
Clinical Profile, Arrhythmias, and Adverse Cardiac Outcomes in Emery–Dreifuss Muscular Dystrophies: A Systematic Review of the Literature


 ,
, 
 , and
, and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
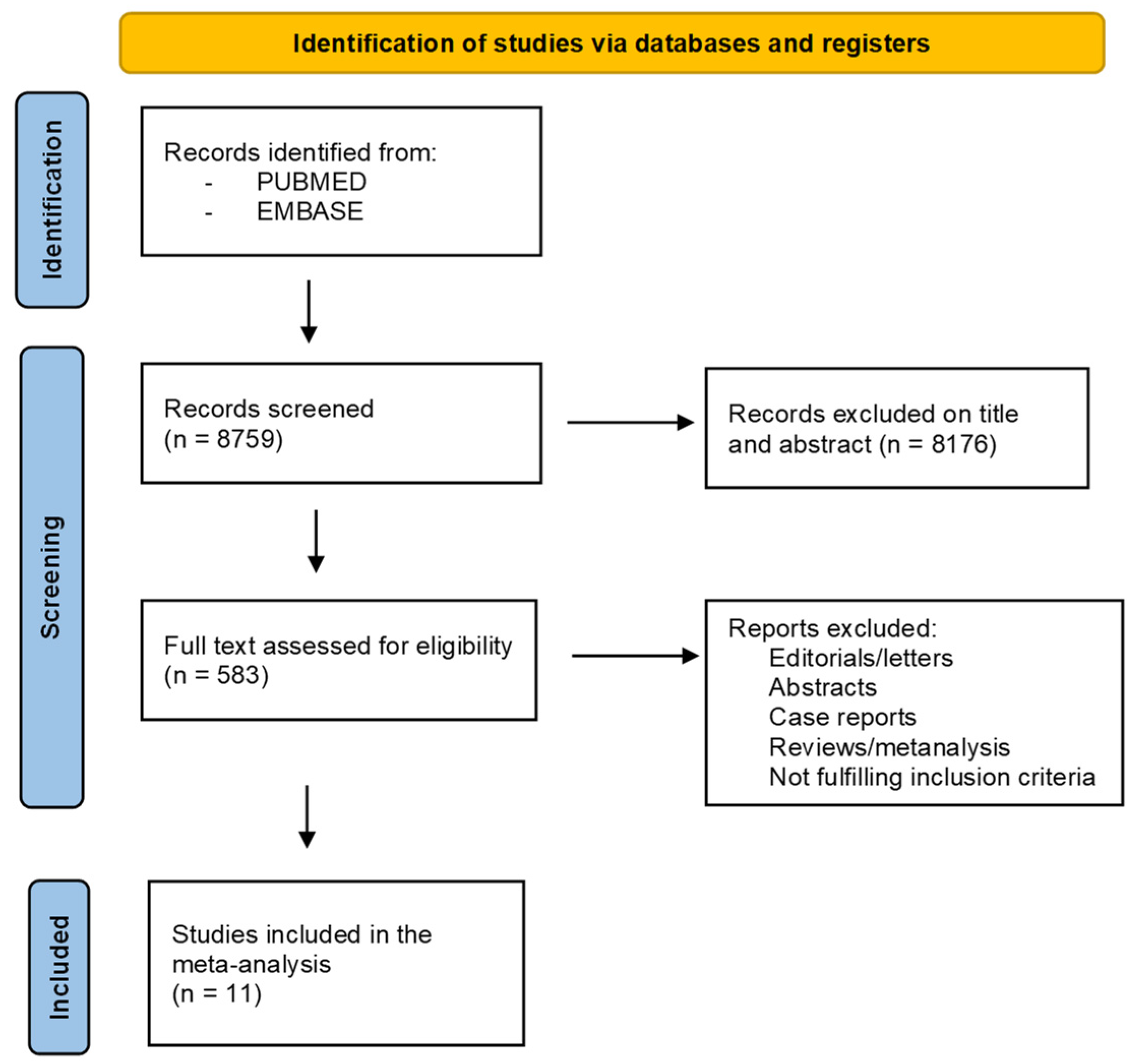
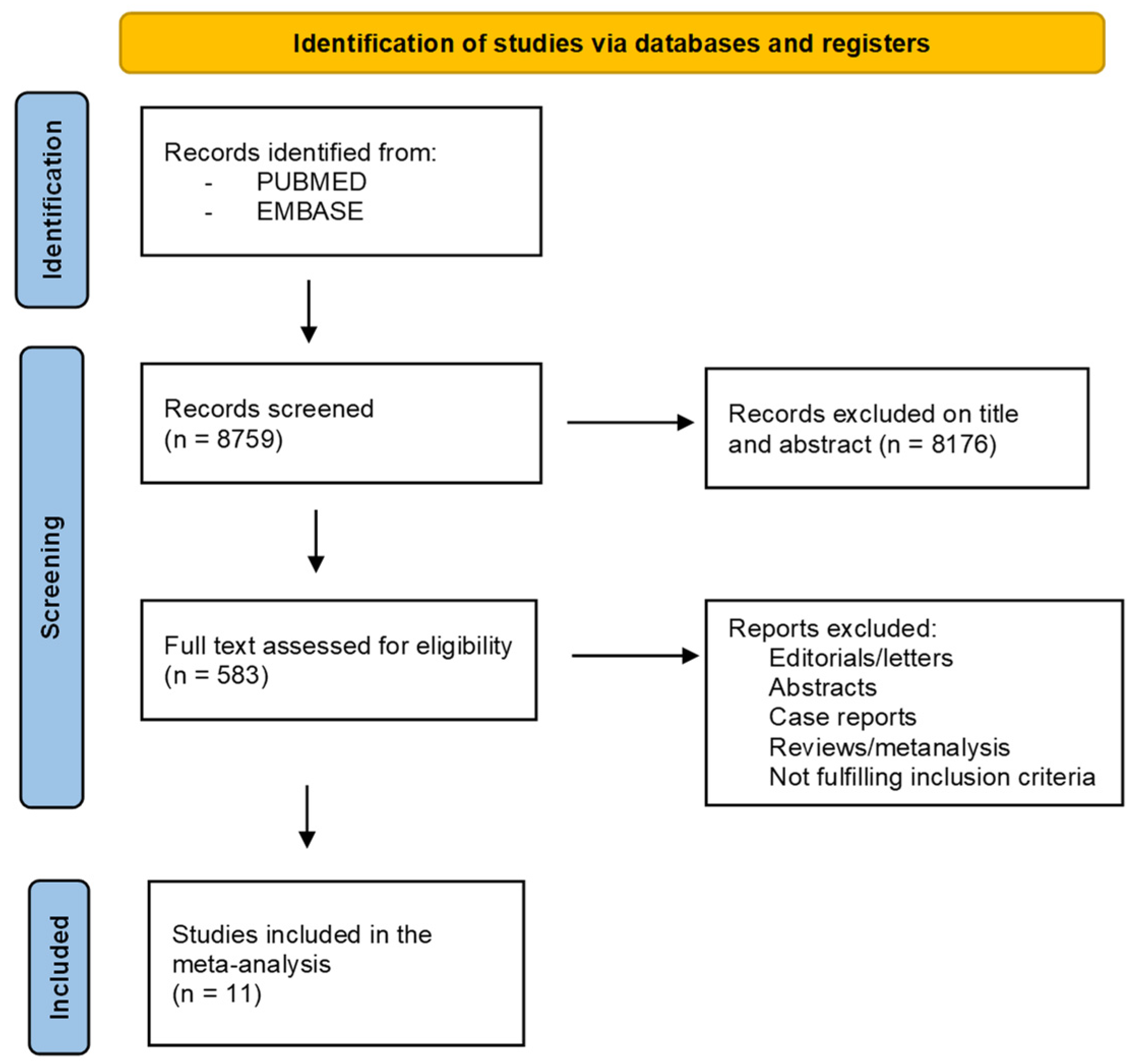
3.1. Literature Search and Study Screening
3.2. Methodological Quality of the Data
3.3. Atrial Arrhythmias
3.4. Ventricular Arrhythmias and Sudden Cardiac Death
3.5. Conduction Disturbances
3.6. Thromboembolic Events and Stroke
3.7. Ventricular Dysfunction, Heart Failure and Heart Transplantation
3.8. All-Cause Death and Cardiovascular Mortality
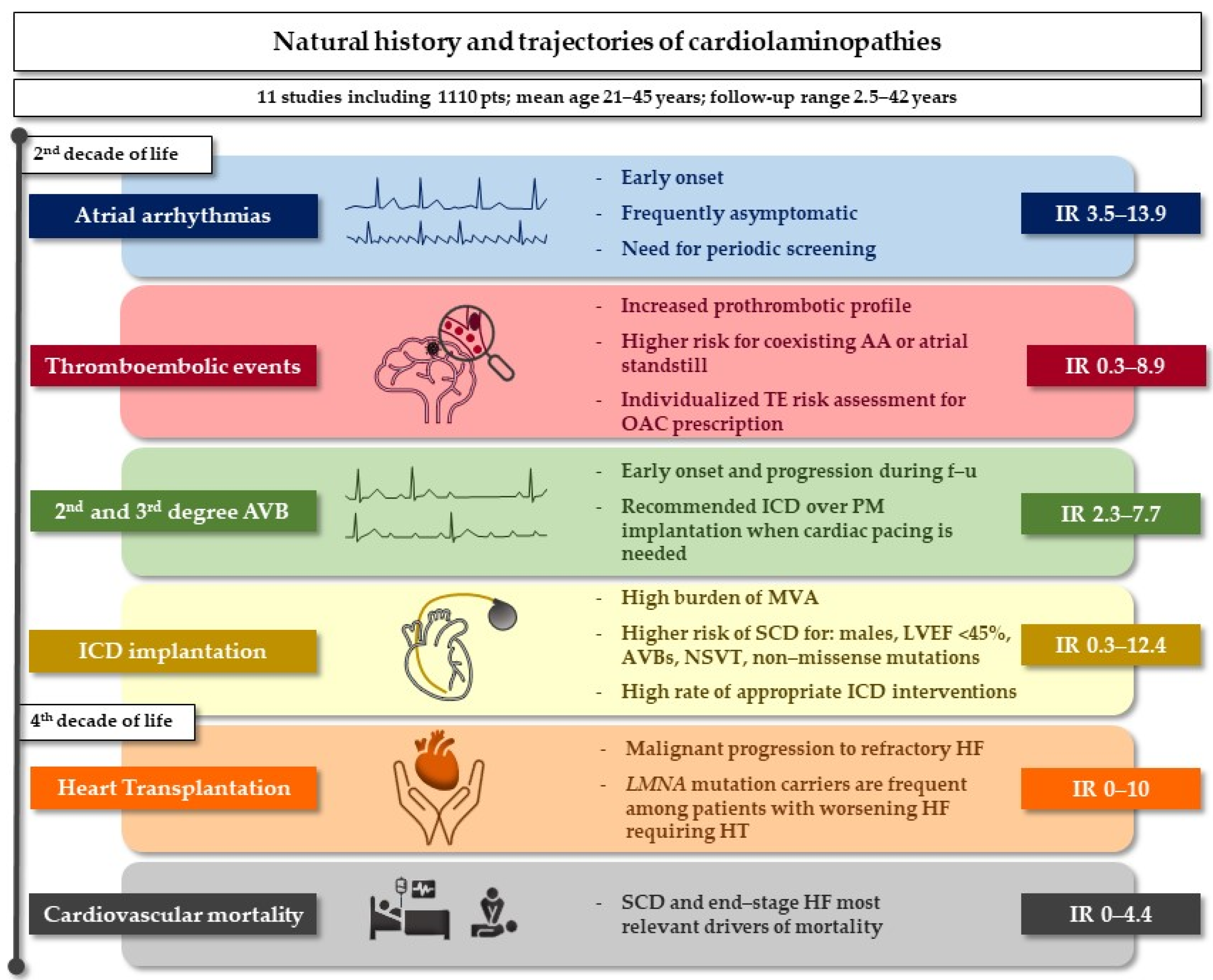
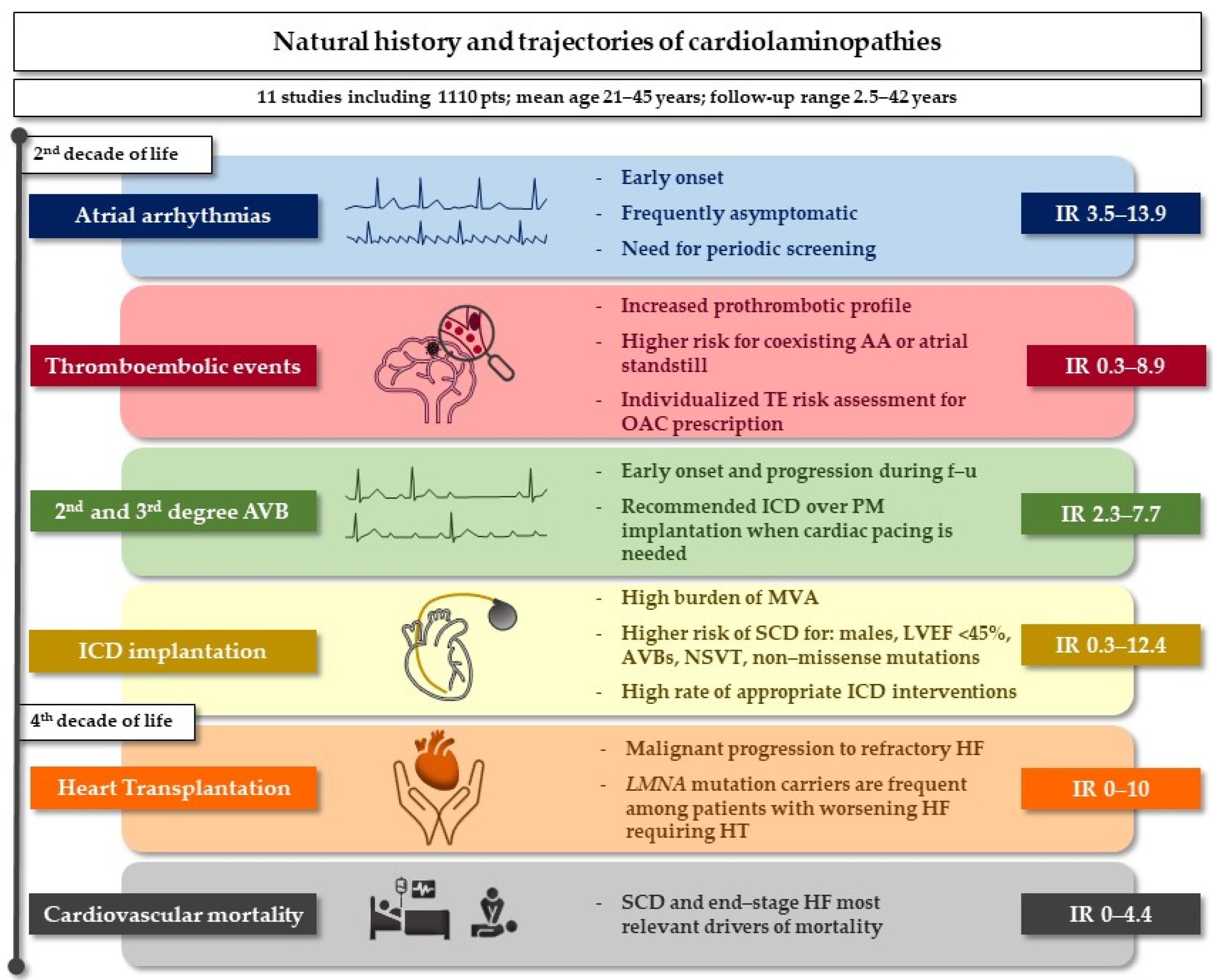
4. Discussion
4.1. Atrial Arrhythmias
4.2. Thromboembolic Risk
4.3. Conduction Disturbances and Malignant Ventricular Arrhythmias
4.4. Systolic Dysfunction and Heart Transplantation
4.5. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Emery, A.E.; Dreifuss, F.E. Unusual type of benign x-linked muscular dystrophy. J. Neurol. Neurosurg. Psychiatry 1966, 29, 338–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bione, S.; Maestrini, E.; Rivella, S.; Mancini, M.; Regis, S.; Romeo, G.; Toniolo, D. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat. Genet. 1994, 8, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Bonne, G.; Di Barletta, M.R.; Varnous, S.; Bécane, H.M.; Hammouda, E.H.; Merlini, L.; Muntoni, F.; Greenberg, C.R.; Gary, F.; Urtizberea, J.A.; et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet. 1999, 21, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Fatkin, D.; MacRae, C.; Sasaki, T.; Wolff, M.R.; Porcu, M.; Frenneaux, M.; Atherton, J.; Vidaillet, H.J.; Spudich, S.; De Girolami, U.; et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 1999, 341, 1715–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Bethmann, C.; Worth, N.F.; Davies, J.D.; Wasner, C.; Feuer, A.; Ragnauth, C.D.; Yi, Q.; Mellad, J.A.; Warren, D.T.; et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum. Mol. Genet. 2007, 16, 2816–2833. [Google Scholar] [CrossRef]
- Zhou, C.; Li, C.; Zhou, B.; Sun, H.; Koullourou, V.; Holt, I.; Puckelwartz, M.J.; Warren, D.T.; Hayward, R.; Lin, Z.; et al. Novel nesprin-1 mutations associated with dilated cardiomyopathy cause nuclear envelope disruption and defects in myogenesis. Hum. Mol. Genet. 2017, 26, 2258–2276. [Google Scholar] [CrossRef]
- Meinke, P.; Mattioli, E.; Haque, F.; Antoku, S.; Columbaro, M.; Straatman, K.R.; Worman, H.J.; Gundersen, G.G.; Lattanzi, G.; Wehnert, M.; et al. Muscular dystrophy-associated SUN1 and SUN2 variants disrupt nuclear-cytoskeletal connections and myonuclear organization. PLoS Genet. 2014, 10, e1004605. [Google Scholar] [CrossRef] [Green Version]
- Gueneau, L.; Bertrand, A.T.; Jais, J.P.; Salih, M.A.; Stojkovic, T.; Wehnert, M.; Hoeltzenbein, M.; Spuler, S.; Saitoh, S.; Verschueren, A.; et al. Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. Am. J. Hum. Genet. 2009, 85, 338–353. [Google Scholar] [CrossRef] [Green Version]
- Muchir, A.; Worman, H.J. Emery-Dreifuss muscular dystrophy: Focal point nuclear envelope. Curr. Opin. Neurol. 2019, 32, 728–734. [Google Scholar] [CrossRef]
- Lammerding, J.; Schulze, P.C.; Takahashi, T.; Kozlov, S.; Sullivan, T.; Kamm, R.D.; Stewart, C.L.; Lee, R.T. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin. Investig. 2004, 113, 370–378. [Google Scholar] [CrossRef] [Green Version]
- Hutchison, C.J. Lamins: Building blocks or regulators of gene expression? Nat. Rev. Mol. Cell Biol. 2002, 3, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.; Ostlund, C.; Stewart, C.L.; Man, N.; Worman, H.J.; Morris, G.E. Effect of pathogenic mis-sense mutations in lamin A on its interaction with emerin in vivo. J. Cell Sci. 2003, 116, 3027–3035. [Google Scholar] [CrossRef] [Green Version]
- Cenni, V.; Squarzoni, S.; Loi, M.; Mattioli, E.; Lattanzi, G.; Capanni, C. Emerin Phosphorylation during the Early Phase of the Oxidative Stress Response Influences Emerin-BAF Interaction and BAF Nuclear Localization. Cells 2020, 9, 1415. [Google Scholar] [CrossRef] [PubMed]
- Camozzi, D.; Capanni, C.; Cenni, V.; Mattioli, E.; Columbaro, M.; Squarzoni, S.; Lattanzi, G. Diverse lamin-dependent mechanisms interact to control chromatin dynamics. Focus on laminopathies. Nucleus 2014, 5, 427–440. [Google Scholar] [CrossRef] [Green Version]
