Mitochondria’s Role in Skin Ageing


{kind=link}
Abstract
1. Skin Structure
2. Mitochondria’s Role in Skin
3. Hallmarks of Skin Ageing
4. Mitochondria and Ageing
5. Pigmentation
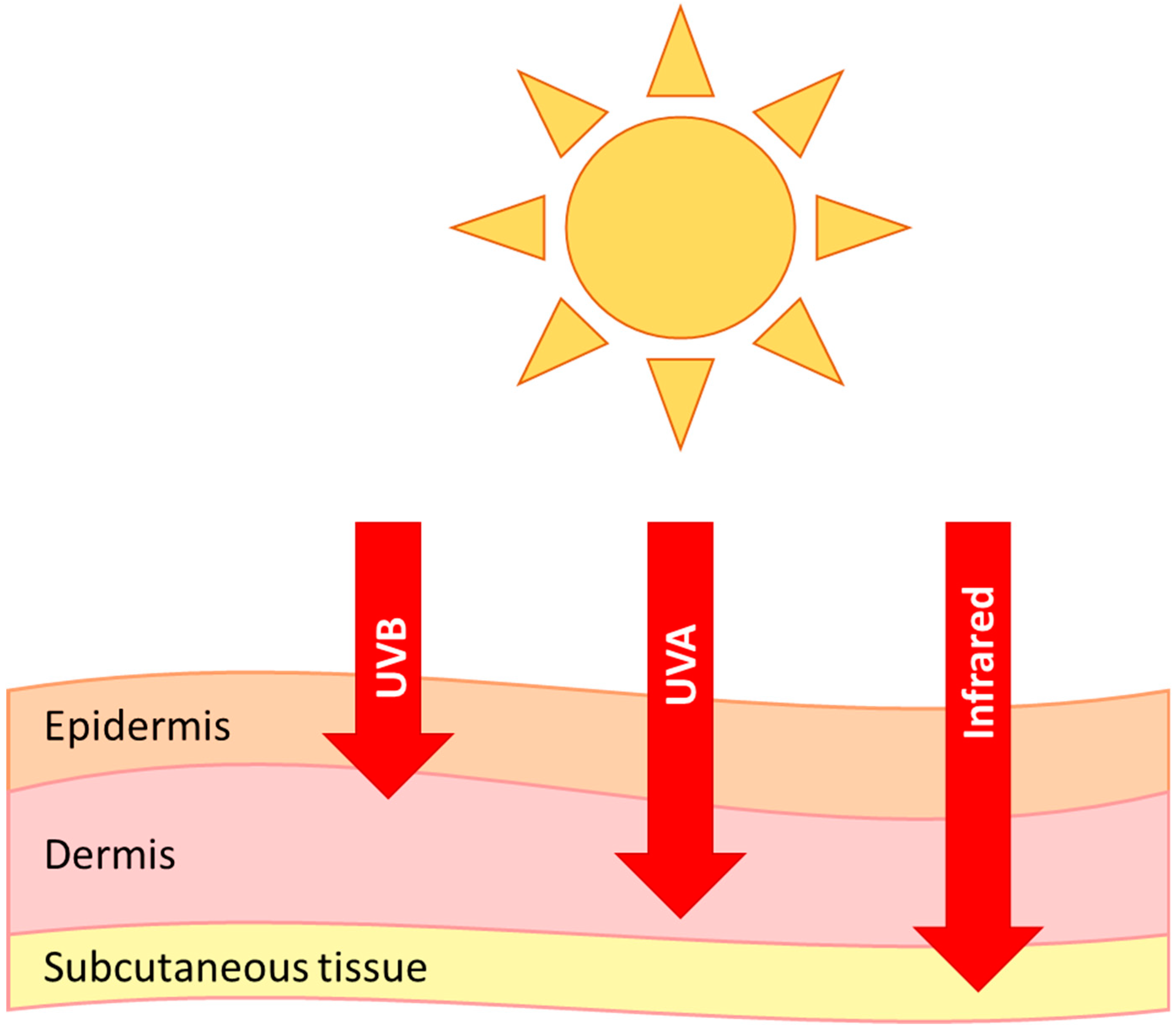
6. Photoageing
7. Pollution
8. Hair Follicles
9. Summary
Funding
Conflicts of Interest
Abbreviations
| UVA | Ultraviolet A |
| UVB | Ultraviolet B |
| IR | Infrared |
| MMPs | Matrix metalloproteinases |
| ROS | Reactive oxygen species |
| AP-1 | activator protein-1 |
| NFκB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| TGF-β | Transforming growth factor beta |
| PM2.5 | Small particulate matter |
| AhR | Aryl hydrocarbon receptor |
| Bcl-2 | B-cell lymphoma 2 |
| ETC | Electron transport chain |
References
- Thingnes, J.; Lavelle, T.J.; Hovig, E.; Omholt, S.W. Understanding the Melanocyte Distribution in Human Epidermis: An Agent-Based Computational Model Approach. PLoS ONE 2012, 7, e40377. [Google Scholar] [CrossRef]
- Koster, M.I. Making an epidermis. Ann. N. Y. Acad. Sci. 2009, 1170, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Poon, F.; Kang, S.; Chien, A.L. Mechanisms and treatments of photoaging. Photodermatol. Photoimmunol. Photomed. 2015, 31, 65–74. [Google Scholar] [CrossRef]
- Wickersham, M.; Wachtel, S.; Wong Fok Lung, T.; Soong, G.; Jacquet, R.; Richardson, A.; Parker, D.; Prince, A. Metabolic Stress Drives Keratinocyte Defenses against Staphylococcus aureus Infection. Cell Rep. 2017, 18, 2742–2751. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr. Opin. Cell Biol. 2009, 21, 894–899. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial metabolism as a regulator of keratinocyte differentiation. Cell. Logist. 2013, 3, e25456. [Google Scholar] [CrossRef]
- Feichtinger, R.G.; Sperl, W.; Bauer, J.W.; Kofler, B. Mitochondrial dysfunction: A neglected component of skin diseases. Exp. Dermatol. 2014, 23, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Birch-Machin, M.A. Mitochondria and skin disease. Clin. Exp. Dermatol. 2000, 25, 141–146. [Google Scholar] [CrossRef]
- Plummer, C.; Spring, P.J.; Marotta, R.; Chin, J.; Taylor, G.; Sharpe, D.; Athanasou, N.A.; Thyagarajan, D.; Berkovic, S.F. Multiple Symmetrical Lipomatosis—A mitochondrial disorder of brown fat. Mitochondrion 2013, 13, 269–276. [Google Scholar] [CrossRef]
- Boulton, S.J.; Bowman, A.; Koohgoli, R.; Birch-Machin, M.A. Skin manifestations of mitochondrial dysfunction: More important than previously thought. Exp. Dermatol. 2015, 24, 12–13. [Google Scholar] [CrossRef]
- Farage, M.A.; Miller, K.W.; Elsner, P.; Maibach, H.I. Intrinsic and extrinsic factors in skin ageing: A review. Int. J. Cosmet. Sci. 2008, 30, 87–95. [Google Scholar] [CrossRef]
- Naidoo, K.; Birch-Machin, M. Oxidative Stress and Ageing: The Influence of Environmental Pollution, Sunlight and Diet on Skin. Cosmetics 2017, 4, 4. [Google Scholar] [CrossRef]
- Wey, S.-J.; Chen, D.-Y. Common cutaneous disorders in the elderly. J. Clin. Gerontol. Geriatr. 2010, 1, 36–41. [Google Scholar] [CrossRef]
