1. Introduction
Skin aging is a complex process characterized by cumulative changes, including reduced cellular proliferation, increased cellular senescence, and heightened production of mitochondrial reactive oxygen species (ROS). These changes impact both the function and structure of the skin, resulting in noticeable clinical signs, such as increased skin laxity, dryness (xerosis), wrinkles, and uneven pigmentation [
1]. Additionally, skin aging leads to increased fragility, diminished healing capabilities, and greater susceptibility to infections and skin cancers.
At the cellular level, the clinical manifestations of aging reflect numerous functional alterations. For example, reduced keratinocyte proliferation leads to a thinner stratum corneum, which results in a weakened barrier function [
1]. The dermis is also affected, with a reduced fibroblast population exhibiting lower activity, leading to a less robust extracellular matrix that undergoes fragmentation and accelerated degradation. These changes ultimately result in altered biomechanical properties of the skin [
2,
3]. These modifications are primarily induced by a normal physiological and genetically programmed process known as intrinsic aging. However, since the skin serves as the primary interface with the environment, it is exposed to various environmental assaults, including ultraviolet and infrared irradiation, as well as environmental pollutants. These external factors contribute to what is termed extrinsic aging [
4]. Both aging mechanisms significantly interact and affect the same targets, ultimately contributing to the aging phenotype [
5]. Notably, a key shared feature is the generation of reactive oxygen species (ROS) [
6].
While environmental assaults are known to generate ROS, several intrinsic sources also exist. Mitochondria, in particular, are often considered the primary intrinsic source of ROS due to the routine operation of the electron transport chain [
6]. Although ROS serve as essential signaling molecules in normal cellular processes [
7,
8], they can also cause cellular damage when not adequately neutralized by antioxidant systems. In addition to their harmful effects on proteins and lipids, ROS can affect mitochondrial components, including mitochondrial DNA. These damages can ultimately lead to impaired mitochondrial function, reduced ATP production, and further ROS production [
9,
10]. This cycle of altered metabolism leading to additional damage is known as the mitochondrial free radical theory of aging (MFRTA), which posits that mitochondrial free radical production is the primary driving force behind the aging process.
Given their significance, efforts have been made to target skin mitochondria in antiaging strategies aimed at neutralizing excessive free radicals. One frequently used ingredient is coenzyme Q10 (CoQ10), a component of the electron transport chain. CoQ10 is also part of the network of small molecules with antioxidant properties, primarily found in the epidermis, where it neutralizes ROS generated by environmental stressors. Topical application of a CoQ10-containing cream enhances its availability, boosts ROS neutralization, and counteracts the effects of photoaging [
11,
12]. Another commonly found ingredient in antiwrinkle cosmetics is vitamin C, a potent antioxidant used to reverse UV-induced oxidative damage by scavenging ROS [
13].
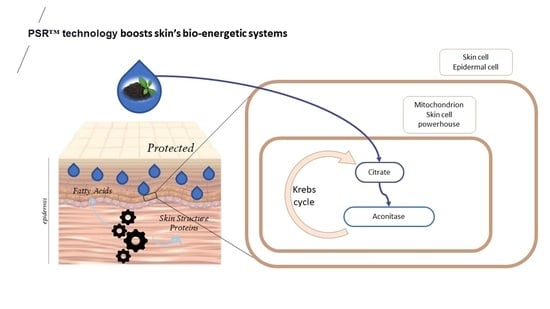
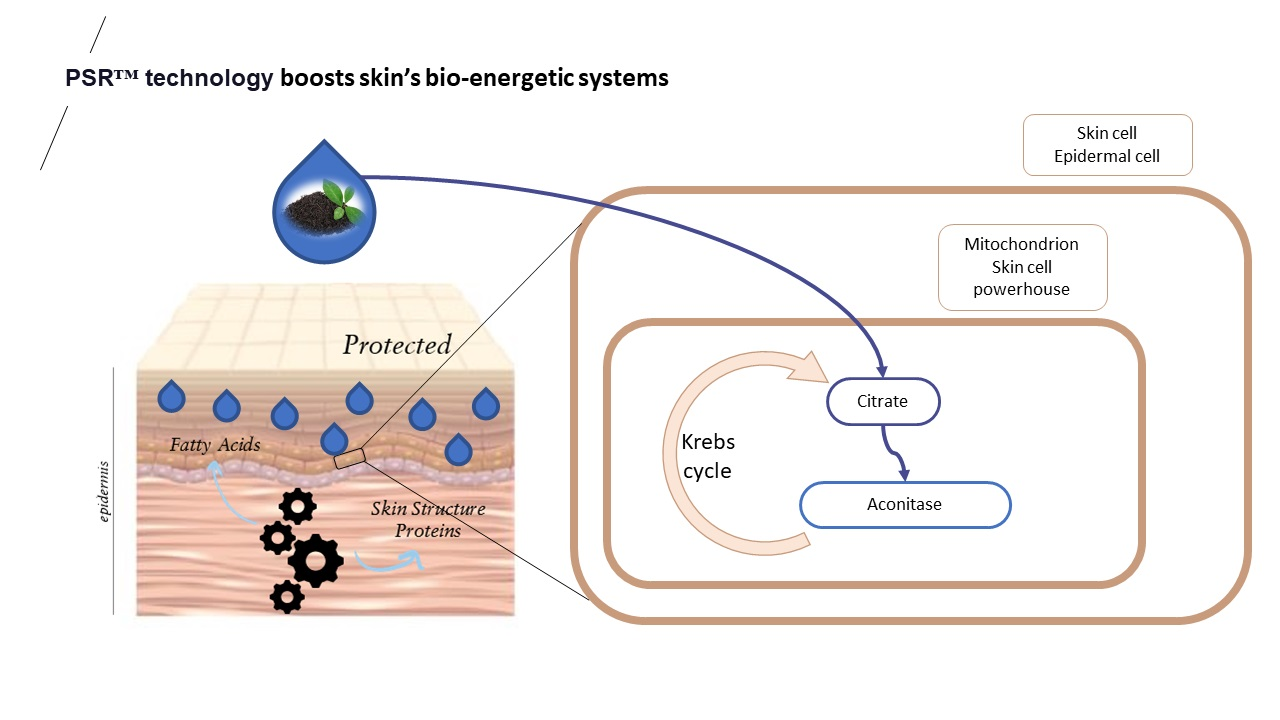
In our quest to identify natural ingredients with potential antiaging effects, we conducted tests on a black tea extract prepared using plant small RNA technology (PSRTM), a process designed to preserve and enrich extracts in small molecules. In this report, we share the findings of our experiments primarily centered on evaluating whether the mentioned extract has the potential to counteract the mitochondrial alterations induced by reactive oxygen species (ROS) associated with aging. These alterations contribute to compromised mitochondrial function, diminished ATP production, and heightened ROS generation., Our findings suggest that this extract exhibits properties that activate and enhance mitochondrial function.
2. Materials and Methods
2.1. Tea Extract Preparation and Characterization
All the plant extracts were produced by Ashland (Ashland, Sophia Antipolis, France). Camelia sinensis leaves were cultivated in Mauritius at the Domaine de Bois Chéri. The plantation is located on the South Mauritius plateau, at about 500 m altitude. Plants are grown in a volcanic soil under a tropical climate subject to marine air influence, being only 10 km away from the seashore. This corresponds to rather harsh conditions favoring the synthesis of several potent phytocompounds.
Two extracts were prepared. A conventional extract was obtained by a one-hour maceration in deionized water heated at 45 °C that was then filtered. A PSRTM extract was also prepared using a patented extraction process plant small RNA technology (Ashland, patent WO2017084958A1) that, thanks to polar solvents, enables the preservation and enrichment of the extract in polyphenols, amino acids, sugar, minerals, and small RNAs. Tea extracts (conventional or PSR extract, final concentration: 0.1% or 1% v/v) were used for the experiments.
2.1.1. Analysis of the Composition of Mauritius Tea Extracts
Dry Matter
Dry matter was quantified after heating extracts at 105 °C for 12 h to evaporate all water and volatile compounds.
Total Amount of Sugar, Polyphenol, and Amino Acids Quantification
Total sugar amount was determined colorimetrically. Briefly, the method consists of the addition of concentrated sulfuric acid that reacts with phenol and forms a colored complex whose absorbance is measured at 490 nm. The sugar content is determined using a glucose standard curve.
The amount of total polyphenol was quantified using the Folin–Ciocalteu assay. Upon oxidation, the Folin–Ciocalteu reagent produces a blue color whose absorbance is measured at 760 nm. The content is expressed as gallic acid equivalents using a gallic acid standard curve.
Quantification of total amino acid of extracts was assessed by the formation of a colored complex, following the rupture of the amine and carboxylic functions by the ninhydrin reagent [
14]. The absorbance of the complex was read at 570 nm and quantified using a standard curve of amino acids pool.