- Mattioli, E.; Columbaro, M.; Capanni, C.; Maraldi, N.M.; Cenni, V.; Scotlandi, K.; Marino, M.T.; Merlini, L.; Squarzoni, S.; Lattanzi, G. Prelamin A-mediated recruitment of SUN1 to the nuclear envelope directs nuclear positioning in human muscle. Cell Death Differ. 2011, 18, 1305–1315. [Google Scholar] [CrossRef]
- Roncarati, R.; Viviani Anselmi, C.; Krawitz, P.; Lattanzi, G.; von Kodolitsch, Y.; Perrot, A.; di Pasquale, E.; Papa, L.; Portararo, P.; Columbaro, M.; et al. Doubly heterozygous LMNA and TTN mutations revealed by exome sequencing in a severe form of dilated cardiomyopathy. Eur. J. Hum. Genet. 2013, 21, 1105–1111. [Google Scholar] [CrossRef] [Green Version]
- Cenni, V.; Capanni, C.; Mattioli, E.; Schena, E.; Squarzoni, S.; Bacalini, M.G.; Garagnani, P.; Salvioli, S.; Franceschi, C.; Lattanzi, G. Lamin A involvement in ageing processes. Ageing Res. Rev. 2020, 62, 101073. [Google Scholar] [CrossRef]
- Chen, S.N.; Lombardi, R.; Karmouch, J.; Tsai, J.Y.; Czernuszewicz, G.; Taylor, M.R.G.; Mestroni, L.; Coarfa, C.; Gurha, P.; Marian, A.J. DNA Damage Response/TP53 Pathway Is Activated and Contributes to the Pathogenesis of Dilated Cardiomyopathy Associated with LMNA (Lamin A/C) Mutations. Circ. Res. 2019, 124, 856–873. [Google Scholar] [CrossRef]
- González, J.M.; Navarro-Puche, A.; Casar, B.; Crespo, P.; Andrés, V. Fast regulation of AP-1 activity through interaction of lamin A/C, ERK1/2, and c-Fos at the nuclear envelope. J. Cell Biol. 2008, 183, 653–666. [Google Scholar] [CrossRef] [Green Version]
- Le Dour, C.; Macquart, C.; Sera, F.; Homma, S.; Bonne, G.; Morrow, J.P.; Worman, H.J.; Muchir, A. Decreased WNT/β-catenin signalling contributes to the pathogenesis of dilated cardiomyopathy caused by mutations in the lamin a/C gene. Hum. Mol. Genet. 2017, 26, 333–343. [Google Scholar] [CrossRef]
- Shin, J.Y.; Worman, H.J. Molecular Pathology of Laminopathies. Annu. Rev. Pathol. 2022, 17, 159–180. [Google Scholar] [CrossRef]
- Sullivan, T.; Escalante-Alcalde, D.; Bhatt, H.; Anver, M.; Bhat, N.; Nagashima, K.; Stewart, C.L.; Burke, B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 1999, 147, 913–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fidziańska, A.; Toniolo, D.; Hausmanowa-Petrusewicz, I. Ultrastructural abnormality of sarcolemmal nuclei in Emery-Dreifuss muscular dystrophy (EDMD). J. Neurol. Sci. 1998, 159, 88–93. [Google Scholar] [CrossRef]
- Nikolova, V.; Leimena, C.; McMahon, A.C.; Tan, J.C.; Chandar, S.; Jogia, D.; Kesteven, S.H.; Michalicek, J.; Otway, R.; Verheyen, F.; et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J. Clin. Investig. 2004, 113, 357–369. [Google Scholar] [CrossRef]
- Wong, X.; Stewart, C.L. The Laminopathies and the Insights They Provide into the Structural and Functional Organization of the Nucleus. Annu. Rev. Genom. Hum. Genet. 2020, 21, 263–288. [Google Scholar] [CrossRef] [PubMed]
- Salvarani, N.; Crasto, S.; Miragoli, M.; Bertero, A.; Paulis, M.; Kunderfranco, P.; Serio, S.; Forni, A.; Lucarelli, C.; Dal Ferro, M.; et al. The K219T-Lamin mutation induces conduction defects through epigenetic inhibition of SCN5A in human cardiac laminopathy. Nat. Commun. 2019, 10, 2267. [Google Scholar] [CrossRef] [Green Version]
- Boriani, G.; Gallina, M.; Merlini, L.; Bonne, G.; Toniolo, D.; Amati, S.; Biffi, M.; Martignani, C.; Frabetti, L.; Bonvicini, M.; et al. Clinical relevance of atrial fibrillation/flutter, stroke, pacemaker implant, and heart failure in Emery-Dreifuss muscular dystrophy: A long-term longitudinal study. Stroke 2003, 34, 901–908. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Baldinger, S.H.; Gandjbakhch, E.; Maury, P.; Sellal, J.M.; Androulakis, A.F.; Waintraub, X.; Charron, P.; Rollin, A.; Richard, P.; et al. Long-Term Arrhythmic and Nonarrhythmic Outcomes of Lamin A/C Mutation Carriers. J. Am. Coll. Cardiol. 2016, 68, 2299–2307. [Google Scholar] [CrossRef]
