
Using Mechanical Alloying to Create Bimetallic Catalysts for Vapor-Phase Carbon Nanofiber Synthesis

Abstract
:
{kind=link}
{kind=link}
{kind=link}
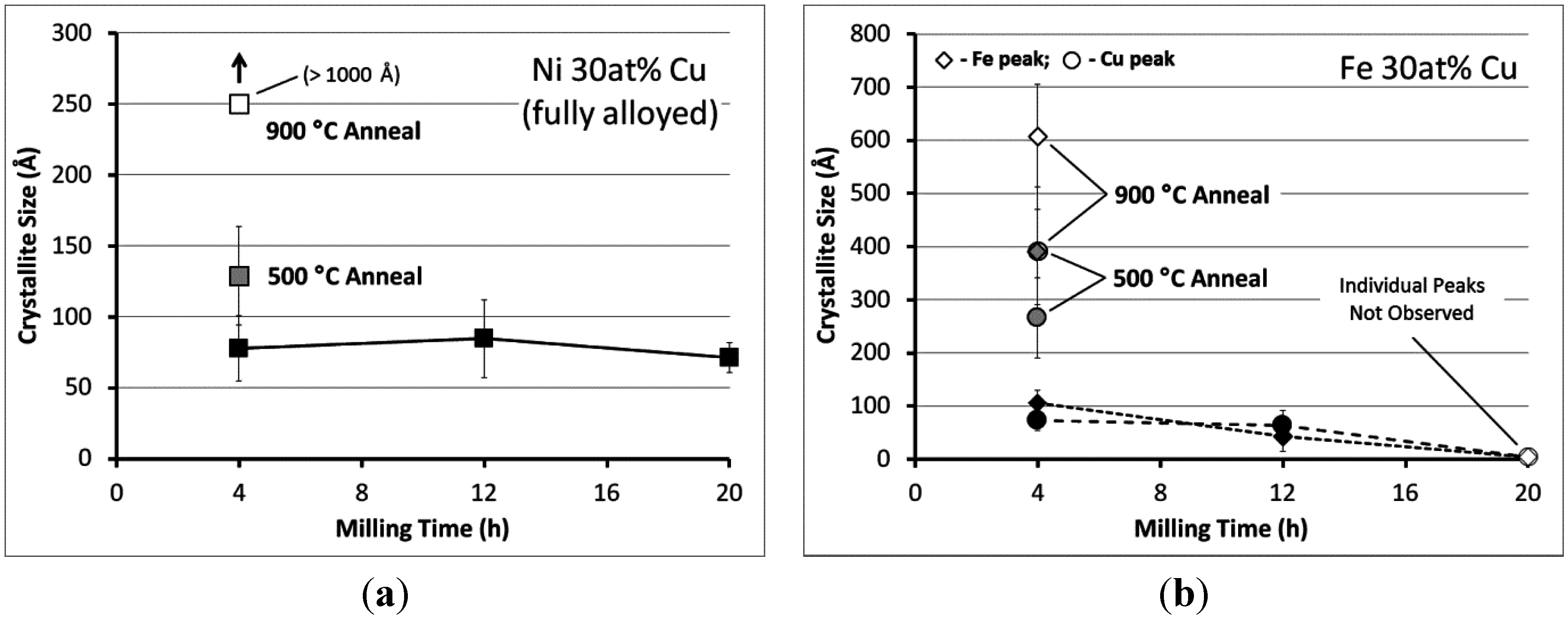