Thin Layer Chromatography Analysis of the Sugar Composition
The sugar profile was also analyzed using thin-layer chromatography on a silica plate. The mobile phase was a solution of butanol/ethanol/water in a ratio of 5/2/1. After migration, plates were revealed using an orcinol-containing solution and heat treatment (105 °C for 2 min).
HPLC Analysis of the Polyphenol Composition
Polyphenol composition was analyzed by liquid chromatography. Samples were separated on a Uptisphere® CS EvolutionTM C18-AQ column (100 mm × 4.6 mm × 2.6 μm, Interchim, Montluçon, France) by an Agilent 1100 HPLC system (Agilent Technologies, Santa Clara, CA, USA) using a mobile phase (0.8 mL/minute) consisting of 0.1% TFA (trifluoroacetic acids) in water. To facilitate elution, a 29 min gradient of increasing methanol concentration was used. Detection wavelengths were set at 255 nm, 280 nm, 290 nm, and 324 nm using a UV detector.
HPLC Analysis of the Organic Acid Composition
Organic acid composition (citric, lactic, malic, succinic, and tartaric acids) was analyzed by HPLC using an EC 150/4,6 Nucleoshell RP 18 plus 5 μm column (150 × 4.6 mm, Macherey Nagel, Allentown, PA, USA) and an Agilent 1260 HPLC system (Agilent Technologies, CA, USA). The mobile phase (0.3 mL/minute) consisted of formic acid 0.01% solution from T0 until 9 min, then 95% acetonitrile and 5% formic acid 0.01% solution between 13 and 14 min and, finally, back to a 0.01% formic acid solution for 6 min. Detection was performed using an ACQUITY Qda mass spectrometer detector ACQUITY Qda (Waters, Milford, MA, USA) with an electrospray ion source in negative mode. The spray voltage was 0.8 kV, and the capillary temperature was set to 600 °C.
Quantification of L-Theanine and Theine
L-theanine was quantified by HPLC (Agilent 1200 HPLC system, Agilent Technologies, Santa Clara, CA, USA) using an Uptisphere strategy C18-2 column (250 mm × 4.6 mm × 5 μm, Interchim, Montluçon, France). The mobile phase (0.8 mL/minute) was made of 0.1% phosphoric acid for 8 min, 95% acetonitrile and 5% of 0.1% phosphoric acid between 10 and 15 min and then 0.1% phosphoric acid until 16 min.
Theine was also analyzed by HPLC (Agilent 1100 HPLC system, Agilent Technologies, Santa Clara, CA, USA) using an Uptisphere CS evolution C18-AQ column (100 mm × 4.6 mm × 2.6 μm, Interchim, France). The mobile phase (0.8 mL/min) was a gradient starting with 0.1% TFA (trifluoroacetic acids) in water upon injection and reaching 100% methanol after 18.8 min until 21 min. Detection was performed at 272 nm using a UV detector.
Identification and Quantification of HPLC Peaks
For all HPLC analyses, the identification of peaks was achieved by comparing retention times to those of authentic standards (Sigma-Aldrich, Burlington, MA, USA). Quantification was performed by comparing the surface area of peaks to those of these standards.
Analysis of the Mineral Content
To quantify the mineral content of extracts, samples were prepared using an UltraWAVE Single Reaction Chamber Microwave Digestion System (Milestone, Sorisole, Italy). For each sample, 1 g was weighed into a digestion tube, and 7 mL concentrated HNO3 added. The temperature was ramped to 240 °C and maintained for 15 min. After cooling, the samples were diluted to a final volume of 25 mL with ultrapure water and analyzed by inductively coupled plasma–optical emission spectroscopy (ICP–OES) using a Varian Vista-Pro ICP (Varian Inc., Palo Alto, CA, USA).
2.2. Skin Sample Procurement Skin Sample and Cell Culture
Experimental design was carried out with respect to the ethical permissions. The principal requirements of the Declaration of Helsinki were considered to protect the rights, safety, and wellbeing of subjects participating in the study. Before initiating the studies, the investigator had obtained written consent from the participants and full approval from the Freiburg Ethics Commission International for the protocol, protocol amendment(s), if applicable. All participants who provided their skin biopsies for this research provided their written informed consent to participate in this study and for their data to be used for research purposes. Normal human skin samples were obtained from the surgical discard of anonymous healthy patients with informed consent of adult donors or children’s parents in accordance with ethical guidelines (French Bioethics law of 2004) and declared to the French research ministry (Declaration no. agreement DC 2011-1323 delivered to Ashland, France). Approval was not required as per the local legislation where the study was conducted. This study uses strains obtained from deidentified (human samples. Ethical guidelines (French Bioethics law of 2004) did not require the study to be reviewed or approved by an ethics committee because the samples are the surgical discard of anonymous healthy patients. Skin cell cultures of keratinocytes and fibroblasts were established individually from abdominal skin biopsies obtained from a 20-year-old and a 40-year-old healthy Caucasian female undergoing plastic surgery.
2.3. Culture and Treatments of Skin Explants
Six millimeters of punch biopsies were prepared from human skin fragments obtained. They were placed in a culture medium containing 50% of DMEM 1 g/L glucose (Lonza, Switzerland) and 50% of Ham’s-F12 (Lonza, Switzerland) supplemented with 10% FBS (Gibco, Carlsbad, CA, USA), 2 mM of L-glutamine (Lonza, Switzerland) and 100 µg/mL of Primocin® (InvivoGen, Carlsbad, CA, USA), and maintained at 37 °C in a humidified atmosphere containing 5% of CO2.
Tea extract (conventional or PSR extract, final concentration: 1% v/v) or PBS (control) was added to the culture medium twice a day for 24 h. Explants were then stressed with 5 J/cm2 UVA (oven type BLX-E365, Fisher Scientific, Waltham, MA, USA) followed by 200 mJ/cm2 UVB (oven type BLXE312, Fisher Scientific, Waltham, MA, USA) and retreated twice a day for 24 h before further analysis.
2.4. Isolation and Preparation of Keratinocytes and Fibroblasts
NHFs were extracted from normal human dermis. NHKs were cultivated in serum-free DMEM medium containing 20% FBS, 2 mM glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin at 37 °C in a humidified atmosphere containing 5% of CO
2 as previously described [
15]. Primary cultures of human keratinocytes were prepared from skin biopsies and were grown in Epilife medium (Thermofischer, Saint Aubin, France).
2.5. Culture and Treatments of Isolated Keratinocytes and Fibroblast
Keratinocytes were grown in Keratinocyte Serum Free Medium (KSFM, Gibco, Waltham, MA, USA) supplemented with 5 ng/mL of human recombinant EGF (Gibco, MA, USA), 50 µg/mL of bovine pituitary extract (Gibco, Waltham, MA, USA), and 100 µg/mL of Primocin® (InvivoGen, Carlsbad, CA, USA). Fibroblasts were grown in DMEM 1 g/l glucose (Lonza, Switzerland) supplemented with 10% of FBS (Gibco, Waltham, MA, USA), 2 mM of L-Glutamine (Lonza, Switzerland), and 100 µg/mL of Primocin® (InvivoGen, Carlsbad, CA, USA). All cultures were maintained at 37 °C under 5% of CO2.
Tea extract (final concentration: 0.1% v/v), either the conventional extract or the PSRTM extract, was directly diluted in the culture medium. For the negative control, a similar amount of water was used. The addition of extract, or water, was performed twice a day for 48 h.
When applicable, keratinocytes were subjected to UV stress. Cells were subjected to 2 J/cm2 UVA (oven type BLX-E365, Fisher Scientific, Waltham, MA, USA) followed by 60 mJ/cm2 UVB (oven type BLX-E312, Fisher Scientific, Waltham, MA, USA). For immunohistochemical analysis, keratinocytes were reincubated 24 h before staining. To analyze the DNA repair inducing capacities of the extracts, a reverse order was used. The UV stress was applied first (2 J/cm2 UVA followed by 60 mJ/cm2 UVB), and then extracts were added (0.1% v/v) twice a day for 24 h.
2.6. Enzymatic Assay of Aconitase Activity
Cells were resuspended in isolation buffer (210 mM mannitol, 70 mM sucrose, 1 mM EDTA, 5 mM HEPES, pH 7.2). Digitonin (10% w/v in DMSO) was added slowly to a final concentration of 0.2 to 0.4 mg/mL. Permeabilized cells were collected by centrifugation and disrupted in a glass Dounce homogenizer. Cell debris and nuclei were removed by centrifugation for 5 min at 625× g. The final supernatant was spun at 10,000× g for 20 min to obtain the mitochondrial pellet. Mitochondria were diluted to 0.05 mg/mL in 25 mM KH2PO4, pH 7.25, containing 0.05% Triton X-100. Aconitase activity was assayed as the rate of NADP reduction (340 nm) by isocitrate dehydrogenase upon addition of 1.0 mM sodium citrate, 0.6 mM MnCl2, 0.2 mM NADP, and 1.0 unit/mL isocitrate dehydrogenase.