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.J.; Turner, R.; Nikaido, O.; Rees, J.L.; Birch-Machin, M.A. The Spectrum of Mitochondrial DNA Deletions is a Ubiquitous Marker of Ultraviolet Radiation Exposure in Human Skin. J. Investig. Dermatol. 2000, 115, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Michikawa, Y.; Mazzucchelli, F.; Bresolin, N.; Scarlato, G.; Attardi, G. Aging-Dependent Large Accumulation of Point Mutations in the Human mtDNA Control Region for Replication. Science (New York N. Y.) 1999, 286, 774–779. [Google Scholar] [CrossRef]
- Wang, Y.; Michikawa, Y.; Mallidis, C.; Bai, Y.; Woodhouse, L.; Yarasheski, K.E.; Miller, C.A.; Askanas, V.; Engel, W.K.; Bhasin, S.; et al. Muscle-specific mutations accumulate with aging in critical human mtDNA control sites for replication. Proc. Natl. Acad. Sci. USA 2001, 98, 4022–4027. [Google Scholar] [CrossRef] [PubMed]
- Del Bo, R.; Crimi, M.; Sciacco, M.; Malferrari, G.; Bordoni, A.; Napoli, L.; Prelle, A.; Biunno, I.; Moggio, M.; Bresolin, N.; et al. High mutational burden in the mtDNA control region from aged muscles: A single-fiber study. Neurobiol. Aging 2003, 24, 829–838. [Google Scholar] [CrossRef]
- Pavicic, W.H.; Richard, S.M. Correlation analysis between mtDNA 4977-bp deletion and ageing. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2009, 670, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; Flint Beal, M.; Wallace, D.C. Mitochondrial DNA deletions in human brain: Regional variability and increase with advanced age. Nat. Genet. 1992, 2, 324. [Google Scholar] [CrossRef]
- Yang, J.H.; Lee, H.C.; Lin, K.J.; Wei, Y.H. A specific 4977-bp deletion of mitochondrial DNA in human ageing skin. Arch. Dermatol. Res. 1994, 286, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.M.; Murphy, G.; Ralph, N.; O’Gorman, S.M.; Murphy, J.E.J. Mitochondrial DNA deletion percentage in sun exposed and non sun exposed skin. J. Photochem. Photobiol. B Biol. 2016, 165, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Eshaghian, A.; Vleugels, R.A.; Canter, J.A.; McDonald, M.A.; Stasko, T.; Sligh, J.E. Mitochondrial DNA Deletions Serve as Biomarkers of Aging in the Skin, but Are Typically Absent in Nonmelanoma Skin Cancers. J. Investig. Dermatol. 2006, 126, 336–344. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Birket, M.J.; Birch-Machin, M.A. Ultraviolet radiation exposure accelerates the accumulation of the aging-dependent T414G mitochondrial DNA mutation in human skin. Aging Cell 2007, 6, 557–564. [Google Scholar] [CrossRef]
- Coskun, P.E.; Beal, M.F.; Wallace, D.C. Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc. Natl. Acad. Sci. USA 2004, 101, 10726–10731. [Google Scholar] [CrossRef] [PubMed]
- Kassem, A.M.; El-Guendy, N.; Tantawy, M.; Abdelhady, H.; El-Ghor, A.; Wahab, A.H.A. Mutational Hotspots in the Mitochondrial D-Loop Region of Cancerous and Precancerous Colorectal Lesions in Egyptian Patients. DNA Cell Biol. 2011, 30, 899–906. [Google Scholar] [CrossRef]