- Hasselberg, N.E.; Haland, T.F.; Saberniak, J.; Brekke, P.H.; Berge, K.E.; Leren, T.P.; Edvardsen, T.; Haugaa, K.H. Lamin A/C cardiomyopathy: Young onset, high penetrance, and frequent need for heart transplantation. Eur. Heart J. 2018, 39, 853–860. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Stang, A. Critical evaluation of the Newcastle-Ottawa scale for the assessment of the quality of nonrandomized studies in meta-analyses. Eur. J. Epidemiol 2010, 25, 603–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Rijsingen, I.A.; Arbustini, E.; Elliott, P.M.; Mogensen, J.; Hermans-van Ast, J.F.; van der Kooi, A.J.; van Tintelen, J.P.; van den Berg, M.P.; Pilotto, A.; Pasotti, M.; et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J. Am. Coll. Cardiol. 2012, 59, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Anselme, F.; Moubarak, G.; Savouré, A.; Godin, B.; Borz, B.; Drouin-Garraud, V.; Gay, A. Implantable cardioverter-defibrillators in lamin A/C mutation carriers with cardiac conduction disorders. Heart Rhythm 2013, 10, 1492–1498. [Google Scholar] [CrossRef] [PubMed]
- van Rijsingen, I.A.; Bakker, A.; Azim, D.; Hermans-van Ast, J.F.; van der Kooi, A.J.; van Tintelen, J.P.; van den Berg, M.P.; Christiaans, I.; Lekanne Dit Deprez, R.H.; Wilde, A.A.; et al. Lamin A/C mutation is independently associated with an increased risk of arterial and venous thromboembolic complications. Int. J. Cardiol. 2013, 168, 472–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, K.; Aiba, T.; Makiyama, T.; Nishiuchi, S.; Ohno, S.; Kato, K.; Yamamoto, Y.; Doi, T.; Shizuta, S.; Onoue, K.; et al. Clinical Manifestations and Long-Term Mortality in Lamin A/C Mutation Carriers from a Japanese Multicenter Registry. Circ. J. 2018, 82, 2707–2714. [Google Scholar] [CrossRef] [Green Version]
- Peretto, G.; Di Resta, C.; Perversi, J.; Forleo, C.; Maggi, L.; Politano, L.; Barison, A.; Previtali, S.C.; Carboni, N.; Brun, F.; et al. Cardiac and Neuromuscular FeatuRes. of Patients with LMNA-Related Cardiomyopathy. Ann. Intern. Med. 2019, 171, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Ditaranto, R.; Boriani, G.; Biffi, M.; Lorenzini, M.; Graziosi, M.; Ziacchi, M.; Pasquale, F.; Vitale, G.; Berardini, A.; Rinaldi, R.; et al. Differences in cardiac phenotype and natural history of laminopathies with and without neuromuscular onset. Orphanet J. Rare Dis. 2019, 14, 263. [Google Scholar] [CrossRef]
- Marchel, M.; Madej-Pilarczyk, A.; Tymińska, A.; Steckiewicz, R.; Ostrowska, E.; Wysińska, J.; Russo, V.; Grabowski, M.; Opolski, G. Cardiac Arrhythmias in Muscular Dystrophies Associated with Emerinopathy and Laminopathy: A Cohort Study. J. Clin. Med. 2021, 10, 732. [Google Scholar] [CrossRef]
- Barriales-Villa, R.; Ochoa, J.P.; Larrañaga-Moreira, J.M.; Salazar-Mendiguchía, J.; Díez-López, C.; Restrepo-Córdoba, M.A.; Álvarez-Rubio, J.; Robles-Mezcua, A.; Olmo-Conesa, M.C.; Nicolás-Rocamora, E.; et al. Risk predictors in a Spanish cohort with cardiac laminopathies. The REDLAMINA registry. Rev. Esp. Cardiol. 2021, 74, 216–224. [Google Scholar] [CrossRef]
- Mitani, A.A.; Haneuse, S. Small Data Challenges of Studying Rare Diseases. JAMA Netw. Open 2020, 3, e201965. [Google Scholar] [CrossRef] [Green Version]
- Schnabel, R.B.; Yin, X.; Gona, P.; Larson, M.G.; Beiser, A.S.; McManus, D.D.; Newton-Cheh, C.; Lubitz, S.A.; Magnani, J.W.; Ellinor, P.T.; et al. 50 year trends in atrial fibrillation prevalence, incidence, risk factors, and mortality in the Framingham Heart Study: A cohort study. Lancet 2015, 386, 154–162. [Google Scholar] [CrossRef] [Green Version]
- Boriani, G.; Diemberger, I.; Martignani, C.; Biffi, M.; Branzi, A. The epidemiological burden of atrial fibrillation: A challenge for clinicians and health care systems. Eur. Heart J. 2006, 27, 893–894. [Google Scholar] [CrossRef] [PubMed]
- Goette, A.; Kalman, J.M.; Aguinaga, L.; Akar, J.; Cabrera, J.A.; Chen, S.A.; Chugh, S.S.; Corradi, D.; D’Avila, A.; Dobrev, D.; et al. EHRA/HRS/APHRS/SOLAECE expert consensus on atrial cardiomyopathies: Definition, characterization, and clinical implication. Europace 2016, 18, 1455–1490. [Google Scholar] [CrossRef] [PubMed]
- Goette, A.; Lendeckel, U. Atrial Cardiomyopathy: Pathophysiology and Clinical Consequences. Cells 2021, 10, 2605. [Google Scholar] [CrossRef]
- van Tintelen, J.P.; Tio, R.A.; Kerstjens-Frederikse, W.S.; van Berlo, J.H.; Boven, L.G.; Suurmeijer, A.J.; White, S.J.; den Dunnen, J.T.; te Meerman, G.J.; Vos, Y.J.; et al. Severe myocardial fibrosis caused by a deletion of the 5′ end of the lamin A/C gene. J. Am. Coll. Cardiol. 2007, 49, 2430–2439. [Google Scholar] [CrossRef] [Green Version]
- Eldin, A.J.; Akinci, B.; da Rocha, A.M.; Meral, R.; Simsir, I.Y.; Adiyaman, S.C.; Ozpelit, E.; Bhave, N.; Gen, R.; Yurekli, B.; et al. Cardiac phenotype in familial partial lipodystrophy. Clin. Endocrinol. 2021, 94, 1043–1053. [Google Scholar] [CrossRef]