2.7. Measure of Keratinocyte Respiration and Quantification of ATP Production
Mitochondrial respiration was evaluated using a Seahorse XF96 extracellular flux analyzer from Agilent (Les Ulis, France). A total of 4 × 104 cells were plated on an XF96 cell-culture microplate. On the following day, the medium was substituted with XF assay medium, and sequential injections of 1 μM oligomycin A, 1 μM FCCP, and 2 μM antimycin/rotenone (Sigma-Aldrich, Saint Quentin Falavier, France) were administered. The obtained results were normalized to the total protein content in each well, determined using the DC Protein Assay Reagent from Sigma-Aldrich (Saint Quentin Falavier, France).
2.8. Measure of Skin Explants Respiration and Quantification of ATP Production
The XF24 Extracellular Flux Analyzer from Agilent (Les Ullis, France) was employed to measure the change in oxygen consumption rate (OCR). XF Islet Capture Microplates (Agilent, Les Ullis, France) were utilized for the measurements, featuring a net to prevent the oxygen sensor from making direct contact with the skin explants. The RHE (reconstructed human epidermis) was coated with XF assay medium, followed by the sequential injection of 4 μM oligomycin A, 4 μM FCCP, and 2 μM antimycin/rotenone (Sigma-Aldrich, Saint Quentin Falavier, France). The results were normalized to the total protein content in each RHE.
2.9. Quantification of Reactive Oxygen Species (ROS) in Mitochondria
Quantification of mitochondrial ROS was performed in isolated keratinocytes using the MitoSOXTM reagent (Invitrogen, Waltham, MA, USA) following the manufacturer’s instructions. After 15 min of incubation at 37 °C, cells were fixed with 3.7% formaldehyde for 10 min, and the red fluorescence (excitation/emission 510/580 nm) obtained upon oxidation of the reagent within the mitochondria was analyzed using an Axiovert 200M microscope (Zeiss, Oberkochen, Germany). Photos were taken with an EXI blue camera (Teledyne, Tucson, AZ, USA) coupled to the Volocity® 6.1 acquisition software (PerkinElmer, Stockbridge, GA, USA). Five photos per condition, each performed in duplicate (n = 10), were considered.
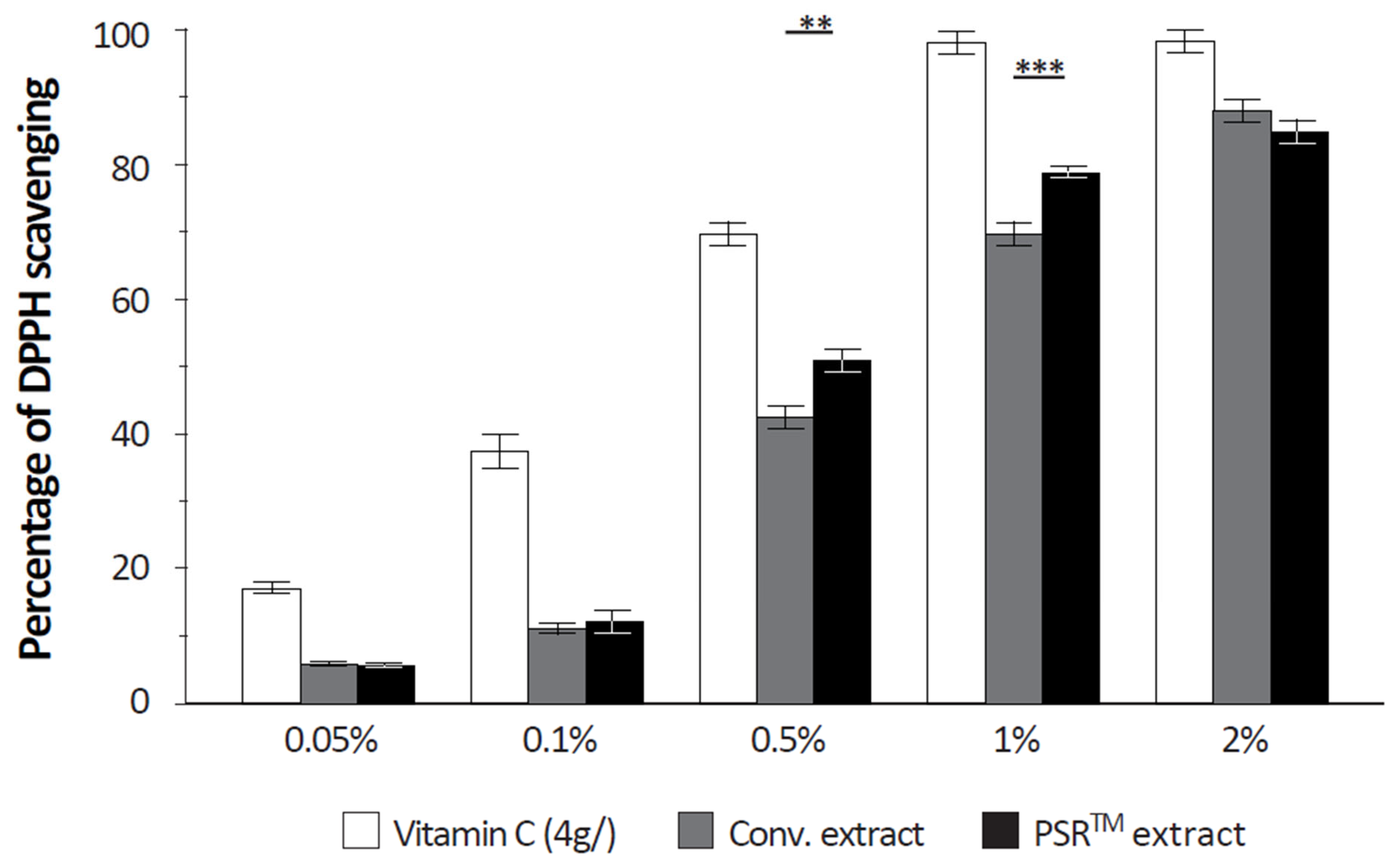
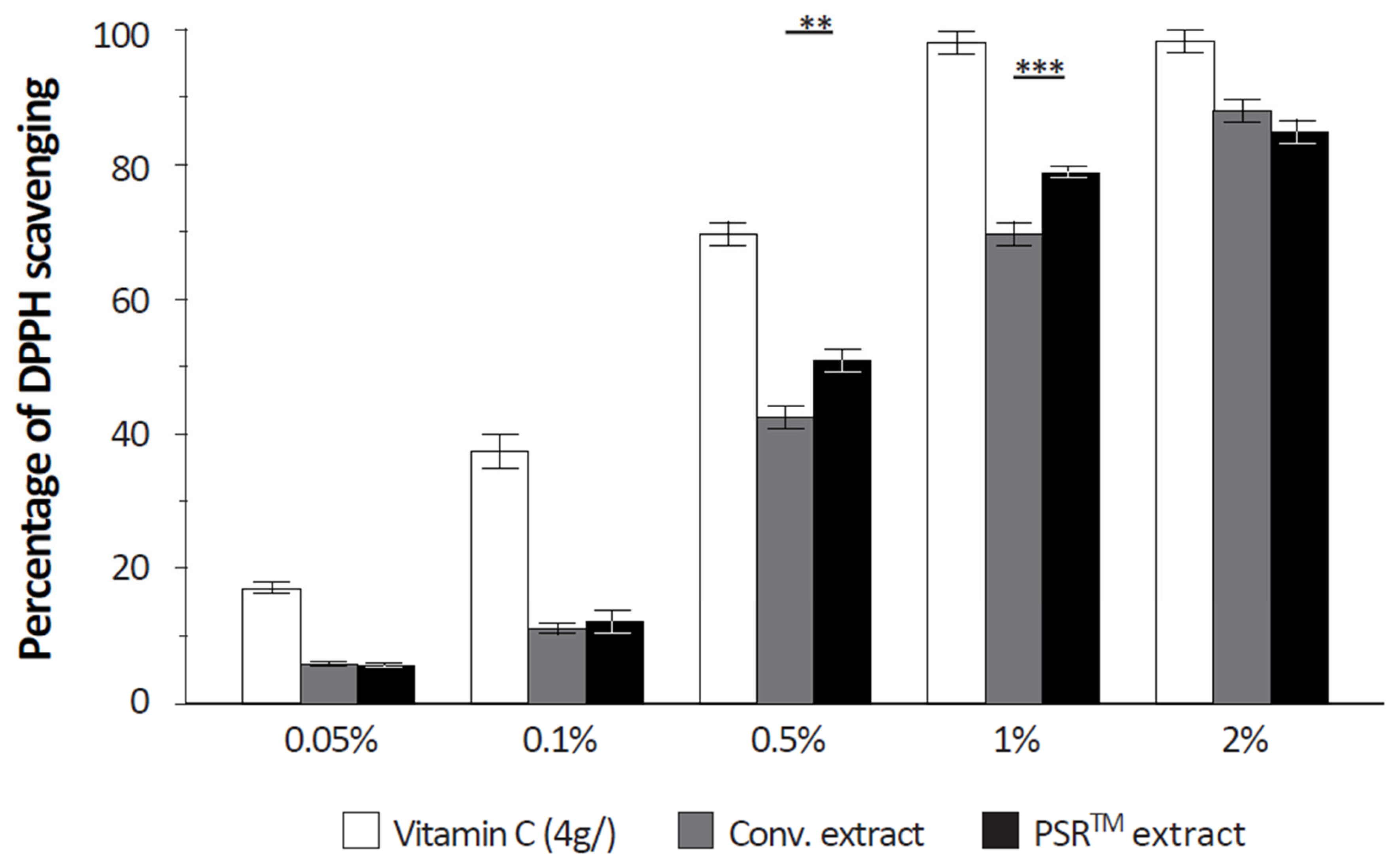
2.10. Quantification of the Free Radical Scavenging Capacity of Extracts
The free radical scavenging capacity was analyzed using a DPPH (α,α-diphenyl-β-picrylhydrazyl) assay. Upon reaction with an antioxidant that can donate hydrogen, DPPH is reduced and changes color (from deep violet to light yellow). Color change was quantified at 517 nm. Vitamin C (4 g/L) was used as a positive reference.