{kind=link}
{kind=link}
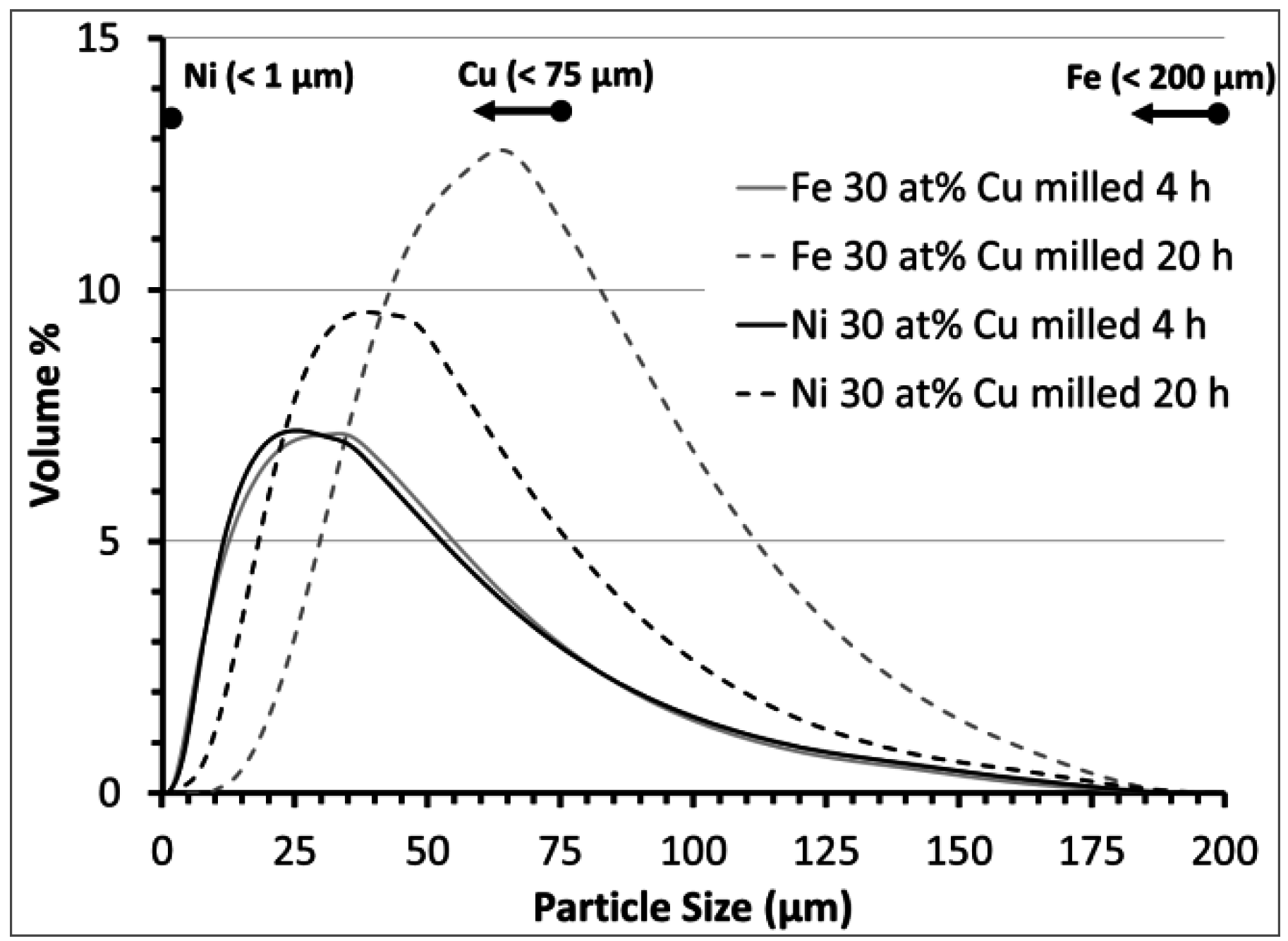{kind=link}