- Boriani, G.; Vitolo, M.; Diemberger, I.; Proietti, M.; Valenti, A.C.; Malavasi, V.L.; Lip, G.Y.H. Optimizing indices of AF susceptibility and burden to evaluate AF severity, risk and outcomes. Cardiovasc. Res. 2021, 117, 1–21. [Google Scholar] [CrossRef]
- Gourraud, J.B.; Khairy, P.; Abadir, S.; Tadros, R.; Cadrin-Tourigny, J.; Macle, L.; Dyrda, K.; Mondesert, B.; Dubuc, M.; Guerra, P.G.; et al. Atrial fibrillation in young patients. Expert Rev. Cardiovasc. Ther. 2018, 16, 489–500. [Google Scholar] [CrossRef]
- Chugh, S.S.; Havmoeller, R.; Narayanan, K.; Singh, D.; Rienstra, M.; Benjamin, E.J.; Gillum, R.F.; Kim, Y.H.; McAnulty, J.H.; Zheng, Z.J.; et al. Worldwide epidemiology of atrial fibrillation: A Global Burden of Disease 2010 Study. Circulation 2014, 129, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Steckiewicz, R.; Stolarz, P.; Świętoń, E.; Madej-Pilarczyk, A.; Grabowski, M.; Marchel, M.; Pieniak, M.; Filipiak, K.J.; Hausmanowa-Petrusewicz, I.; Opolski, G. Cardiac pacing in 21 patients with Emery-Dreifuss muscular dystrophy: A single-centre study with a 39-year follow-up. Kardiol. Pol. 2016, 74, 576–583. [Google Scholar] [CrossRef]
- Boriani, G.; Schnabel, R.B.; Healey, J.S.; Lopes, R.D.; Verbiest-van Gurp, N.; Lobban, T.; Camm, J.A.; Freedman, B. Consumer-led screening for atrial fibrillation using consumer-facing wearables, devices and apps: A survey of health care professionals by AF-SCREEN international collaboration. Eur. J. Intern. Med. 2020, 82, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Guo, J.; Shi, X.; Yao, Y.; Sun, Y.; Xia, Y.; Yu, B.; Liu, T.; Chen, Y.; Lip, G.Y.H.; et al. Mobile health technology-supported atrial fibrillation screening and integrated care: A report from the mAFA-II trial Long-term Extension Cohort. Eur. J. Intern. Med. 2020, 82, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, Z.T.; Anderson, K.C.; Quintana, J.A.; O’Neill, M.J.; Sims, R.A.; Glazer, A.M.; Shaffer, C.M.; Crawford, D.M.; Stricker, T.; Ye, F.; et al. Early-Onset Atrial Fibrillation and the Prevalence of Rare Variants in Cardiomyopathy and Arrhythmia Genes. JAMA Cardiol. 2021, 6, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, R.B.; Haeusler, K.G.; Healey, J.S.; Freedman, B.; Boriani, G.; Brachmann, J.; Brandes, A.; Bustamante, A.; Casadei, B.; Crijns, H.J.G.M.; et al. Searching for Atrial Fibrillation Poststroke: A White Paper of the AF-SCREEN International Collaboration. Circulation 2019, 140, 1834–1850. [Google Scholar] [CrossRef]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association of Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2020, 42, 373–498. [Google Scholar] [CrossRef]
- Boriani, G.; Vitolo, M.; Lane, D.A.; Potpara, T.S.; Lip, G.Y. Beyond the 2020 guidelines on atrial fibrillation of the European society of cardiology. Eur. J. Intern Med. 2021, 86, 1–11. [Google Scholar] [CrossRef]
- Boriani, G.; Biagini, E.; Ziacchi, M.; Malavasi, V.L.; Vitolo, M.; Talarico, M.; Mauro, E.; Gorlato, G.; Lattanzi, G. Cardiolaminopathies from bench to bedside: Challenges in clinical decision-making with focus on arrhythmia-related outcomes. Nucleus 2018, 9, 442–459. [Google Scholar] [CrossRef] [Green Version]
- Meune, C.; Van Berlo, J.H.; Anselme, F.; Bonne, G.; Pinto, Y.M.; Duboc, D. Primary prevention of sudden death in patients with lamin A/C gene mutations. N. Engl. J. Med. 2006, 354, 209–210. [Google Scholar] [CrossRef]
- van Berlo, J.H.; de Voogt, W.G.; van der Kooi, A.J.; van Tintelen, J.P.; Bonne, G.; Yaou, R.B.; Duboc, D.; Rossenbacker, T.; Heidbüchel, H.; de Visser, M.; et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: Do lamin A/C mutations portend a hig5757h risk of sudden death? J. Mol. Med. 2005, 83, 79–83. [Google Scholar] [CrossRef]
- Glikson, M.; Nielsen, J.C.; Kronborg, M.B.; Michowitz, Y.; Auricchio, A.; Barbash, I.M.; Barrabés, J.A.; Boriani, G.; Braunschweig, F.; Brignole, M.; et al. 2021 ESC Guidelines on cardiac pacing and cardiac resynchronization therapy. Eur. Heart J. 2021, 42, 3427–3520. [Google Scholar] [CrossRef]