2.11. Real-Time qPCR Analysis of the Matrix Metalloprotease 1 (MMP1) mRNA
Total fibroblast mRNAs were prepared using the mirVanaTM miRNA Isolation Kit (Invitrogen, MA, USA) according to the manufacturer’s instructions. Total RNAs (1.5 µg) were reverse-transcribed with the High-Capacity cDNA Reverse Transcription Kit (Fisher Scientific, MA, USA). Finally, real-time qPCR was performed on a StepOnePlusTM thermocycler (Applied Biosystems, Waltham, MA, USA) with TaqManTM Gene Expression Master Mix (Fisher Scientific, Waltham, MA, USA) and TaqManTM Gene Expression Assays (Fisher Scientific, MA, USA). The 18S ribosomal gene was used as an internal standard. Results were analyzed using relative quantification (2−∆∆Ct) with the StepOne 2.3 software (Applied Biosystems, MA, USA). Every sample was analyzed in triplicate, and negative control real-time qPCR with water instead of DNA was performed.
2.12. Immunohistochemical Analysis of Isolated Keratinocytes or Fibroblasts
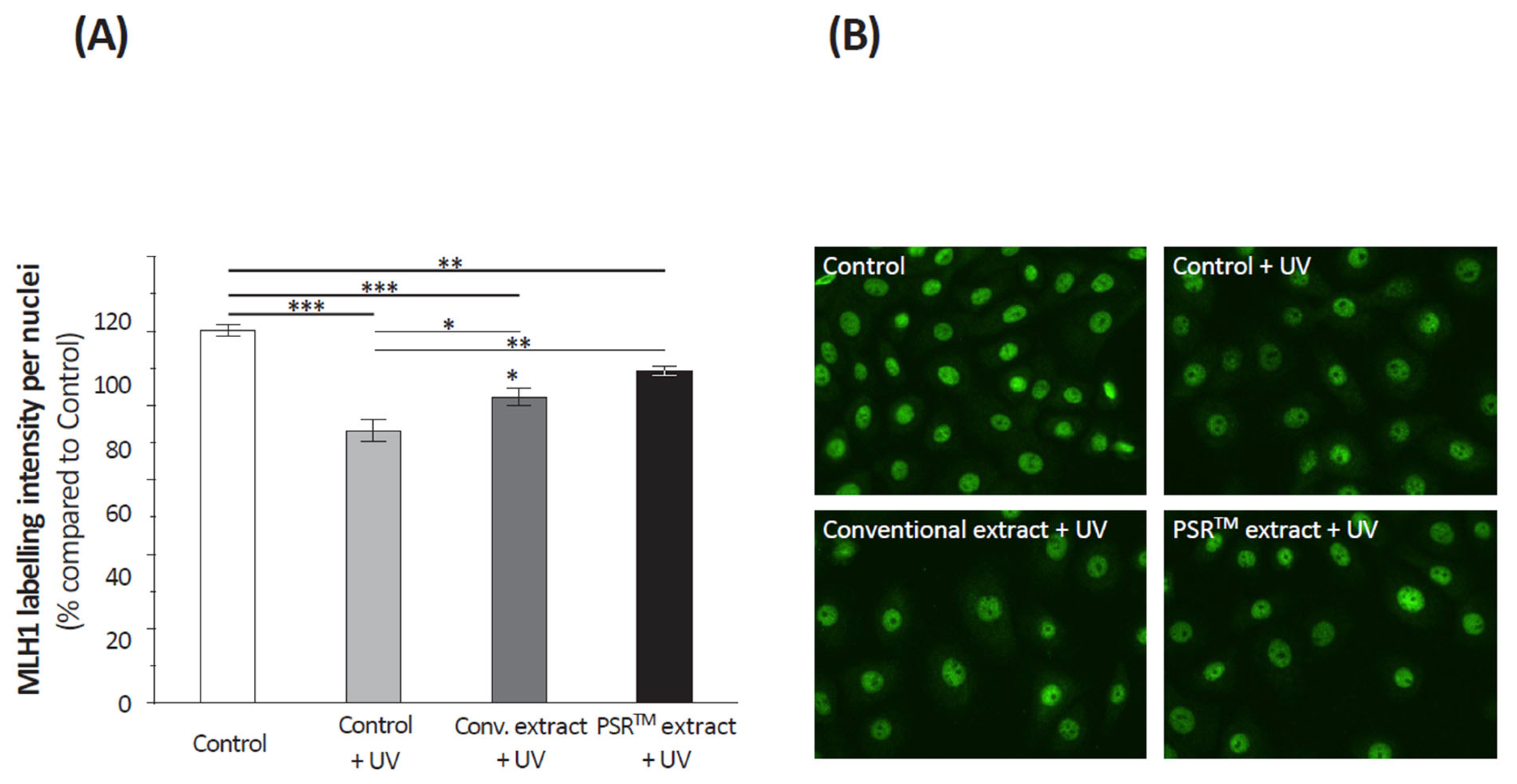
For the labeling of cyclobutane pyrimidine dimers (CPDs), cells were fixed with 3.7% paraformaldehyde (15 min), membranes were permeabilized with 0.5% Triton X-100 (10 min) followed by a 2 mol/L HCl solution (30 min), and unspecific binding sites were blocked with 20% FBS (Gibco, Waltham, MA, USA, 30 min). For the labeling of Mut Homolog 1 (MLH1) in keratinocytes and type I procollagen labeling in fibroblasts, cells were fixed with cold methanol for 4 min at 4 °C.
CPDs were labeled using an anti-CPDs mouse monoclonal antibody (Euromedex, France—1/500, 90 min) revealed by an Alexa Fluor® 488 donkey anti-mouse IgG polyclonal antibody (Invitrogen, Waltham, MA, USA—1/1000, 60 min). For MLH1 labeling, the primary antibody was an anti-MLH1 rabbit polyclonal antibody (Abcam, United Kingdom—1/200, 90 min) and the secondary antibody an Alexa Fluor® 488 donkey anti-rabbit IgG antibody (Invitrogen, Waltham, MA, USA—1/1000, 60 min). Finally, type I procollagen was labeled thanks to an anti-type I procollagen rat monoclonal antibody (Merck Millipore, Burlington, MA, USA—1/100, 90 min) and an Alexa Fluor® 488 donkey anti-rat secondary antibody (Invitrogen, Waltham, MA, USA—1/1000, 60 min). For all immunolabeling, a nuclear counterstaining with DAPI was performed and slides mounted in Fluoromount-GTM (Electron Microscopy Sciences, Hatfield, PA, USA).
Slides were examined using an Axiovert 200M microscope (Zeiss, Oberkochen, Germany). Pictures were taken with an EXI blue camera (Teledyne, Tucson, AZ, USA) and analyzed using the Volocity® acquisition 6.1 software (PerkinElmer, Stockbridge, GA, USA). Five photos per condition, each performed in duplicate (n = 10), were considered. Results are expressed as the mean of the sum of fluorescent pixel intensities in each photo, considering nuclei only in the case of CPDs and MLH1 labeling.
2.13. Immunohistochemical Analysis of Skin Explants
For fibrillin 1 labeling, skin explants were embedded in OCT and snap-frozen before being cut into 6 µm thick sections. In the case of elastin labeling, skin explants were fixed in 10% formalin, dehydrated in ethanol, embedded in paraffin, and cut into 4 µm thick sections that were deparaffinized and rehydrated.
