involvement of lysosomal dysfunction in autophagosome accumulation and early pathologies in adipose tissue of obese mice. Autophagy 2017, 13, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, C.E.; Rodrigues, V.S.; Gomes, F.S.; De Moura, R.F.; Victorio, S.C.; Bombassaro, B.; Chaim, E.A.; Pareja, J.C.; Geloneze, B.; Velloso, L.A.; et al. Defective regulation of adipose tissue autophagy in obesity. Int. J. Obes. 2013, 37, 1473–1480. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Rodríguez, A.; Gómez-Ambrosi, J.; Catalán, V.; Rotellar, F.; Valentí, V.; Silva, C.; Mugueta, C.; Pulido, M.R.; Vázquez, R.; Salvador, J.; et al. The ghrelin O-Acyltransferase-Ghrelin system reduces TNF-α-Induced apoptosis and autophagy in human visceral adipocytes. Diabetologia 2012, 55, 3038–3050. [Google Scholar] [CrossRef]
- Soussi, H.; Reggio, S.; Alili, R.; Prado, C.; Mutel, S.; Pini, M.; Rouault, C.; Clément, K.; Dugail, I. DAPK2 downregulation associates with attenuated adipocyte autophagic clearance in human obesity. Diabetes 2015, 64, 3452–3463. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef]
- Camargo, A.; Rangel-Zúñiga, O.A.; Alcalá-Díaz, J.; Gomez-Delgado, F.; Delgado-Lista, J.; García-Carpintero, S.; Marín, C.; Almadén, Y.; Yubero-Serrano, E.M.; López-Moreno, J.; et al. Dietary fat may modulate adipose tissue homeostasis through the processes of autophagy and apoptosis. Eur. J. Nutr. 2017, 56, 1621–1628. [Google Scholar] [CrossRef]
- Peña-Orihuela, P.; Camargo, A.; Rangel-Zuñiga, O.A.; Perez-Martinez, P.; Cruz-Teno, C.; Delgado-Lista, J.; Yubero-Serrano, E.M.; Paniagua, J.A.; Tinahones, F.J.; Malagon, M.M.; et al. Antioxidant system response is modified by dietary fat in adipose tissue of metabolic syndrome patients. J. Nutr. Biochem. 2013, 24, 1717–1723. [Google Scholar] [CrossRef]


{kind=link}
| Study [Reference] | Design | WAT Depot | Tissue/Cells/Explant | Methodological Approach | Autophagic Markers | Main Results |
|---|---|---|---|---|---|---|
| Öst et al. [123] | Study groups: (1) T2D and BMI > 27 kg/m2 (n = 7) vs. (2) Non-T2D (non-BMI matched) (n = 7) | Subcutaneous | - Isolated adipocytes | - TEM - Immunofluorescence - Immunoblotting - Real-time qPCR | LC3 mTORC Autophagosome | T2D led to: - ↑ autophagosome number (TEM). - ↑ autophagic flux (accumulation of LC3). - ↓ lipofuscin particles. - = mTOR protein expression. |
| Kovsan et al. [173] | Cohort 1: Non-obese (BMI < 30; n = 15) vs. Obese (BMI > 30; n = 50) Cohort 2: Lean (BMI < 25; n = 66); Subcutaneous obesity (BMI > 30; n = 88) vs. Visceral obesity (BMI > 30; n = 42). Cohort 3: IS obese (BMI > 40; n = 30) vs. IR obese (BMI > 40; n = 30) | -Subcutaneous -Visceral | - Total tissue - Explant (n = 1 Ow) - SVF and adipocytes (only vWAT) (n = 24). | - Real-time qPCR - Immunoblotting - Immunofluorescence (explant). - Autophagic flux analysis | ATG5 ATG12 LC3A LC3B LC3-I & II p62 NBR1 | Total WAT: - ↑ ATG5 & LC3- II protein expression in vWAT vs. sWAT in obesity. - ↑ ATG12-ATG5 complex protein expression in vWAT vs. sWAT, regardless of BMI. - ↑ ATG5 and LC3B mRNA levels in vWAT vs. sWAT. - ↑ mRNA ATG5, LC3A and LC3B levels in obese vs. lean. - ↑ mRNA ATG5, LC3A and LC3B levels in IR obese vs. IS obese. mRNA/protein levels of ATG5, LC3B/LC3-II higher in adipocytes vs. SVF. Explant: - ↑ LC3-positive dots vWAT vs. sWAT. - ↑ autophagic flux in obesity (p62 accumulation). |
| Jansen et al. [172] | Study groups: (1) Obese (BMI = 27–35; n = 16) (2) Lean (BMI = 20–25; n = 17) | - Subcutaneous - Visceral | - Total tissue - Explant | - Immunoblotting | LC3-II | Total sWAT - ↑ LC3-II in obesity. - Positive correlation between LC3-II levels and BMI, HOMA-IR, macrophage infiltration. Explant (vWAT & sWAT): - Autophagy inhibition increased proinflammatory response. |