{kind=link}
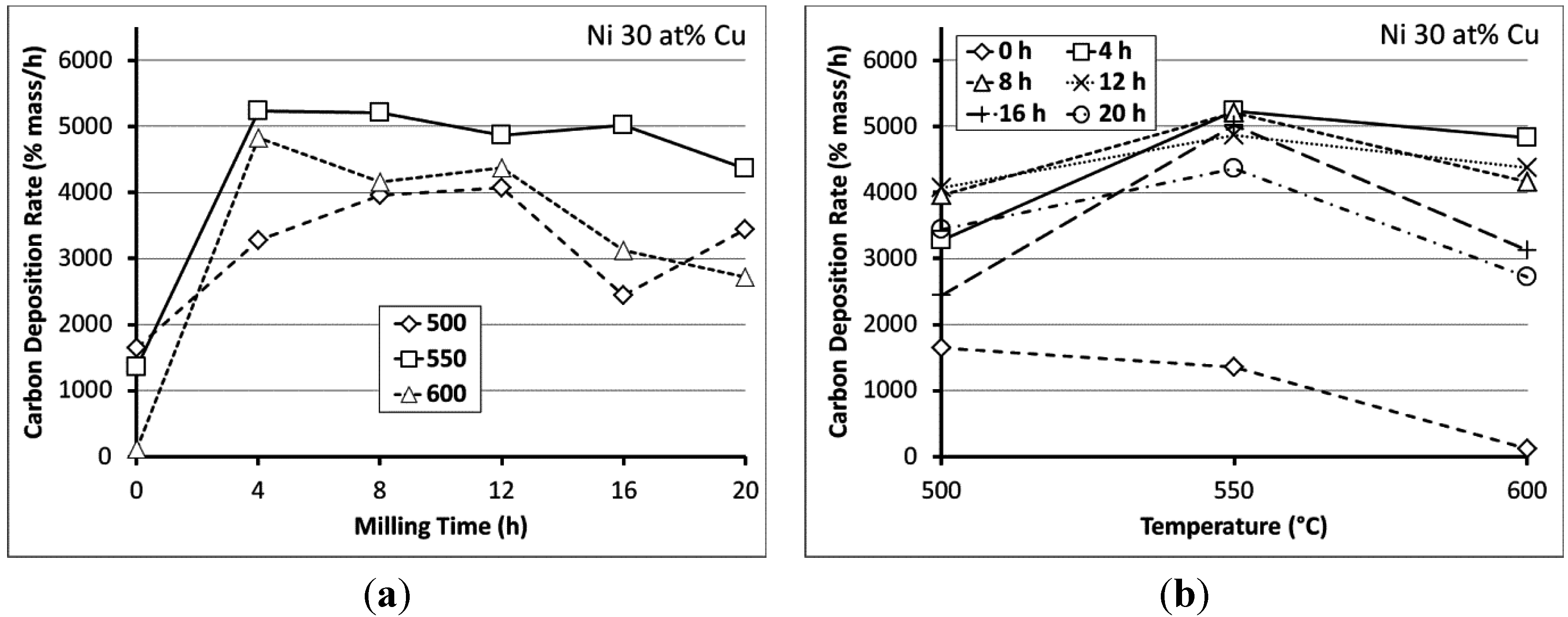
{kind=link}