Fibrillin 1 was detected thanks to an anti-fibrillin 1 rabbit polyclonal primary antibody (Bio-Techne, MN, USA—1/300, 90 min). For elastin, an anti-elastin rabbit polyclonal primary antibody was used (Abcam, United Kingdom—1/2000, 90 min). Both were revealed by an Alexa Fluor® 488 donkey anti-rabbit antibody (Invitrogen, MA, USA—1/1000, 60 min). For all immunolabeling, a nuclear counterstaining with DAPI was performed and slides mounted in Fluoromount-GTM (Electron Microscopy Sciences, PA, USA).
Labeled slides were analyzed using an Eclipse Ni-E microscope (Nikon, Tokyo, Japan) equipped with a DS-Fi3 camera (Nikon, Tokyo, Japan). Images were processed using the NiS-AR (Nikon, Tokyo, Japan) acquisition software 4.5. Image analysis was performed with ImageJ 2023 software. Whatever condition, it enabled the selection of a constant depth of the upper dermis fibers where the total length of fibers was measured and normalization of the length of the dermal–epidermal junction. Five photos per condition were analyzed, and each condition was tested in duplicate.
2.14. Statistical Analysis
After checking the normality of data distribution using the Shapiro–Wilk test (p < 0.05), statistical differences were analyzed using the one-way analysis of variance (ANOVA) followed by a Tukey–Kramer test. Results are presented as mean ± standard error of the mean (SEM). p ≤ 0.05 was considered significant, p ≤ 0.01 very significant, and p ≤ 0.005 highly significant.
4. Discussion
As anticipated when using the PSR
TM extraction technology, the composition of the PSR
TM Mauritius black tea extract was found to be rich in small molecules (as shown in
Table 1). Notably, it exhibited a high concentration of organic compounds, with lactic and citric acids being the primary components of interest. Contrary to the conventional belief that lactic acid is solely a byproduct of glycolysis, it plays various essential roles, including controlling the cytosolic NADH-to-NAD+ ratio, which is involved in the regulation of oxidative phosphorylation. On the other hand, citric acid is a well-known antioxidant and serves as a substrate in the tricarboxylic acid (TCA) cycle, playing a crucial role in NAD
+ to NADH conversion and subsequent ATP production. It also regulates glycolysis, gluconeogenesis, and fatty acid synthesis.
Additionally, prior research demonstrated that when mitochondria face prooxidants, there is an interaction between aconitase and the mitochondrial iron-binding protein frataxin [
16,
17,
18]. This interaction is dependent on the enzyme’s substrate, citrate, and it safeguards the [4Fe-4S]
2+ cluster of aconitase from oxidant-induced disassembly. This interaction is necessary for enzyme reactivation. A decrease in aconitase activity could result in reduced NADH production, limiting electron flow and, consequently, oxygen radical production through a compromised electron transport chain. The potential beneficial effects of redox regulation may be overlooked if oxidative stress is short-lived or leads to irreversible functional changes due to prolonged exposure [
17].
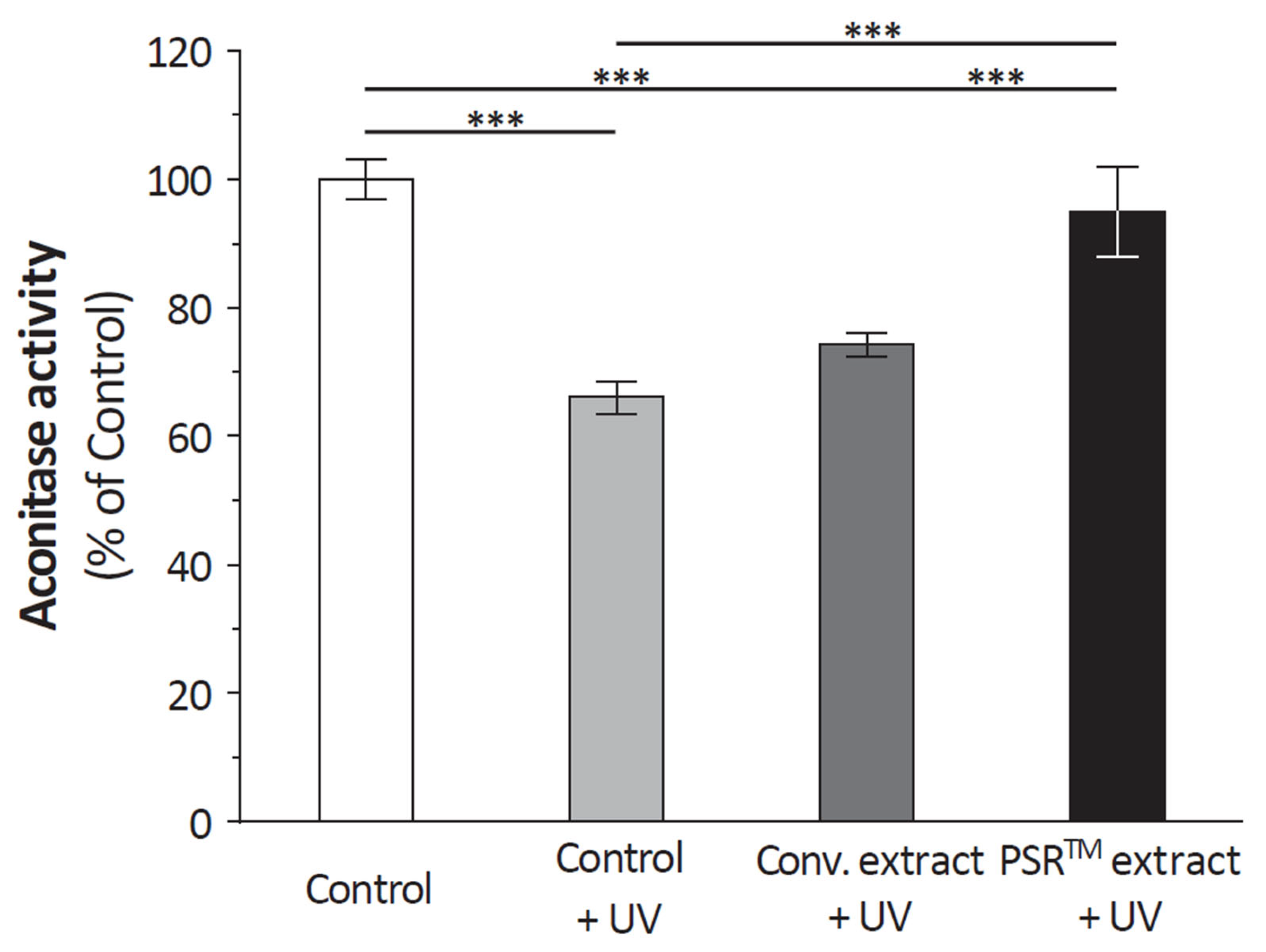
These findings prompted an investigation into the impact of the PSR
TM extract on mitochondria. In UV-stressed cells, aconitase activity resembles that of unstressed conditions when the PSR
TM extract is added to the culture medium (as depicted in
Figure 1). Improved basal respiration and ATP production are also observed, suggesting that the high citric acid content in the extract enhances the entire TCA cycle in vitro, ultimately leading to increased energy production (shown in
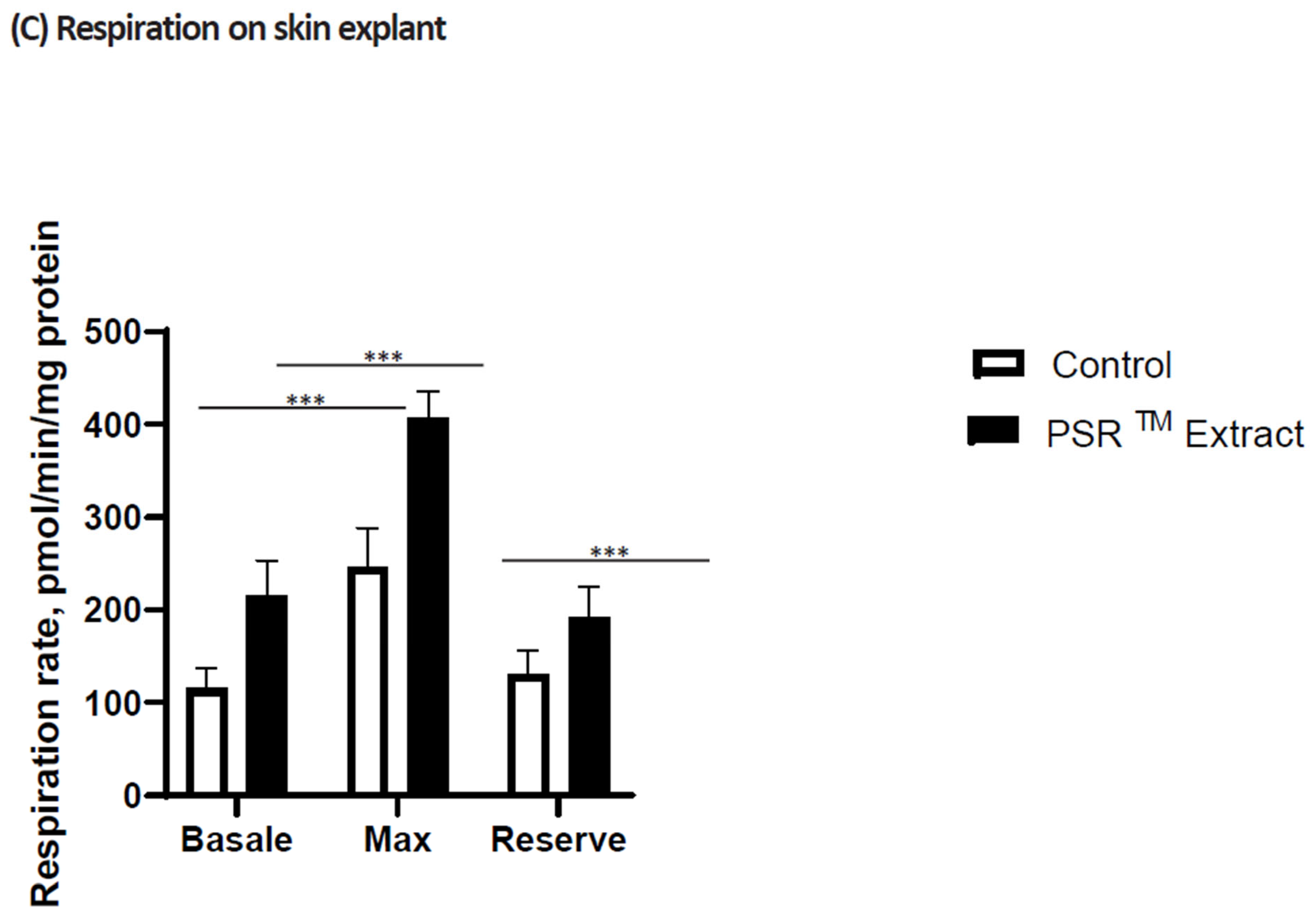
Figure 2A,B). Additionally, treating skin explants with the PSR
TM extract for 24 h increases mitochondrial spare respiratory capacity and basal respiration. These observations indicate that treatment with the tea extract induces biogenesis of mitochondrial respiratory complexes, enhancing the mitochondrial respiratory reserve and, consequently, improving the skin’s mitochondrial fitness (as illustrated in
Figure 2C).