| Rodríguez, A. et al. [180] | Study groups: (1) NG Lean (BMI < 25; n = 55) (2) NG Obese (n = 66)* (3) IGT Obese (n = 37) * (4) T2D Obese (n = 36)* * Obese: BMI ≥ 30 | - Visceral | - Total tissue - Adipocytes - SVF | - Real-time qPCR | BECN1 ATG5 ATG7 | Total vWAT: ↑ BECN1 and ATG7 mRNA levels in T2D Obese vs. NG lean and obese. Adipocytes vs. SVF: No significant differences. Adipocytes: - ↑ BECN1, ATG5 and ATG7 mRNA levels upon TNFα stimulus. - ↓ ATGs mRNA upon acylated ghrelin stimulus. |
| Nuñez, C.E et al. [178] | Study groups: (1) Non-T2D obese(BMI = 43 ± 4.3; n = 9)* (2) T2D (BMI = 32 ± 2.2; n = 6)* (3) Lean control (BMI = 23 ± 2.7; n = 8) * Prospective follow-up ~1 year after surgery | Subcutaneous | - Total tissue - Explant | - Immunoblotting - - TEM | Beclin Autophagosome | - ↑ Beclin in obese groups vs. lean group. - ↓ Beclin after surgery (non-differences between T2D and non-T2D). - ↓ autophagosome after surgery. |
| Kosacka, J. et al. [174] | Study groups: (1) Lean (BMI < 25; n = 20). (2) Non-T2D Obese (BMI > 30; n = 20). (3) T2D Obese (BMI > 30; n = 20) | - Subcutaneous - Visceral | - Total tissue | - Immunoblotting - Immunofluorescence - Real-time qPCR - TEM | LC3-I & II ATG5/12 p62 mTOR (mRNA: LC3A & B, ATG5) Autophagosome | General trends: - gradual increase in LC3, ATGs & p62 from group (1) to (3) in vWAT and sWAT. - Parallel increase in inflammatory and caspase-dependent apoptotic markers. - Higher number of autophagosomes in obese vs. lean. |
| Soussi, H. et al. [181] | Study groups: (1) Lean (BMI = 20–23; n = 12). (2) Obese (BMI = 34–79; n = 24). * Subcohort from group 2: Obese for bariatric surgery follow up (3–12 months) (n = 9). | - Subcutaneous | -Isolated adipocytes | - Real-time qPCR. - Immunoblotting. - Autophagic flux. | LC3-I & II p62 | - ↑ p62 mRNA and protein levels in obesity. - ↓ absolute LC3-II levels in obese vs. lean after lysosomal inhibition. - ↑ absolute LC3-II levels post-surgery vs. pre-surgery after lysosomal inhibition. |
| Xu, Q. et al. [175] | Study groups: (1) Ow/Obese patients (BMI > 25–41.7; n = 17) with or without IGT. (2) NG lean (BMI ≤ 25; n = 9). | - Subcutaneous | - Total tissue. - Differentiated adipocytes from hMADS (non-clinical phenotype indicated). | - Real-time PCR - Autophagic flux | mRNA: ULK1-2 BECN1 ATG 5, 7 & 12 LC3A-B Protein: LC3-I & II p62 | Total sWAT: ↑ ATGs mRNA levels in Ow/obese group. Adipocytes: ↑ LC3-II and = p62 protein levels after lysosomal and lipolysis inhibition vs. non-lipolysis inhibition. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clemente-Postigo, M.; Tinahones, A.; El Bekay, R.; Malagón, M.M.; Tinahones, F.J. The Role of Autophagy in White Adipose Tissue Function: Implications for Metabolic Health. Metabolites 2020, 10, 179. https://doi.org/10.3390/metabo10050179
Clemente-Postigo M, Tinahones A, El Bekay R, Malagón MM, Tinahones FJ. The Role of Autophagy in White Adipose Tissue Function: Implications for Metabolic Health. Metabolites. 2020; 10(5):179. https://doi.org/10.3390/metabo10050179
Chicago/Turabian StyleClemente-Postigo, Mercedes, Alberto Tinahones, Rajaa El Bekay, María M. Malagón, and Francisco J. Tinahones. 2020. "The Role of Autophagy in White Adipose Tissue Function: Implications for Metabolic Health" Metabolites 10, no. 5: 179. https://doi.org/10.3390/metabo10050179
APA StyleClemente-Postigo, M., Tinahones, A., El Bekay, R., Malagón, M. M., & Tinahones, F. J. (2020). The Role of Autophagy in White Adipose Tissue Function: Implications for Metabolic Health. Metabolites, 10(5), 179. https://doi.org/10.3390/metabo10050179