- Køber, L.; Thune, J.J.; Nielsen, J.C.; Haarbo, J.; Videbæk, L.; Korup, E.; Jensen, G.; Hildebrandt, P.; Steffensen, F.H.; Bruun, N.E.; et al. Defibrillator Implantation in Patients with Nonischemic Systolic Heart Failure. N. Engl. J. Med. 2016, 375, 1221–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatzoulis, K.A.; Vouliotis, A.I.; Tsiachris, D.; Salourou, M.; Archontakis, S.; Dilaveris, P.; Gialernios, T.; Arsenos, P.; Karystinos, G.; Sideris, S.; et al. Primary prevention of sudden cardiac death in a nonischemic dilated cardiomyopathy population: Reappraisal of the role of programmed ventricular stimulation. Circ. Arrhythm. Electrophysiol. 2013, 6, 504–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobar-Lopez, L.; Ochoa, J.P.; Mirelis, J.G.; Espinosa, M.; Navarro, M.; Gallego-Delgado, M.; Barriales-Villa, R.; Robles-Mezcua, A.; Basurte-Elorz, M.T.; Gutiérrez García-Moreno, L.; et al. Association of Genet. ic Variants with Outcomes in Patients with Nonischemic Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2021, 78, 1682–1699. [Google Scholar] [CrossRef] [PubMed]
- Wahbi, K.; Ben Yaou, R.; Gandjbakhch, E.; Anselme, F.; Gossios, T.; Lakdawala, N.K.; Stalens, C.; Sacher, F.; Babuty, D.; Trochu, J.N.; et al. Development and Validation of a New Risk Prediction Score for Life-Threatening Ventricular Tachyarrhythmias in Laminopathies. Circulation 2019, 140, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Kayvanpour, E.; Sedaghat-Hamedani, F.; Amr, A.; Lai, A.; Haas, J.; Holzer, D.B.; Frese, K.S.; Keller, A.; Jensen, K.; Katus, H.A.; et al. Genotype-phenotype associations in dilated cardiomyopathy: Meta-analysis on more than 8000 individuals. Clin. Res. Cardiol. 2017, 106, 127–139. [Google Scholar] [CrossRef]
- Taylor, M.R.; Fain, P.R.; Sinagra, G.; Robinson, M.L.; Robertson, A.D.; Carniel, E.; Di Lenarda, A.; Bohlmeyer, T.J.; Ferguson, D.A.; Brodsky, G.L.; et al. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J. Am. Coll. Cardiol. 2003, 41, 771–780. [Google Scholar] [CrossRef] [Green Version]
- Sanna, T.; Dello Russo, A.; Toniolo, D.; Vytopil, M.; Pelargonio, G.; De Martino, G.; Ricci, E.; Silvestri, G.; Giglio, V.; Messano, L.; et al. Cardiac featuRes. of Emery-Dreifuss muscular dystrophy caused by lamin A/C gene mutations. Eur. Heart J. 2003, 24, 2227–2236. [Google Scholar] [CrossRef] [Green Version]
- Gigli, M.; Merlo, M.; Graw, S.L.; Barbati, G.; Rowland, T.J.; Slavov, D.B.; Stolfo, D.; Haywood, M.E.; Dal Ferro, M.; Altinier, A.; et al. Genet. ic Risk of Arrhythmic Phenotypes in Patients with Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2019, 74, 1480–1490. [Google Scholar] [CrossRef]
- Atalaia, A.; Ben Yaou, R.; Wahbi, K.; De Sandre-Giovannoli, A.; Vigouroux, C.; Bonne, G. Laminopathies’ Treatments Systematic Review: A Contribution Towards a ‘Treatabolome’. J. Neuromuscul. Dis. 2021, 8, 419–439. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Study, Year | Study Design | Population (n) | Age (y), Median or Mean ± SD | Median F-U (y) | Women, n (%) | Unaffected or Asymptomatic, n (%) | Neuromuscular Involvement, n (%) | Main Findings |
|---|---|---|---|---|---|---|---|---|
| LMNAmutation carriers | ||||||||
| Boriani et al. [27], 2003 | Retrospective | 8 (18 total cohort) | 29.5 | 9.5 ± 9 | 2 (25) | 1 (12.5) | 6 (75) | Pts with EMD mutations are at great risk to develop AF, AFL, bradycardia, ASS and stroke |
| Van Rijsingen et al. [32], 2012 | Retrospective | 269 | 36 | 3.5 | 121 (45) | 56/248 (23) | 41/198 (21) | Among LMNA mutation carriers, male sex, EF < 45%, NSVT and non-missense mutations portend a greater risk of MVA |
| Anselme et al. [33], 2013 | Prospective | 47 | 38 ± 11 | 7.9 (5.1 ICD carriers) | 21 (45) | N/A | [isolated nm involv.] 18 (38) | MVA are frequent in LMNA carriers with cardiac conduction disorders irrespective of EF |
| Van Rijsingen et al. [34], 2013 | Retrospective | 76 | 45 | 42 ± 12 (mean ± SD) | 35 (46) | N/A | 25 (33) | LMNA mutation is an independent predictor of arterial and venous TE |
| Kumar et al. [28], 2016 | Retrospective | 122 | 41 | 7 | 52 (43) | 18 (9) | 18 (15) | LMNA-related heart disease is associated with a high frequency of phenotypic progression and index phenotype does not predict adverse events |
| Hasselberg et al. [29], 2018 | Retrospective | 79 | 42 ± 16 | 7.8 | 36 (46) | N/A | N/A | Among DCM, the prevalence of LMNA mutation is 6.2% with high penetration in asymptomatic young genotype-positive members. LMNA carriers have a high incidence of HT. |
| Nakajima et al. [35], 2018 | Retrospective | 110 | 43 ± 15 | 5 | 42 (38) | N/A | N/A | Several cardiac presentations are age-related in LMNA-related heart disease, LVD is the only predictor for mortality |