These initial results share similarities with the effects of CoQ10, a compound commonly used in cosmetics to target mitochondria and reduce age-related skin alterations. CoQ10 enhances oxygen consumption in isolated keratinocytes, signifying improved mitochondrial activity and energy metabolism. In vivo, CoQ10′s beneficial effect is evident through enhanced mitochondrial activity, as evidenced by mitochondrial membrane potential in UV-stressed keratinocytes [
11,
19].
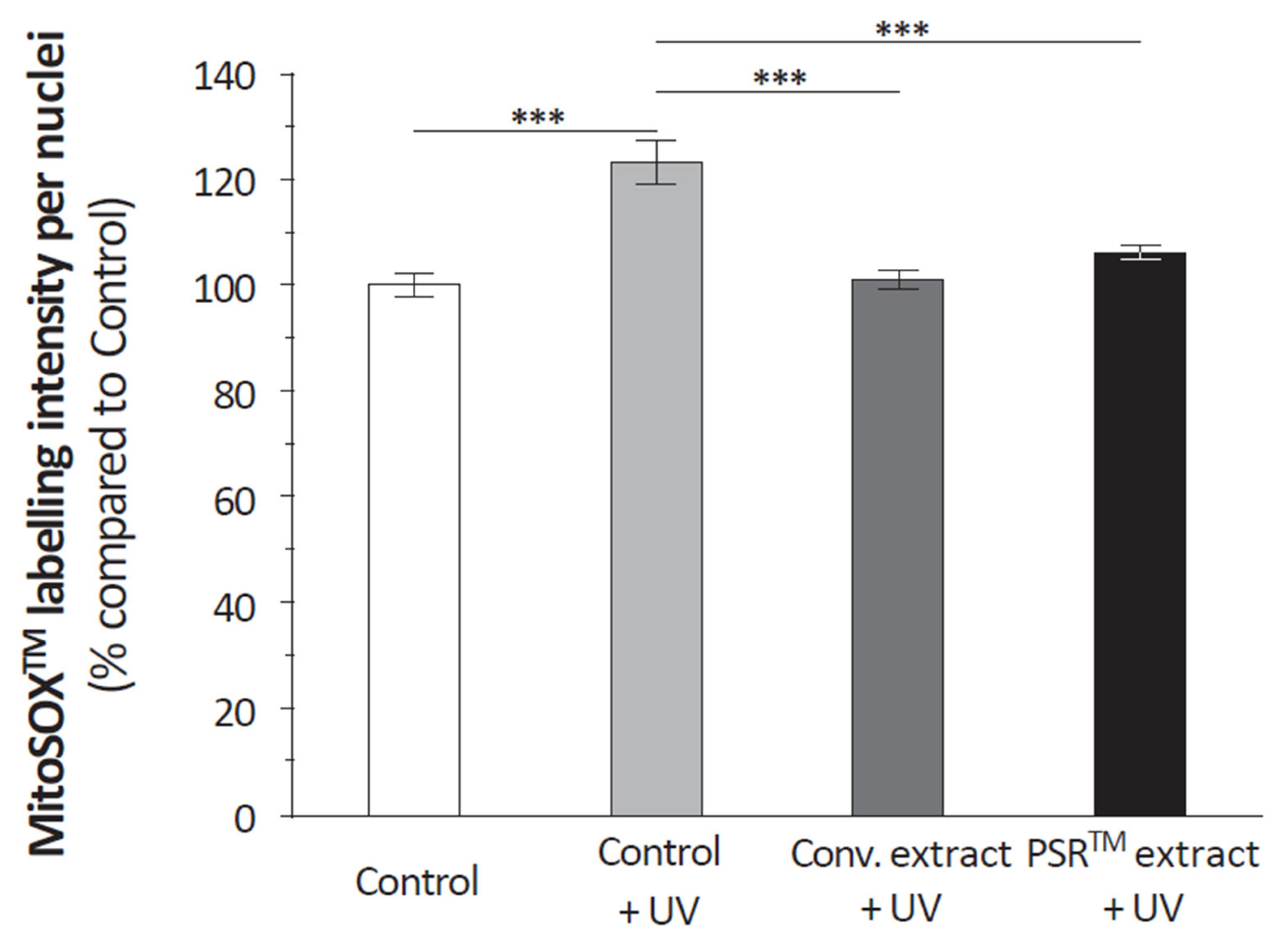
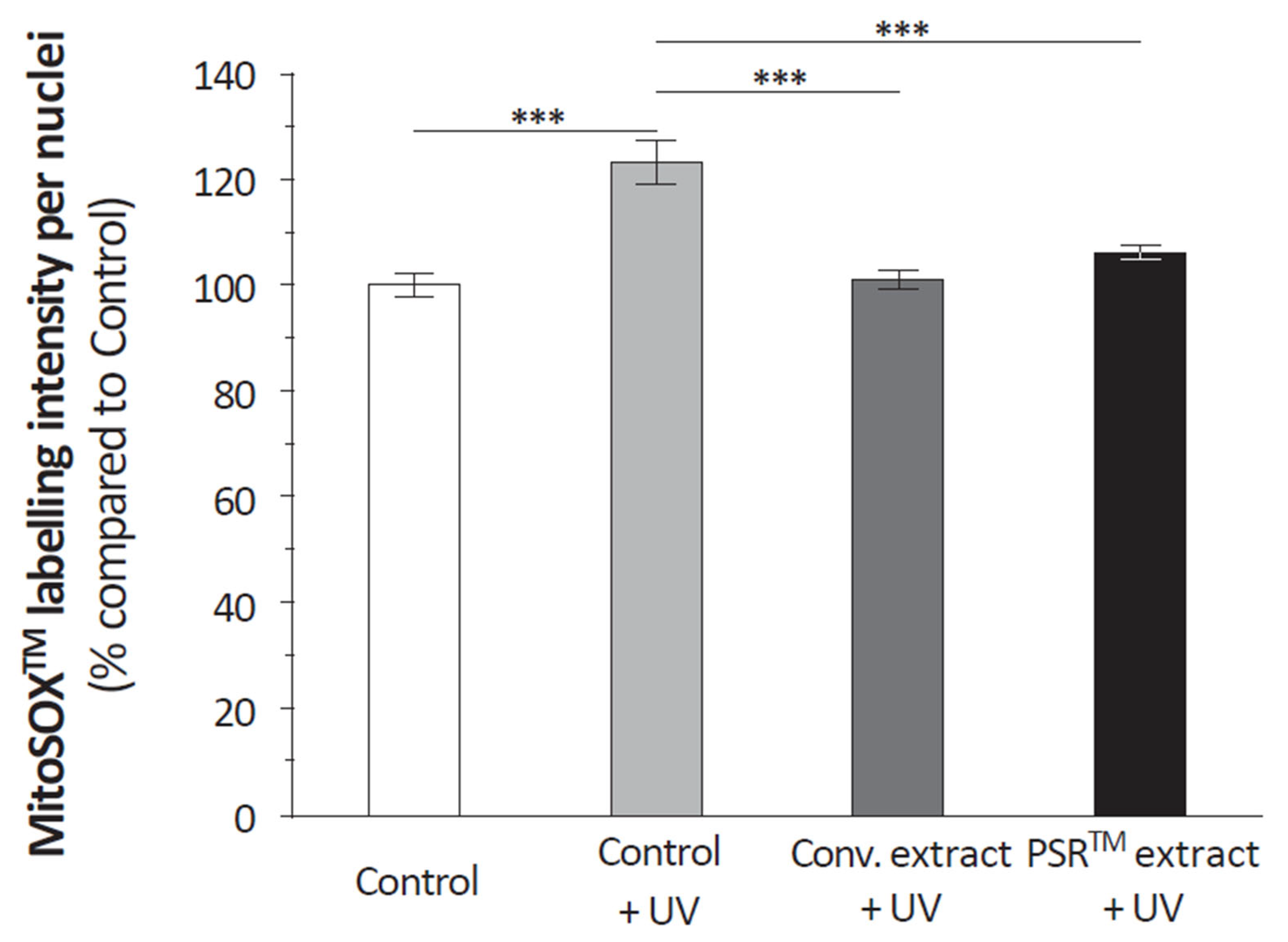
Another important aspect of the PSR
TM extract is its antioxidant properties, which are demonstrated by its ability to scavenge reactive oxygen species (ROS), as depicted in
Figure 4. Citric acid, present in significant quantities, likely contributes to this antioxidative effect by modulating ROS production through its impact on aconitase activity [