- Murdock, D.G.; Christacos, N.C.; Wallace, D.C. The age-related accumulation of a mitochondrial DNA control region mutation in muscle, but not brain, detected by a sensitive PNA-directed PCR clamping based method. Nucleic Acids Res. 2000, 28, 4350–4355. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, Y.; Ye, K.; Picard, M.; Gu, Z. Independent impacts of aging on mitochondrial DNA quantity and quality in humans. BMC Genom. 2017, 18, 890. [Google Scholar] [CrossRef]
- Payne, B.A.I.; Chinnery, P.F. Mitochondrial dysfunction in aging: Much progress but many unresolved questions. Biochim. Biophys. Acta 2015, 1847, 1347–1353. [Google Scholar] [CrossRef]
- Rinnerthaler, M.; Bischof, J.; Streubel, M.K.; Trost, A.; Richter, K. Oxidative stress in aging human skin. Biomolecules 2015, 5, 545–589. [Google Scholar] [CrossRef]
- Lin, S.-J.; Kaeberlein, M.; Andalis, A.A.; Sturtz, L.A.; Defossez, P.-A.; Culotta, V.C.; Fink, G.R.; Guarente, L. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature 2002, 418, 344. [Google Scholar] [CrossRef]
- Bartke, A.; Wright, J.C.; Mattison, J.A.; Ingram, D.K.; Miller, R.A.; Roth, G.S. Extending the lifespan of long-lived mice. Nature 2001, 414, 412. [Google Scholar] [CrossRef]
- Lane, M.A.; Ingram, D.K.; Ball, S.S.; Roth, G.S. Dehydroepiandrosterone Sulfate: A Biomarker of Primate Aging Slowed by Calorie Restriction. J. Clin. Endocrinol. Metab. 1997, 82, 2093–2096. [Google Scholar] [CrossRef]
- Lanza, I.R.; Zabielski, P.; Klaus, K.A.; Morse, D.M.; Heppelmann, C.J.; Bergen, H.R., 3rd; Dasari, S.; Walrand, S.; Short, K.R.; Johnson, M.L.; et al. Chronic caloric restriction preserves mitochondrial function in senescence without increasing mitochondrial biogenesis. Cell Metab. 2012, 16, 777–788. [Google Scholar] [CrossRef]
- Mattison, J.A.; Roth, G.S.; Beasley, T.M.; Tilmont, E.M.; Handy, A.M.; Herbert, R.L.; Longo, D.L.; Allison, D.B.; Young, J.E.; Bryant, M.; et al. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature 2012, 489, 318. [Google Scholar] [CrossRef]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric Restriction Delays Disease Onset and Mortality in Rhesus Monkeys. Science (New York N. Y.) 2009, 325, 201–204. [Google Scholar] [CrossRef]
- Forni, M.F.; Peloggia, J.; Braga, T.T.; Chinchilla, J.E.O.; Shinohara, J.; Navas, C.A.; Camara, N.O.S.; Kowaltowski, A.J. Caloric Restriction Promotes Structural and Metabolic Changes in the Skin. Cell Rep. 2017, 20, 2678–2692. [Google Scholar] [CrossRef]
- Stier, A.; Bize, P.; Habold, C.; Bouillaud, F.; Massemin, S.; Criscuolo, F. Mitochondrial uncoupling prevents cold-induced oxidative stress: A case study using UCP1 knockout mice. J. Exp. Biol. 2014, 217, 624–630. [Google Scholar] [CrossRef]
- Weyemi, U.; Parekh, P.R.; Redon, C.E.; Bonner, W.M. SOD2 deficiency promotes aging phenotypes in mouse skin. Aging 2012, 4, 116–118. [Google Scholar] [CrossRef]
- Singh, B.; Schoeb, T.R.; Bajpai, P.; Slominski, A.; Singh, K.K. Reversing wrinkled skin and hair loss in mice by restoring mitochondrial function. Cell Death Dis. 2018, 9, 735. [Google Scholar] [CrossRef]