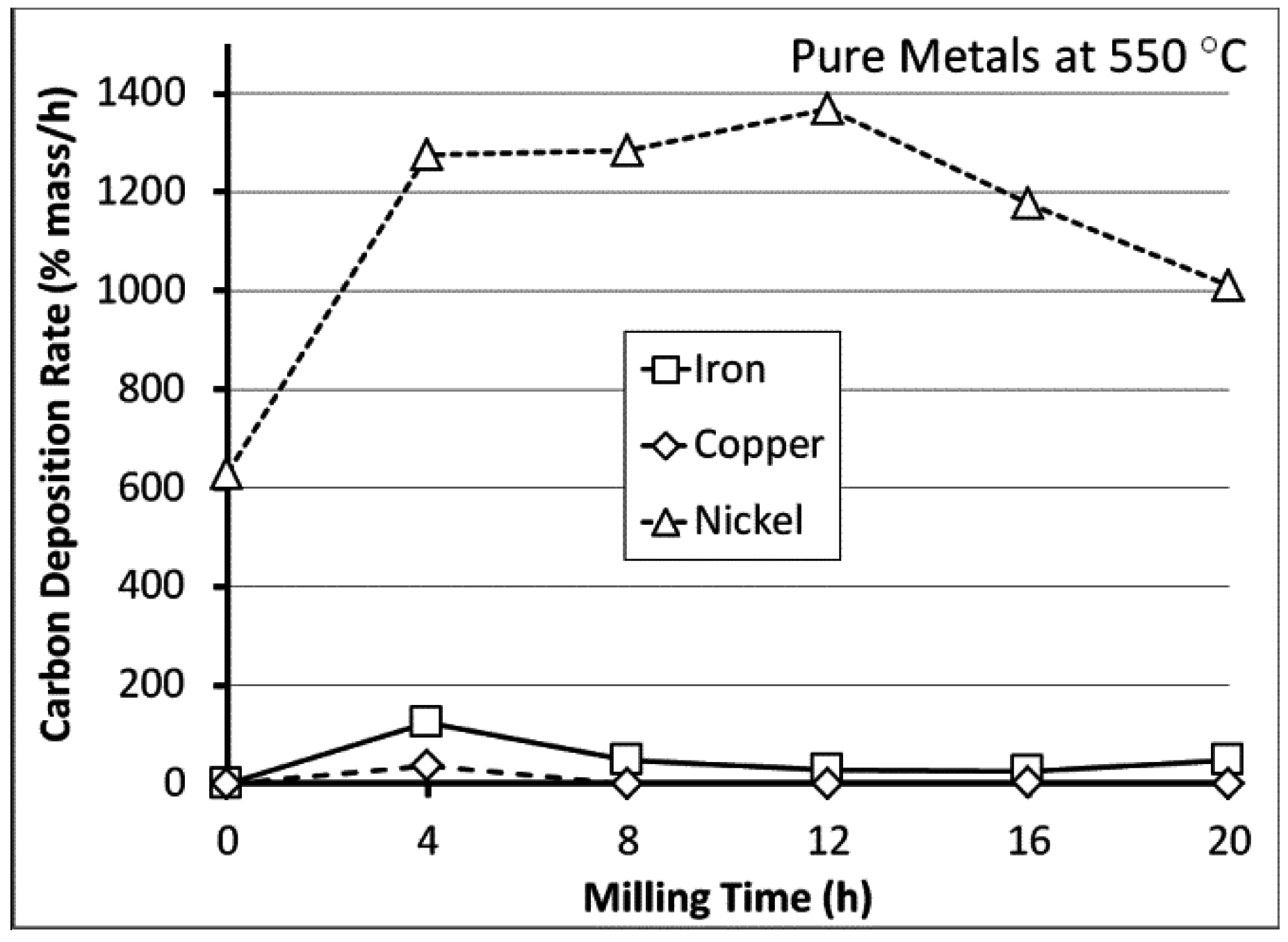{kind=link}
{kind=link}
1. Introduction
2. Experimental