16,
17]. However, it is challenging to rule out the role of other small molecules, as the conventional extract also reduces ROS in UV-stressed keratinocytes to a similar extent as the PSR
TM extract (as shown in
Figure 3). Regardless of the specific compound(s) involved (as detailed in
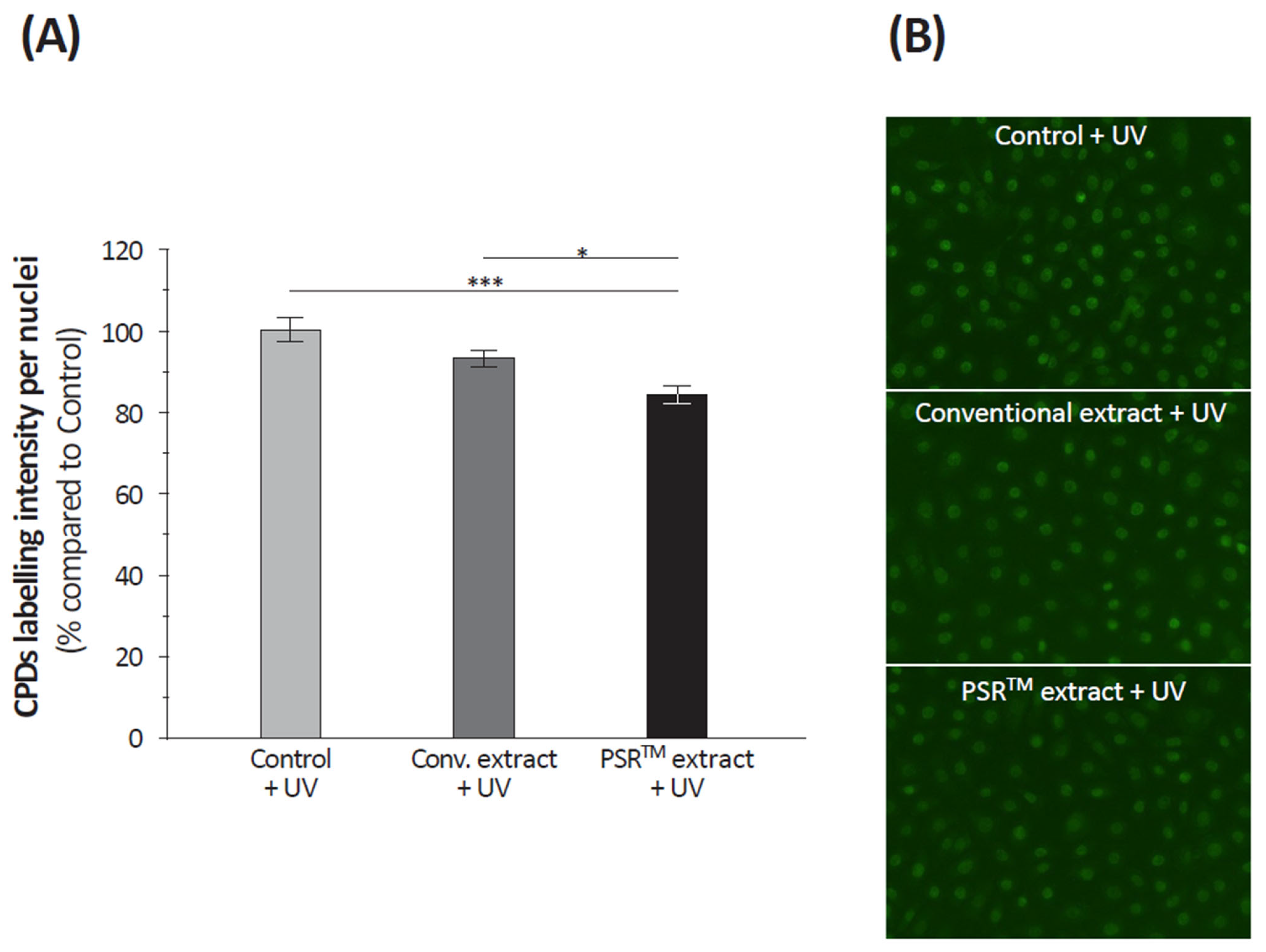
Table 1), ROS scavenging is crucial for mitigating the pro-oxidative state caused by regular mitochondrial operation. One indication of this protective effect is the lower level of cyclobutane pyrimidine dimers (CPDs) observed in UV-treated keratinocytes (depicted in
Figure 5 and
Figure 6). This suggests that the PSR
TM extract may reduce damage to mitochondrial DNA, thus helping maintain a pool of healthy and efficient mitochondria, which can halt the cycle of mitochondrial function deterioration, a key factor in the aging process.