| Peretto et al. [36], 2019 | Prospective/ Retrospective | 164 | 38 | 10 | 84 (51) | N/A | 104 (63) | Many LMNA mutation carriers develop neurological disease in their 30s and cardiac manifestation in the next decade |
| Ditaranto et al. [37], 2019 | Prospective/ Retrospective | 40 | 39 | 2.5 | 18 (45) | N/A | 14 (35) | Pts with neuromuscular presentation have an earlier cardiac involvement (from AF and/or AVB to cardiomyopathy) |
| Marchel et al. [38], 2021 | Prospective | 15 (45 total cohort) | 26 | 11 | 11 (73) | N/A | 15 (100) | Atrial arrhythmias are common in EMD/LMNA mutation carriers; they occur earlier in EMD pts. VA are common (60%) and earlier in LMNA compared to the EMD group |
| Barriales-Villa et al. [39], 2021 | Retrospective | 140 | 42.8 M, 38 F | 5 (probands), 3 (relatives) | 69 (49.3) | N/A | 34 (24.3) | Among LMNA mutation carriers, NSVT and EF < 45% were the only independent predictors of MVA |
| EMDmutation carriers | ||||||||
| Boriani et al. [27], 2003 | Retrospective | 10 (18 total cohort) | 24.5 (affected males) | 16 | 3 (30) | 0 (0) | 6 (60) | Pts with EMD mutations are at great risk to develop AF, AFL, bradycardia, ASS and stroke |
| Marchel et al. [38], 2021 | Prospective | 30 (45 total cohort) | 21 | 11 | 6 (20) | N/A | 30 (100) | Atrial arrhythmias are common findings in EMD/LMNA mutation carriers, they occurred earlier in EMD pts. VA were very common (60%) in LMNA and occurred definitely earlier compared to the EMD group |
| Study | Selection | Comparability | Outcome | Total |
|---|---|---|---|---|
| Boriani et al. [27], 2003 | ** | - | *** | 5 |
| Van Rijsingen et al. [32], 2012 | ** | ** | *** | 7 |
| Anselme et al. [33], 2013 | ** | ** | *** | 7 |
| Van Rijsingen et al. [34], 2013 | ** | ** | *** | 7 |
| Kumar et al. [28], 2016 | ** | - | *** | 5 |
| Hasselberg et al. [29], 2018 | ** | ** | *** | 7 |
| Nakajima et al. [35], 2018 | ** | ** | *** | 7 |
| Peretto et al. [36], 2019 | ** | - | *** | 5 |
| Ditaranto et al. [37], 2019 | ** | - | *** | 5 |
| Marchel et al. [38], 2021 | ** | - | *** | 5 |
| Barriales-Villa et al. [39], 2021 | ** | ** | *** | 7 |
| Population (n) | Age (y), Median or Mean ± SD | AF, AFL, AT, n (%) | SSS/SAB, n (%) | ASS, n (%) | Median F-U (y) | AF, AFL, AT, n (%) | SSS/SAB, n (%) | ASS, n (%) | IR AF/AFl/AT | IR SSS/SAB | IR ASS | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline Prevalence | Incident Events or Final Prevalence | |||||||||||
| LMNAmutation carriers | ||||||||||||
| Boriani [27], 2003 | 8 | 29.5 | 1 (12.5) | 0 (0) | N/A | 7 | 3 (42.8) | 3 (60) | 1 (12.5) | 6.1 | 8.6 | 1.8 |
| Van Rijsingen IA [32], 2012 | 269 | 36 | 86/239 (36) | N/A | N/A | 3.5 | N/A | N/A | N/A | N/A | N/A | N/A |
| Anselme [33], 2013 | 47 | 38±11 | 12 (26) | N/A | N/A | 7.9 | 31 (88.6) | N/A | N/A | 11.2 | N/A | N/A |
| Van Rijsingen [34], 2013 | 76 | 45 | 48 (63) | N/A | N/A | 42 ± 12 (mean ± SD) | N/A | N/A | N/A | N/A | N/A | N/A |
| Kumar [28], 2016 | 122 | 41 ± 14 | 52 (42.7) | N/A | N/A | 7 | 62 (88.6) | N/A | N/A | 12.7 | N/A | N/A |
| Hasselberg [29], 2018 | 79 | 42 ± 16 | N/A | N/A | N/A | 7.8 | * 48 (no population at risk) | N/A | N/A | N/A | N/A | N/A |
| Nakajima [35], 2018 | 110 baseline/90 end of f-U | 43 ± 15 | 31 (34.4) | 27/110 (25) | N/A | 5 | 27 (45.7) | 30 (no population at risk) | N/A | 9.2 | N/A | N/A |
| Peretto [36], 2019 | 164 | 38 | 19/137 (14) | N/A | N/A | 10 | 103 (no population at risk) | 13 (no population at risk) | N/A | N/A | N/A | N/A |
| Ditaranto [37], 2019 | 40 | 39 | 17 (42.5) | 4 (10) | N/A | 2.5 | 8 (34.8) | N/A | 2 (5) | 13.9 | N/A | 2 |
| Marchel [38], 2021 | 15 | 26 | N/A | N/A | 0 (0) | 11 | 10 (66.6) | N/A | 0 (0) | N/A | N/A | 0 |
| Barriales-Villa [39], 2021 | 140 | 40.4 | 42 (30) | N/A | N/A | 3.8 | N/A | N/A | N/A | N/A | N/A | N/A |
| EMD mutation carriers | ||||||||||||
| Boriani [27], 2003 | 10 | 24.5 (affected males) | 1 (10) | 1 (10) | N/A | 16 | 5 (55.5) | 4 (44.4) | 4 (40) | 3.5 | 3.5 | 2.5 |
| Marchel [38], 2021 | 30 | 21 | 22 (73.3) | N/A | 3 (10) | 11 | * 11 (36.6) | N/A | 11 (40.7) | N/A | N/A | 3.7 |
| Population (n) | Age (y), Median or Mean ± SD | SVT, n (%) | Median F-U (y) | F-U ICD Carriers | MVA, n (%) | ICD Implantation, n (%) | ICD Appropriate Intervention, n (%) | IR MVA | IR ICD Implantation | IR ICD Intervention | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline Prevalence | Incident Events | ||||||||||