3. Results and Discussion
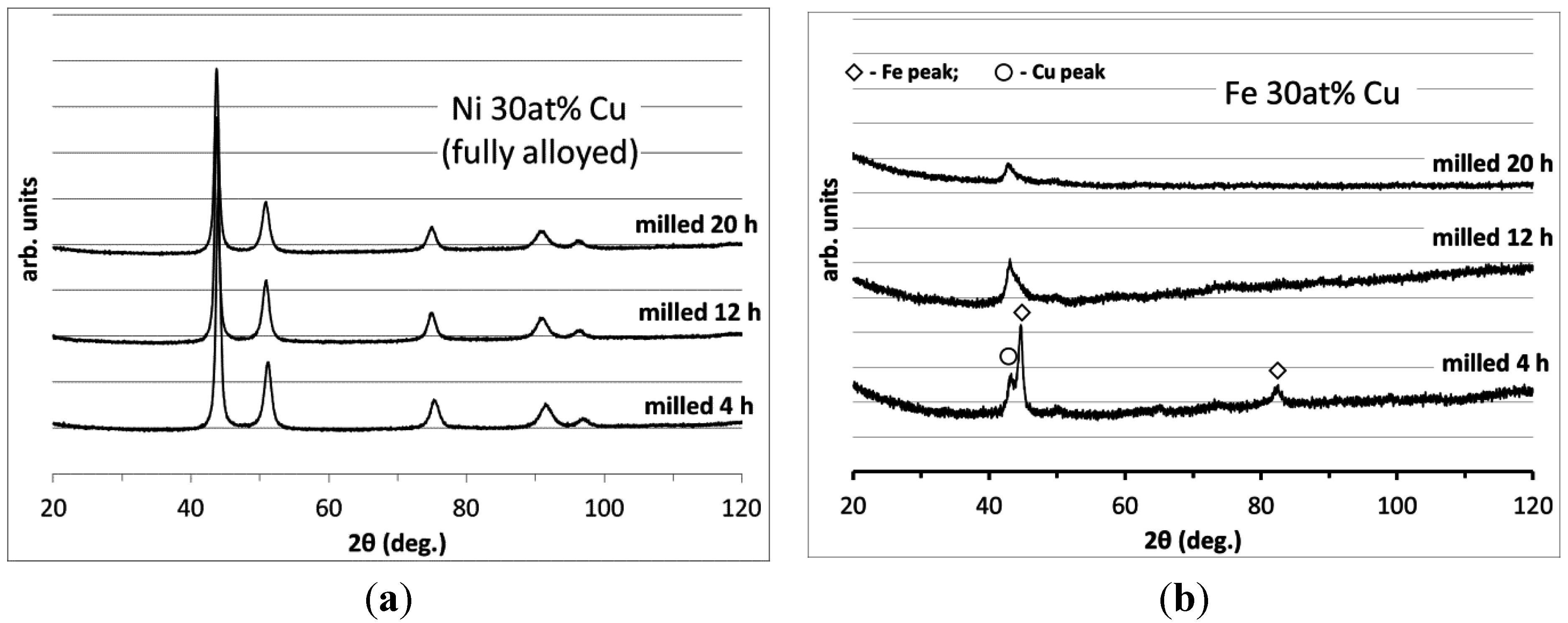