- McCully, K.K.; Fielding, R.A.; Evans, W.J.; Leigh, J.S., Jr.; Posner, J.D. Relationships between in vivo and in vitro measurements of metabolism in young and old human calf muscles. J. Appl. Physiol. (Bethesda Md. 1985) 1993, 75, 813–819. [Google Scholar] [CrossRef]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef]
- Niyazov, D.M.; Kahler, S.G.; Frye, R.E. Primary Mitochondrial Disease and Secondary Mitochondrial Dysfunction: Importance of Distinction for Diagnosis and Treatment. Mol. Syndromol. 2016, 7, 122–137. [Google Scholar] [CrossRef]
- Denat, L.; Kadekaro, A.L.; Marrot, L.; Leachman, S.A.; Abdel-Malek, Z.A. Melanocytes as instigators and victims of oxidative stress. J. Investig. Dermatol. 2014, 134, 1512–1518. [Google Scholar] [CrossRef]
- Song, X.; Mosby, N.; Yang, J.; Xu, A.; Abdel-Malek, Z.; Kadekaro, A.L. alpha-MSH activates immediate defense responses to UV-induced oxidative stress in human melanocytes. Pigment Cell Melanoma Res. 2009, 22, 809–818. [Google Scholar] [CrossRef]
- Liu, F.; Hamer, M.A.; Deelen, J.; Lall, J.S.; Jacobs, L.; van Heemst, D.; Murray, P.G.; Wollstein, A.; de Craen, A.J.; Uh, H.W.; et al. The MC1R Gene and Youthful Looks. Curr. Biol. CB 2016, 26, 1213–1220. [Google Scholar] [CrossRef]
- Böhm, M.; Hill, H.Z. Ultraviolet B, melanin and mitochondrial DNA: Photo-damage in human epidermal keratinocytes and melanocytes modulated by alpha-melanocyte-stimulating hormone. F1000 Res. 2016, 5, 881. [Google Scholar] [CrossRef]
- Snyder, J.R.; Hall, A.; Ni-Komatsu, L.; Khersonsky, S.M.; Chang, Y.-T.; Orlow, S.J. Dissection of Melanogenesis with Small Molecules Identifies Prohibitin as a Regulator. Chem. Biol. 2005, 12, 477–484. [Google Scholar] [CrossRef]
- Daniele, T.; Hurbain, I.; Vago, R.; Casari, G.; Raposo, G.; Tacchetti, C.; Schiaffino, M.V. Mitochondria and melanosomes establish physical contacts modulated by Mfn2 and involved in organelle biogenesis. Curr. Biol. CB 2014, 24, 393–403. [Google Scholar] [CrossRef]
- Slominski, A.T.; Semak, I.; Fischer, T.W.; Kim, T.-K.; Kleszczyński, K.; Hardeland, R.; Reiter, R.J. Metabolism of melatonin in the skin: Why is it important? Exp. Dermatol. 2017, 26, 563–568. [Google Scholar] [CrossRef]
- Slominski, A.; Pruski, D. Melatonin inhibits proliferation and melanogenesis in rodent melanoma cells. Exp. Cell Res. 1993, 206, 189–194. [Google Scholar] [CrossRef]
- Lerner, A.B.; Nordlund, J.J. The Effects of Oral Melatonin on Skin Color and on the Release of Pituitary Hormones. J. Clin. Endocrinol. Metab. 1977, 45, 768–774. [Google Scholar] [CrossRef]
- McElhinney, D.B.; Hoffman, S.J.; Robinson, W.A.; Ferguson, J.A.N. Effect of Melatonin on Human Skin Color. J. Investig. Dermatol. 1994, 102, 258–259. [Google Scholar] [CrossRef]
- Kim, T.-K.; Lin, Z.; Tidwell, W.J.; Li, W.; Slominski, A.T. Melatonin and its metabolites accumulate in the human epidermis in vivo and inhibit proliferation and tyrosinase activity in epidermal melanocytes in vitro. Mol. Cell. Endocrinol. 2015, 404, 1–8. [Google Scholar] [CrossRef]
- Dell’Anna, M.L.; Ottaviani, M.; Kovacs, D.; Mirabilii, S.; Brown, D.A.; Cota, C.; Migliano, E.; Bastonini, E.; Bellei, B.; Cardinali, G.; et al. Energetic mitochondrial failing in vitiligo and possible rescue by cardiolipin. Sci. Rep. 2017, 7, 13663. [Google Scholar] [CrossRef]