| LMNA mutation carriers | |||||||||||
| Boriani [27], 2003 | 8 | 29.5 | N/A | 7 | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| Van Rijsingen IA [32], 2012 | 269 | 36 | N/A | 3.5 | 2.1 | 53 (19.7) | 117 (43.5) [primary prevention 107 (39.8); secondary prevention 10 (3.7)] | 28/117 (24) | 5.6 | 12.4 | 11.4 |
| Anselme [33], 2013 | 47 | 38 ± 11 | N/A | 7.9 | 5.1 | 14 (29.8) | 21 (44.6) | 11/21 (52.4) | 3.8 | 5.7 | 10.3 |
| Van Rijsingen [34], 2013 | 76 | 45 | N/A | 42 ± 12 (mean ± SD) | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| Kumar [28], 2016 | 122 | 41 ± 14 | 21 (17.2) | 7 | 7 | 39 (32) | 59 (48.3) | 29/58 (50) | 4.6 | 6.9 | 5.2 |
| Hasselberg [29], 2018 | 79 | 42 ± 16 | N/A | 7.8 | 7.8 | 14 (17.7) | 49 (62) | N/A | 2.3 | 8 | N/A |
| Nakajima [35], 2018 | 110 baseline/90 end of f-U | 43 ± 15 | VT + VF 21/110 (19) | 5 | 5 | 46 (51.1) | 44 (48.9) | 12/44 (27.3) | 10.2 | 9.8 | 5.2 |
| Peretto [36], 2019 | 164 | 38 | 2/137 (1.5) | 10 | N/A | 32 (19.5) | N/A | N/A | 2 | N/A | N/A |
| Ditaranto [37], 2019 | 40 | 39 | N/A | 2.5 | 2.5 | SVT/storm 7 (17.5) | 10 (25) | 7/18 (38.9) | 7 | 10 | 15.6 |
| Marchel [38], 2021 | 15 | 26 | VT 0 (0) | 11 | 11 | VT 2 (13.3) | 9 (60) | N/A | 1.2 | 5.5 | N/A |
| Barriales-Villa [39], 2021 | 140 | 40.4 | 0 (0) | 3.8 | 3.8 | 24 (17.1) | 62 (44.3) | 17/62 (27.4) | 4.5 | 11.7 | 6.9 |
| EMD mutation carriers | |||||||||||
| Boriani [27], 2003 | 10 | 24.5 (affected males) | N/A | 16 | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| Marchel [38], 2021 | 30 | 21 | 0 (0) | 11 | N/A | 2 (6.7) | 1 (3.3) | N/A | 0.6 | 0.3 | N/A |
| Population (n) | Age (y), Median or Mean ± SD | 2nd–3rd Degree AVB, n (%) | SSS, n (%) | Median F-U (y) | 2nd–3rd Degree AVB, n (%) | PM Implantation, n (%) | IR 2nd-3rd AVB | IR PM Implantation | |
|---|---|---|---|---|---|---|---|---|---|
| Baseline Prevalence | Incident Events or Final Prevalence | ||||||||
| LMNA mutation carriers | |||||||||
| Boriani [27], 2003 | 8 | 29.5 | 1 (12.5) | N/A | 7 | 2 (25) | 3 (37.5) | 7.1 | 5.4 |
| Van Rijsingen IA [32], 2012 | 269 | 36 | 114/244 (47), 1st, 2nd, 3rd AVB | N/A | 3.5 | N/A | N/A | N/A | N/A |
| Anselme [33], 2013 | 47 | 38 ± 11 | 21 (45), significant conduction disorders ** | N/A | 7.9 | 33 (no population at risk) | N/A | N/A | N/A |
| Van Rijsingen [34], 2013 | 76 | 45 | 51 (67), LMNA 1st, 2nd, 3rd AVB | N/A | 42 ± 12 (mean ± SD) | N/A | N/A | N/A | N/A |
| Kumar [28], 2016 | 122 | 41 ± 14 | 18 (15.4) | N/A | 7 | 27 (26) | N/A | 3.7 | N/A |
| Hasselberg [29], 2018 | 79 | 42 ± 16 | N/A | N/A | 7.8 | * 51 (no population at risk), 1st, 2nd, 3rd AVB | N/A | N/A | N/A |
| Nakajima [35], 2018 | 110 baseline/90 end of f-U | 43 ± 15 | 33 (36.7) | 27/110 (25) | 5 | 22 (38.6) | 11 (12.2) | 7.7 | 2.4 |
| Peretto [36], 2019 | 164 | 38 | 16/137 (11.7) | N/A | 10 | 75 (no population at risk) | N/A | N/A | N/A |
| Ditaranto [37], 2019 | 40 | 39 | 15 (37) | 4 (10) | 2.5 | 2 (8) | 4(10) | 3.2 | 4 |
| Marchel [38], 2021 | 15 | 26 | 7 (46.6) | N/A | 11 | N/A | 7 (46) | N/A | 4.2 |
| Barriales-Villa [39], 2021 | 140 | 40.4 | 34 (24.3) | N/A | 3.8 | N/A | 36 (25.7) | N/A | 6.8 |
| EMD mutation carriers | |||||||||
| Boriani [27], 2003 | 10 | 24.5 (affected males) | 2 (20) | 1 (10) | 16 | 2 (25) | 7 (70) | 2.3 | 4.4 |
| Marchel [38], 2021 | 30 | 21 | 14 (46.7) | ASS 3 (10) | 11 | N/A | 23 (76.6) | N/A | 7 |
| Population (n) | Age (y), Median or Mean ± SD | Median F-U (y) | Stroke, n (%) | IR Stroke | |
|---|---|---|---|---|---|
| Incident Events | |||||
| LMNA mutation carriers | |||||
| Boriani et al. [27], 2003 | 8 | 29.5 | 7 | 5 (62.5) | 8.9 |
| Van Rijsingen et al. [32], 2012 | 269 | 36 | 3.5 | N/A | N/A |
| Anselme et al. [33], 2013 | 47 | 38 ± 11 | 7.9 | 4 (8.5) | 1.1 |
| Van Rijsingen et al. [34], 2013 | 76 | 45 | 42 ± 12 y (mean ± SD) | Arterial TE, 11 (14) | 0.3 |
| Kumar et al. [28], 2016 | 122 | 41 ± 14 | 7 | 10 (8) | 1.2 |
| Hasselberg et al. [29], 2018 | 79 | 42 ± 16 | 7.8 | N/A | N/A |
| Nakajima et al. [35], 2018 | 110 baseline/90 end of f-u | 43 ± 15 | 5 | 11 (12.2) | 2.4 |
| Peretto et al. [36], 2019 | 164 | 38 | 10 | N/A | N/A |
| Ditaranto et al. [37], 2019 | 40 | 39 | 2.5 | N/A | N/A |
| Marchel et al. [38], 2021 | 15 | 26 | 11 | N/A | N/A |
| Barriales-Villa et al. [39], 2021 | 140 | 40.4 | 3.8 | Embolism, 14 (10) | 2.6 |
| EMD mutation carriers | |||||