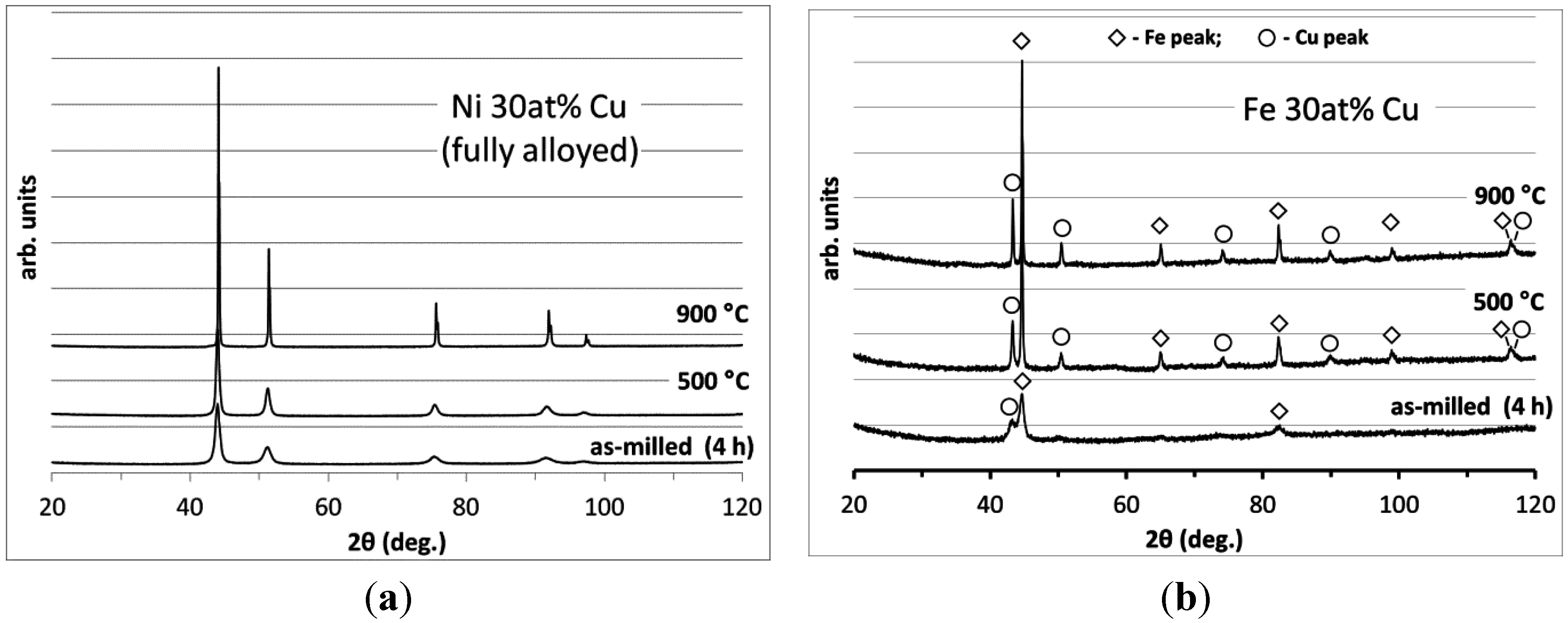
3.1. Catalyst Processing
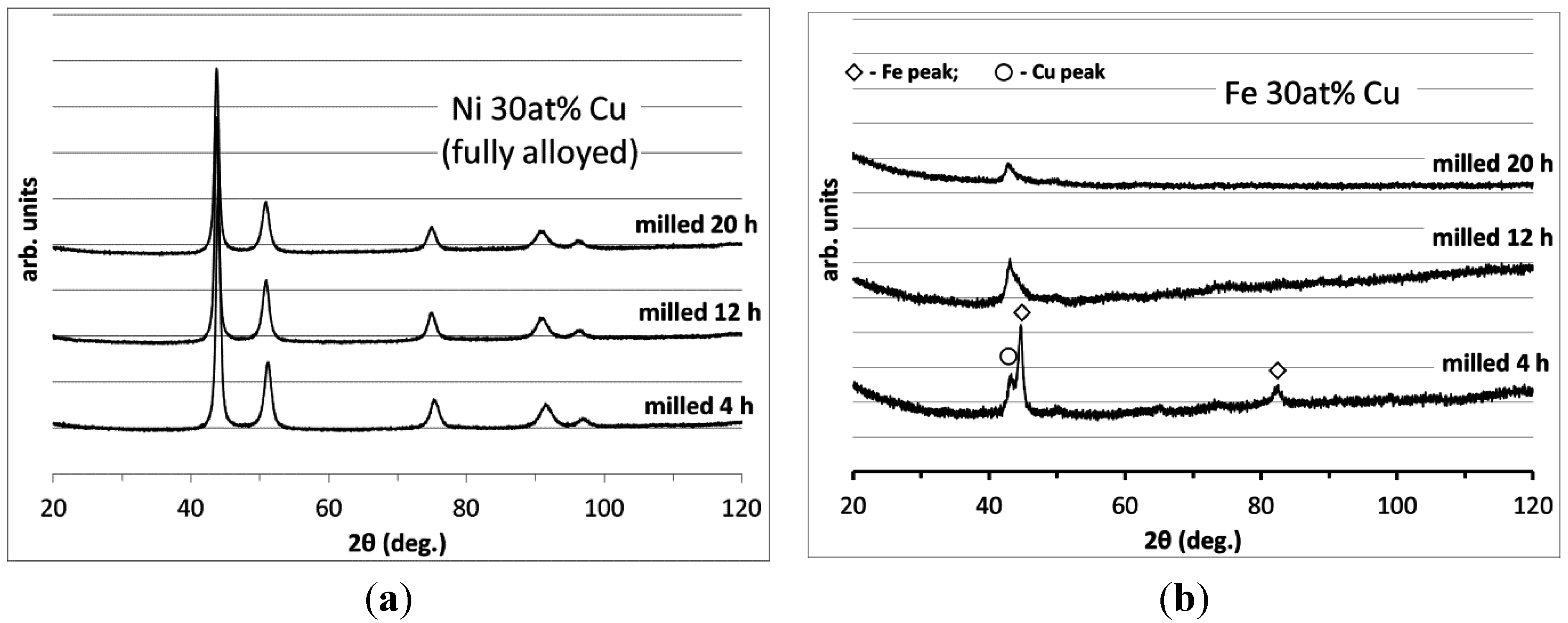