- Karvonen, S.L.; Haapasaari, K.M.; Kallioinen, M.; Oikarinen, A.; Hassinen, I.E.; Majamaa, K. Increased prevalence of vitiligo, but no evidence of premature ageing, in the skin of patients with bp 3243 mutation in mitochondrial DNA in the mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes syndrome (MELAS). Br. J. Dermatol. 1999, 140, 634–639. [Google Scholar] [CrossRef]
- Rünger, T.M.; Epe, B.; Möller, K. Processing of Directly and Indirectly Ultraviolet-Induced DNA Damage in Human Cells. Recent Results Cancer Res. 1995, 139, 31–42. [Google Scholar]
- Sklar, L.R.; Almutawa, F.; Lim, H.W.; Hamzavi, I. Effects of ultraviolet radiation, visible light, and infrared radiation on erythema and pigmentation: A review. Photochem. Photobiol. Sci. 2013, 12, 54–64. [Google Scholar] [CrossRef]
- Swalwell, H.; Latimer, J.; Haywood, R.M.; Birch-Machin, M.A. Investigating the role of melanin in UVA/UVB- and hydrogen peroxide-induced cellular and mitochondrial ROS production and mitochondrial DNA damage in human melanoma cells. Free Radic. Biol. Med. 2012, 52, 626–634. [Google Scholar] [CrossRef]
- Wenczl, E.; van der Schans, G.P.; Roza, L.; Kolb, R.; Smit, N.; Schothorst, A.A. UVA-induced oxidative DNA damage in human melanocytes is related to their (pheo)melanin content. J. Dermatol. Sci. 1998, 16, S221. [Google Scholar] [CrossRef]
- Martinez-Levasseur, L.M.; Birch-Machin, M.A.; Bowman, A.; Gendron, D.; Weatherhead, E.; Knell, R.J.; Acevedo-Whitehouse, K. Whales use distinct strategies to counteract solar ultraviolet radiation. Sci. Rep. 2013, 3, 2386. [Google Scholar] [CrossRef]
- Latimer, J.A.; Lloyd, J.J.; Diffey, B.L.; Matts, P.J.; Birch-Machin, M.A. Determination of the Action Spectrum of UVR-Induced Mitochondrial DNA Damage in Human Skin Cells. J. Investig. Dermatol. 2015, 135, 2512–2518. [Google Scholar] [CrossRef]
- Rashdan, E.; Wilkinson, S.; Birch-Machin, M. British Society for Investigative Dermatology Annual Meeting 2018 26–28 March 2018 Blizard Institute, Barts and The London School of Medicine and Dentistry. Br. J. Dermatol. 2018. [Google Scholar] [CrossRef]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458. [Google Scholar] [CrossRef]
- Saretzki, G.; Zglinicki, T. Replicative Aging, Telomeres, and Oxidative Stress. Ann. N. Y. Acad. Sci. 2006, 959, 24–29. [Google Scholar] [CrossRef]
- Samani, N.J.; Boultby, R.; Butler, R.; Thompson, J.R.; Goodall, A.H. Telomere shortening in atherosclerosis. Lancet 2001, 358, 472–473. [Google Scholar] [CrossRef]
- Ma, H.M.; Liu, W.; Zhang, P.; Yuan, X.Y. Human skin fibroblast telomeres are shortened after ultraviolet irradiation. J. Int. Med. Res. 2012, 40, 1871–1877. [Google Scholar] [CrossRef]
- Nistico, S.; Ehrlich, J.; Gliozzi, M.; Maiuolo, J.; Del Duca, E.; Muscoli, C.; Mollace, V. Telomere and telomerase modulation by bergamot polyphenolic fraction in experimental photoageing in human keratinocytes. J. Biol. Regul. Homeost. Agents 2015, 29, 723–728. [Google Scholar]