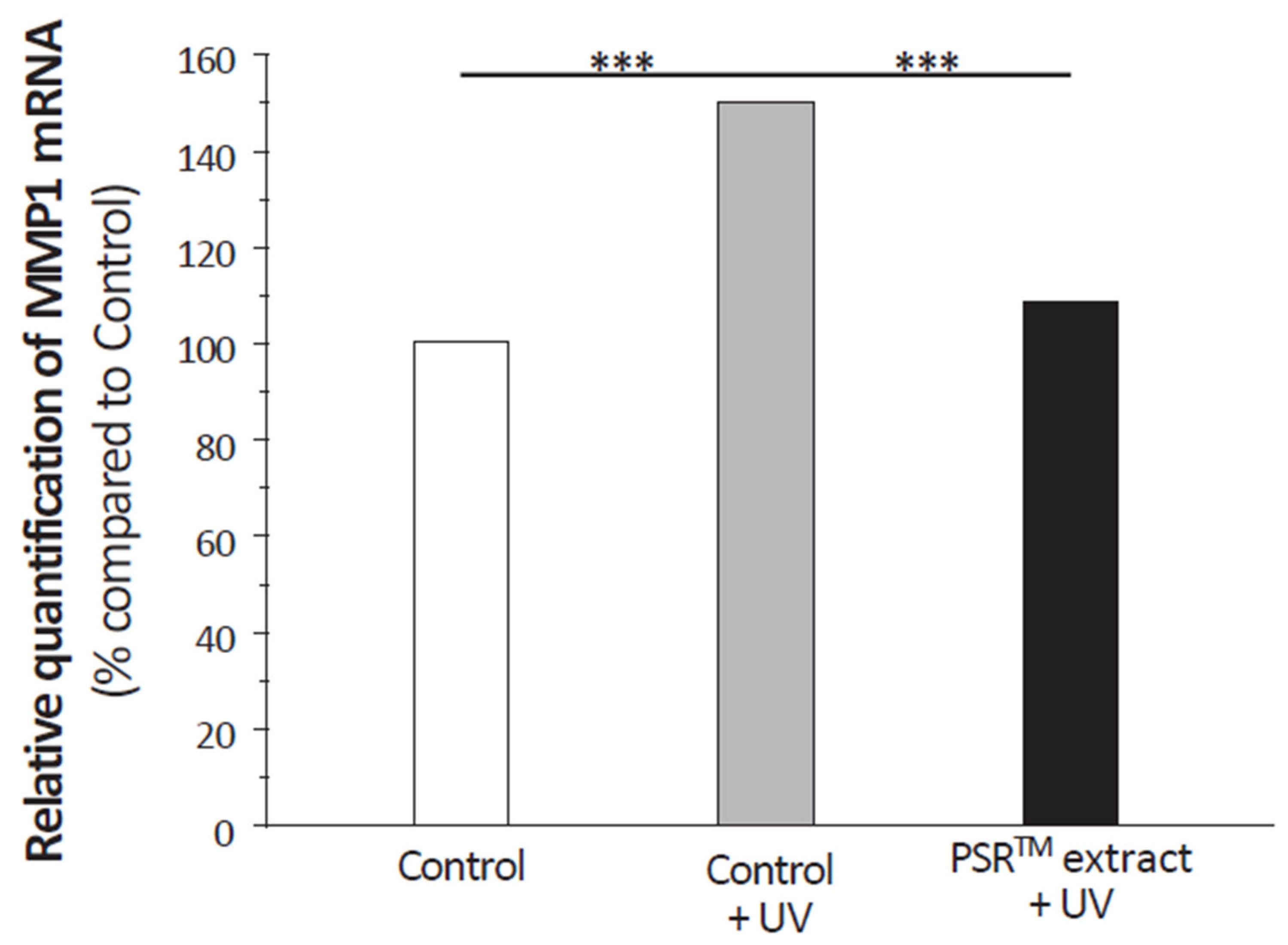
Antioxidative capabilities are not only essential for preserving DNA from damage but also for protecting cellular components. Studies on CoQ10 and vitamin C, both known for their strong antioxidant properties, have shown their effectiveness in protecting the skin from ROS generated by environmental stressors and reducing the signs of aging. In the case of vitamin C, its benefits go beyond its antioxidant properties, as it also plays a crucial role in collagen synthesis and structure. Although the PSRTM extract positively affects the extracellular matrix, it does not contain significant levels of vitamin C. Nevertheless, it reduces MMP1 expression in UV-stressed fibroblasts to levels seen in unstressed conditions, similar to the effect of CoQ10. This is attributed to the antioxidative capacity of CoQ10 in preventing cytokine-mediated MMP1 induction and collagen degradation. The PSRTM extract’s broader impact on the extracellular matrix compared to CoQ10 suggests that it may have a more comprehensive effect on aging signs.
However, it is important to note that this study is preliminary, and the PSRTM Mauritius black tea extract still has limitations. One limitation is the complexity of the extract, which contains various compounds. While citric acid likely plays a major role, other compounds may contribute to the observed effects, making the results more intricate than those of a purified compound. Additionally, all analyses were conducted mostly in vitro, primarily with isolated cells, and it is crucial to test the extract in a clinical study to validate its potential benefits.
As expected upon the use of the PSR
TM extraction technology, the composition of the PSR
TM Mauritius black tea extract revealed its richness in small molecules (
Table 1). It especially highlighted its high content in organic compounds, the main ones being lactic and citric acids, two components with potentially interesting effects. Contrary to what has been long considered, lactic acid is far from only being the dead-end product of glycolysis. Among its many roles, its concentration controls the cytosolic NADH-to-NAD
+ ratio that participates in the regulation of oxidative phosphorylation. On its side, citric acid is a well-known antioxidant, and it is also a substrate of the tricarboxylic acid (TCA) cycle. As such, it Is essential for NAD
+ to NADH conversion and subsequent ATP production. It also regulates glycolysis, glucogenesis, and fatty acids synthesis. Moreover, we have previously shown in vitro that when mitochondria are challenged with prooxidants, aconitase and the mitochondrial iron-binding protein frataxin interact. This interaction requires the enzyme’s substrate citrate, protects the [4Fe-4S]
2+ cluster of aconitase from oxidant-induced disassembly, and is required for enzyme reactivation. Loss of aconitase activity could reduce NADH production, limiting electron flow and thus oxygen radical production by a compromised electron transport chain [
16,
17]. The potential beneficial roles of redox regulation may be easily overlooked if the duration of oxidative stress is short or if prolonged stress results in the progression to irreversible alterations in function. All these elements led us to start investigating the effect of the PSR
TM extract by focusing on mitochondria.
In UV-stressed cells, the aconitase presents an activity similar to that of unstressed conditions when the PSR
TM extract is supplemented to the culture medium (
Figure 1). As an improved basal respiration and ATP production are also observed, these results collectively suggest that the high citric acid content of the extract boosts the entire TCA cycle in vitro, ultimately resulting in increased energy production (
Figure 2A,B). We also found that treatment of skin explants with PSR
TM extract for 24 h increases mitochondria spare respiratory capacity and basal respiration. These data suggest that treatment with our tea extract induced mitochondrial respiratory complexes biogenesis responsible for the increase of mitochondrial respiratory reserve, thus improving mitochondrial fitness of the skin (
Figure 2C).
These first results show some similarities with the effects induced by CoQ10, a compound that frequently enters the composition of cosmetics aimed at reducing age-related skin alterations by targeting mitochondria. CoQ10 increases oxygen consumption in isolated keratinocytes, indicating improved mitochondrial activity and energy metabolism [
20,
21]. The beneficial effect of CoQ10 was evidenced in vivo as the mitochondrial membrane potential of UV-stressed keratinocytes also revealed enhanced mitochondrial activity [
22].
Another critical aspect of the PSR
TM extract is its antioxidant properties, evidenced by the analysis of its ROS scavenging capacity (
Figure 4). Citric acid, present in large amounts, could play a significant role also by modulating ROS production thanks to its role on aconitase activity [
16,
17]. Yet, it seems difficult to exclude the effect of other small molecules, as the conventional extract leads to a similar ROS reduction to the PSR
TM extract in UV-stressed keratinocytes (
Figure 3). Whatever compounds are involved (
Table 1), ROS scavenging is essential to mitigate the deleterious pro-oxidative state induced by the regular operation of mitochondria [
23,
24]. In our case, one of the indications we highlighted for this protective effect is the lower level of CPDs observed in UV-treated keratinocytes (
Figure 5 and
Figure 6). Therefore, we can hypothesize that the PSR
TM extract should reduce damage to mitochondrial DNA, thus helping to preserve a pool of healthy and efficient mitochondria that should help stop the cycle of mitochondrial function alteration, which plays a role in the aging process [
25].
Antioxidative capacities are not only important to preserve DNA from alterations. They also protect cellular components from deleterious effects. Studies of CoQ10 and vitamin C—both having high antioxidative properties—showed that both compounds are particularly potent in protecting the skin, especially against ROS generated by environmental stressors, and reducing the severity of aging signs [
8,
26]. In the case of vitamin C, part of this effect is not only due to antioxidant capacities as, among its many roles, it is essential for collagen synthesis and structure [
26]. If we did observe an induction of type I procollagen synthesis, it seems unlikely to involve vitamin C as none was found at a significant level in the PSR
TM extract. Still, the PSR
TM extract did positively affect the extracellular matrix (
Figure 8). It diminishes MMP1 expression in UV-stressed fibroblasts treated with PSR
TM extract to levels observed in unstressed conditions. A similar effect was shown with CoQ10 [
27]. It was attributed to the antioxidative capacity of CoQ10 to prevent cytokine-mediated MMP1 induction and, therefore, collagen degradation. Such a process might very well be at play with the PSR
TM extract. This is an important aspect to mitigate aging signs as collagen plays an essential role in the severity of wrinkles. Indeed, clinical studies of CoQ10 revealed that it improves wrinkling [
27]. Still, contrary to the PSR
TM extract, CoQ10 was not shown to impact type I collagen synthesis [
19] and its effect on other extracellular matrix components, especially fibrillin 1 (
Figure 8), is poorly characterized. Having a broader, more positive effect on the extracellular matrix than CoQ10, we can expect that the PSR
TM extract will also have an impact on aging signs.
Yet, it is important to keep in mind that this work is an initial study of the PSRTM Mauritius black tea extract. Therefore, it still suffers from limitations. The first one is inherent to our approach, which is working with a complex extract. Even if we can suspect that citric acid plays a major role, part of the effects could be due to other compounds, and results could be more intricate than those induced by a purified compound. Still, the entire study shows that the extract could, similarly to CoQ10, be a mitochondria nutrient as well as an antioxidant. The second limitation is that all analyses were performed in vitro, essentially with isolated cells. It will be essential to test the extract in an appropriate clinical study.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}