| Boriani et al. [27], 2003 | 10 | 24.5 (affected men) | 16 | 1 (10) | 0.6 |
| Marchel et al. [38], 2021 | 30 | 21 | 11 | N/A | N/A |
| Population (n) | Age (y), Median or Mean ± SD | LVEF < 50%, n(%) | LVEF < 45%, n(%) | NYHA ≥ III–IV | Median F-U (y) | NYHA ≥ III–IV | HT, n(%) | IR HT | |
|---|---|---|---|---|---|---|---|---|---|
| Baseline Prevalence | Incident Events or Final Prevalence | ||||||||
| LMNA mutation carriers | |||||||||
| Boriani [27], 2003 | 8 | 29.5 | N/A | N/A | 0 (0) | 7 | 1 (12.5) | 1 (12.5) | 1.3 |
| Van Rijsingen IA [32], 2012 | 269 | 36 | N/A | 89/243 (36.6) | 39/260 (15) | 3.5 | N/A | 36 (13.3) | 3.8 |
| Anselme [33], 2013 | 47 | 38±11 | N/A | 6 (13) | N/A | 7.9 | N/A | 9 (19) | N/A |
| Van Rijsingen [34], 2013 | 76 | 45 | LVEF < 55%, 35(46) | LVEF < 35%, 13(17) | N/A | 42 ± 12 (mean ± SD) | N/A | N/A | N/A |
| Kumar [28], 2016 | 122 | 41 ± 14 | 57 (47) | 0 (0) | N/A | 7 | N/A | 10 (8) | 0.1 |
| Hasselberg [29], 2018 | 79 | 42 ± 16 | N/A | 29 (36.7) | N/A | 7.8 | N/A | 15 (18) | 2.4 |
| Nakajima [35], 2018 | 110 baseline/90 end of f-U | 43 ± 15 | 22/110 (20) | N/A | 8/110 (7.3) | 5 | 30 (34) | N/A | N/A |
| Peretto [36], 2019 | 164 | 38 | N/A | 5/147 (3.5) | N/A | 10 | N/A | 14 (8.5) | 0.9 |
| Ditaranto [37], 2019 | 40 | 39 | N/A | N/A | 7 (17) | 2.5 | N/A | 10 (25) | 10 |
| Marchel [38], 2021 | 15 | 26 | N/A | N/A | 0 (0) | 11 | N/A | N/A | N/A |
| Barriales-Villa [39], 2021 | 140 | 40.4 | N/A | 53 (37.8) | 27 (19.3) | 3.8 | N/A | 29 (20.7) | 5.2 |
| EMD mutation carriers | |||||||||
| Boriani [27], 2003 | 10 | 24.5 (affected males) | N/A | N/A | 0 (0) | 16 | 0 (0) | 0 (0) | 0 |
| Marchel [38], 2021 | 30 | 21 | N/A | N/A | 0 (0) | 11 | N/A | N/A | N/A |
| Population (n) | Age (y), Median or Mean ± SD | Median F-U (y) | All-Cause Death, n (%) | Cardiac Death, n (%) | IR All-Cause Death | IR Cardiac Death | |
|---|---|---|---|---|---|---|---|
| LMNA mutation carriers | |||||||
| Boriani et al. [27], 2003 | 8 | 29.5 | 7 | 1 (12.5) | 1 (12.5) | 1.8 | 1.8 |
| Van Rijsingen et al. [32], 2012 | 269 | 36 | 3.5 | 45 (16.7) | 41 (15.2) | 4.8 | 4.4 |
| Anselme et al. [33], 2013 | 47 | 38 ± 11 | 7.9 | 7 (14.8) | 4 (8.5) | 1.9 | 1.1 |
| Van Rijsingen et al. [34], 2013 | 76 | 45 | 42 ± 12 (mean ± SD) | N/A | N/A | N/A | N/A |
| Kumar et al. [28], 2016 | 122 | 41 ± 14 | 7 | 22 (18) | 21 (17.2) | 2.6 | 2.5 |
| Hasselberg et al. [29], 2018 | 79 | 42 ± 16 | 7.8 | 6 (8) | 6 (8) | 1 | 1 |
| Nakajima et al. [35], 2018 | 110 baseline/90 end of f-U | 43 ± 15 | 5 | 17 (18.9) | 16 (17.7) | 3.8 | 3.6 |
| Peretto et al. [36], 2019 | 164 | 38 | 10 | 10 (6) | 6 (3.6) | 0.6 | 0.4 |
| Ditaranto et al. [37], 2019 | 40 | 39 | 2.5 | N/A | N/A | N/A | N/A |
| Marchel et al. [38], 2021 | 15 | 26 | 11 | N/A | N/A | N/A | N/A |
| Barriales-Villa et al. [39], 2021 | 140 | 40.4 | 3.8 | N/A | 8 (5.7) | N/A | 1.5 |
| EMD mutation carriers | |||||||
| Boriani et al. [27], 2003 | 10 | 24.5 (affected males) | 16 | 1 (10) | 0 (0) | 0.6 | 0 |
| Marchel et al. [38], 2021 | 30 | 21 | 11 | N/A | N/A | N/A | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valenti, A.C.; Albini, A.; Imberti, J.F.; Vitolo, M.; Bonini, N.; Lattanzi, G.; Schnabel, R.B.; Boriani, G. Clinical Profile, Arrhythmias, and Adverse Cardiac Outcomes in Emery–Dreifuss Muscular Dystrophies: A Systematic Review of the Literature. Biology 2022, 11, 530. https://doi.org/10.3390/biology11040530
Valenti AC, Albini A, Imberti JF, Vitolo M, Bonini N, Lattanzi G, Schnabel RB, Boriani G. Clinical Profile, Arrhythmias, and Adverse Cardiac Outcomes in Emery–Dreifuss Muscular Dystrophies: A Systematic Review of the Literature. Biology. 2022; 11(4):530. https://doi.org/10.3390/biology11040530
Chicago/Turabian StyleValenti, Anna Chiara, Alessandro Albini, Jacopo Francesco Imberti, Marco Vitolo, Niccolò Bonini, Giovanna Lattanzi, Renate B. Schnabel, and Giuseppe Boriani. 2022. "Clinical Profile, Arrhythmias, and Adverse Cardiac Outcomes in Emery–Dreifuss Muscular Dystrophies: A Systematic Review of the Literature" Biology 11, no. 4: 530. https://doi.org/10.3390/biology11040530
APA StyleValenti, A. C., Albini, A., Imberti, J. F., Vitolo, M., Bonini, N., Lattanzi, G., Schnabel, R. B., & Boriani, G. (2022). Clinical Profile, Arrhythmias, and Adverse Cardiac Outcomes in Emery–Dreifuss Muscular Dystrophies: A Systematic Review of the Literature. Biology, 11(4), 530. https://doi.org/10.3390/biology11040530