- Bowman, A.; Birch-Machin, M.A. Age-Dependent Decrease of Mitochondrial Complex II Activity in Human Skin Fibroblasts. J. Investig. Dermatol. 2016, 136, 912–919. [Google Scholar] [CrossRef]
- Krishnan, K.J.; Harbottle, A.; Birch-Machin, M.A. The Use of a 3895 bp Mitochondrial DNA Deletion as a Marker for Sunlight Exposure in Human Skin. J. Investig. Dermatol. 2004, 123, 1020–1024. [Google Scholar] [CrossRef]
- Berneburg, M.; Gattermann, N.; Stege, H.; Grewe, M.; Vogelsang, K.; Ruzicka, T.; Krutmann, J. Chronically Ultraviolet-exposed Human Skin Shows a Higher Mutation Frequency of Mitochondrial DNA as Compared to Unexposed Skin and the Hematopoietic System. Photochem. Photobiol. 1997, 66, 271–275. [Google Scholar] [CrossRef]
- Manicone, A.M.; McGuire, J.K. Matrix metalloproteinases as modulators of inflammation. Semin. Cell Dev. Biol. 2008, 19, 34–41. [Google Scholar] [CrossRef]
- Vicentini, F.T.; He, T.; Shao, Y.; Fonseca, M.J.; Verri, W.A., Jr.; Fisher, G.J.; Xu, Y. Quercetin inhibits UV irradiation-induced inflammatory cytokine production in primary human keratinocytes by suppressing NF-kappaB pathway. J. Dermatol. Sci. 2011, 61, 162–168. [Google Scholar] [CrossRef]
- Kang, S.; Chung, J.H.; Lee, J.H.; Fisher, G.J.; Wan, Y.S.; Duell, E.A.; Voorhees, J.J. Topical N-Acetyl Cysteine and Genistein Prevent Ultraviolet-Light-Induced Signaling That Leads to Photoaging in Human Skin in vivo. J. Investig. Dermatol. 2003, 120, 835–841. [Google Scholar] [CrossRef]
- Tewari, A.; Grys, K.; Kollet, J.; Sarkany, R.; Young, A.R. Upregulation of MMP12 and its activity by UVA1 in human skin: Potential implications for photoaging. J. Investig. Dermatol. 2014, 134, 2598–2609. [Google Scholar] [CrossRef]
- Chiang, H.M.; Chen, H.C.; Lin, T.J.; Shih, I.C.; Wen, K.C. Michelia alba extract attenuates UVB-induced expression of matrix metalloproteinases via MAP kinase pathway in human dermal fibroblasts. Food Chem. Toxicol. 2012, 50, 4260–4269. [Google Scholar] [CrossRef]
- Fisher, G.J.; Datta, S.; Wang, Z.; Li, X.Y.; Quan, T.; Chung, J.H.; Kang, S.; Voorhees, J.J. c-Jun-dependent inhibition of cutaneous procollagen transcription following ultraviolet irradiation is reversed by all-trans retinoic acid. J. Clin. Investig. 2000, 106, 663–670. [Google Scholar] [CrossRef]
- Edwards, C.; Pearse, A.; Marks, R.; Nishimori, Y.; Matsumoto, K.; Kawai, M. Degenerative Alterations of Dermal Collagen Fiber Bundles in Photodamaged Human Skin and UV-Irradiated Hairless Mouse Skin: Possible Effect on Decreasing Skin Mechanical Properties and Appearance of Wrinkles. J. Investig. Dermatol. 2001, 117, 1458–1463. [Google Scholar] [CrossRef]
- Schieke, S.M.; Stege, H.; Kürten, V.; Grether-Beck, S.; Sies, H.; Krutmann, J. Infrared-A Radiation-Induced Matrix Metalloproteinase 1 Expression is Mediated Through Extracellular Signal-regulated Kinase 1/2 Activation in Human Dermal Fibroblasts. J. Investig. Dermatol. 2002, 119, 1323–1329. [Google Scholar] [CrossRef]
- Szewczyk, G.; Zadlo, A.; Sarna, M.; Ito, S.; Wakamatsu, K.; Sarna, T. Aerobic photoreactivity of synthetic eumelanins and pheomelanins: Generation of singlet oxygen and superoxide anion. Pigment Cell Melanoma Res. 2016, 29, 669–678. [Google Scholar] [CrossRef]
- Maresca, V.; Flori, E.; Briganti, S.; Camera, E.; Cario-André, M.; Taïeb, A.; Picardo, M. UVA-Induced Modification of Catalase Charge Properties in the Epidermis Is Correlated with the Skin Phototype. J. Investig. Dermatol. 2006, 126, 182–190. [Google Scholar] [CrossRef]
- Elfakir, A.; Ezzedine, K.; Latreille, J.; Ambroisine, L.; Jdid, R.; Galan, P.; Hercberg, S.; Gruber, F.; Malvy, D.; Tschachler, E.; et al. Functional MC1R-Gene Variants Are Associated with Increased Risk for Severe Photoaging of Facial Skin. J. Investig. Dermatol. 2010, 130, 1107–1115. [Google Scholar] [CrossRef]
- Piao, M.J.; Ahn, M.J.; Kang, K.A.; Ryu, Y.S.; Hyun, Y.J.; Shilnikova, K.; Zhen, A.X.; Jeong, J.W.; Choi, Y.H.; Kang, H.K.; et al. Particulate matter 2.5 damages skin cells by inducing oxidative stress, subcellular organelle dysfunction, and apoptosis. Arch. Toxicol. 2018, 92, 2077–2091. [Google Scholar] [CrossRef]
- Valacchi, G.; van der Vliet, A.; Schock, B.C.; Okamoto, T.; Obermuller-Jevic, U.; Cross, C.E.; Packer, L. Ozone exposure activates oxidative stress responses in murine skin. Toxicology 2002, 179, 163–170. [Google Scholar] [CrossRef]
- Ding, A.; Yang, Y.; Zhao, Z.; Hüls, A.; Vierkötter, A.; Yuan, Z.; Cai, J.; Zhang, J.; Gao, W.; Li, J.; et al. Indoor PM2.5 exposure affects skin aging manifestation in a Chinese population. Sci. Rep. 2017, 7, 15329. [Google Scholar] [CrossRef]
- Vierkötter, A.; Schikowski, T.; Ranft, U.; Sugiri, D.; Matsui, M.; Krämer, U.; Krutmann, J. Airborne Particle Exposure and Extrinsic Skin Aging. J. Investig. Dermatol. 2010, 130, 2719–2726. [Google Scholar] [CrossRef]
- Gualtieri, M.; Øvrevik, J.; Mollerup, S.; Asare, N.; Longhin, E.; Dahlman, H.-J.; Camatini, M.; Holme, J.A. Airborne urban particles (Milan winter-PM2.5) cause mitotic arrest and cell death: Effects on DNA, mitochondria, AhR binding and spindle organization. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2011, 713, 18–31. [Google Scholar] [CrossRef]
- Afaq, F.; Zaid, M.A.; Pelle, E.; Khan, N.; Syed, D.N.; Matsui, M.S.; Maes, D.; Mukhtar, H. Aryl Hydrocarbon Receptor Is an Ozone Sensor in Human Skin. J. Investig. Dermatol. 2009, 129, 2396–2403. [Google Scholar] [CrossRef]
- Koohgoli, R.; Hudson, L.; Naidoo, K.; Wilkinson, S.; Chavan, B.; Birch-Machin, M.A. Bad air gets under your skin. Exp. Dermatol. 2017, 26, 384–387. [Google Scholar] [CrossRef]
- Shimada, T.; Fujii-Kuriyama, Y. Metabolic activation of polycyclic aromatic hydrocarbons to carcinogens by cytochromes P450 1A1 and 1B1. Cancer Sci. 2004, 95, 1–6. [Google Scholar] [CrossRef]
- Bastiaens, M.; Hoefnagel, J.; Westendorp, R.; Vermeer, B.-J.; Bouwes Bavinck, J.N. Solar Lentigines are Strongly Related to Sun Exposure in Contrast to Ephelides. Pigment Cell Res. 2004, 17, 225–229. [Google Scholar] [CrossRef]
- Hüls, A.; Vierkötter, A.; Gao, W.; Krämer, U.; Yang, Y.; Ding, A.; Stolz, S.; Matsui, M.; Kan, H.; Wang, S.; et al. Traffic-Related Air Pollution Contributes to Development of Facial Lentigines: Further Epidemiological Evidence from Caucasians and Asians. J. Investig. Dermatol. 2016, 136, 1053–1056. [Google Scholar] [CrossRef]
- Chang, A.L.S. Expanding Our Understanding of Human Skin Aging. J. Investig. Dermatol. 2016, 136, 897–899. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Boulton, S.J.; Birch-Machin, M.A. Impact of hyperpigmentation on superoxide flux and melanoma cell metabolism at mitochondrial complex II. FASEB J. 2015, 29, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Arck, P.C.; Overall, R.; Spatz, K.; Liezman, C.; Handjiski, B.; Klapp, B.F.; Birch-Machin, M.A.; Peters, E.M.J. Towards a “free radical theory of graying”: Melanocyte apoptosis in the aging human hair follicle is an indicator of oxidative stress induced tissue damage. FASEB J. 2006, 20, 1567–1569. [Google Scholar] [CrossRef]
- Commo, S.; Gallard, O.; Bernard, B.A. Human hair greying is linked to a specific depletion of hair follicle melanocytes affecting both the bulb and the outer root sheath. Br. J. Dermatol. 2004, 150, 435–443. [Google Scholar]
- Veis, D.J.; Sorenson, C.M.; Shutter, J.R.; Korsmeyer, S.J. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 1993, 75, 229–240. [Google Scholar] [CrossRef]
- Jo, S.K.; Lee, J.Y.; Lee, Y.; Kim, C.D.; Lee, J.-H.; Lee, Y.H. Three Streams for the Mechanism of Hair Graying. Ann. Dermatol. 2018, 30, 397–401. [Google Scholar] [CrossRef]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, -Y.M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417. [Google Scholar] [CrossRef]
- Trüeb, R.M. Oxidative stress in ageing of hair. Int. J. Trichol. 2009, 1, 6–14. [Google Scholar] [CrossRef]
- Bahta, A.W.; Farjo, N.; Farjo, B.; Philpott, M.P. Premature Senescence of Balding Dermal Papilla Cells In Vitro Is Associated with p16INK4a Expression. J. Investig. Dermatol. 2008, 128, 1088–1094. [Google Scholar] [CrossRef]
- Kloepper, J.E.; Baris, O.R.; Reuter, K.; Kobayashi, K.; Weiland, D.; Vidali, S.; Tobin, D.J.; Niemann, C.; Wiesner, R.J.; Paus, R. Mitochondrial Function in Murine Skin Epithelium Is Crucial for Hair Follicle Morphogenesis and Epithelial–Mesenchymal Interactions. J. Investig. Dermatol. 2015, 135, 679–689. [Google Scholar] [CrossRef]
- Lemasters, J.J.; Ramshesh, V.K.; Lovelace, G.L.; Lim, J.; Wright, G.D.; Harland, D.; Dawson, T.L. Compartmentation of Mitochondrial and Oxidative Metabolism in Growing Hair Follicles: A Ring of Fire. J. Investig. Dermatol. 2017, 137, 1434–1444. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stout, R.; Birch-Machin, M. Mitochondria’s Role in Skin Ageing. Biology 2019, 8, 29. https://doi.org/10.3390/biology8020029
Stout R, Birch-Machin M. Mitochondria’s Role in Skin Ageing. Biology. 2019; 8(2):29. https://doi.org/10.3390/biology8020029
Chicago/Turabian StyleStout, Roisin, and Mark Birch-Machin. 2019. "Mitochondria’s Role in Skin Ageing" Biology 8, no. 2: 29. https://doi.org/10.3390/biology8020029
APA StyleStout, R., & Birch-Machin, M. (2019). Mitochondria’s Role in Skin Ageing. Biology, 8(2), 29. https://doi.org/10.3390/biology8020029