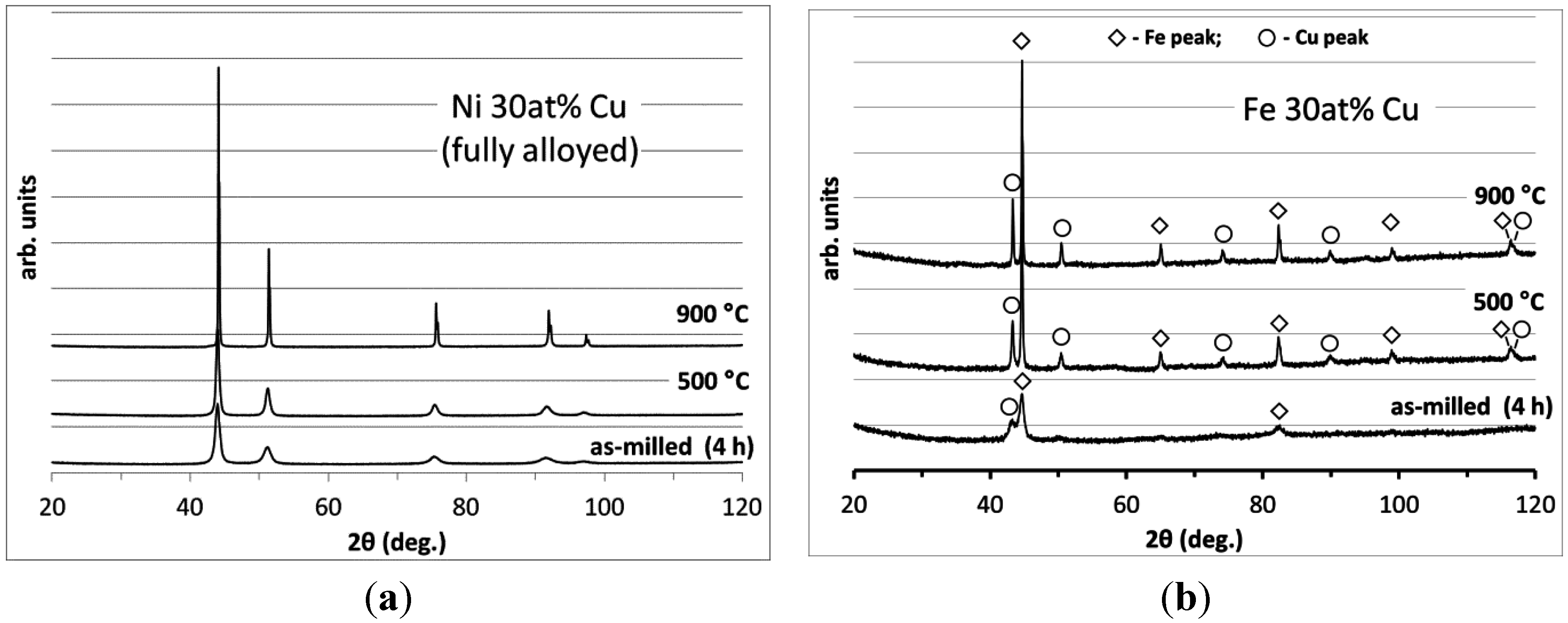
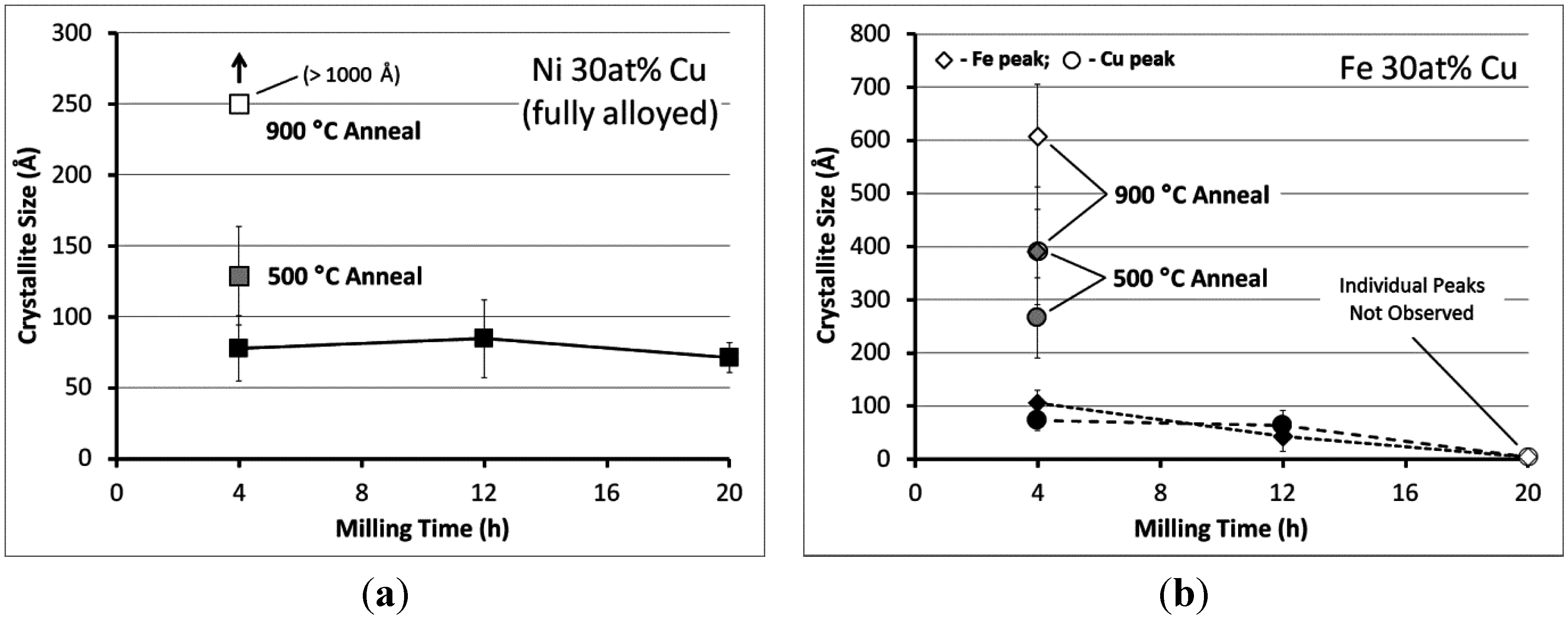

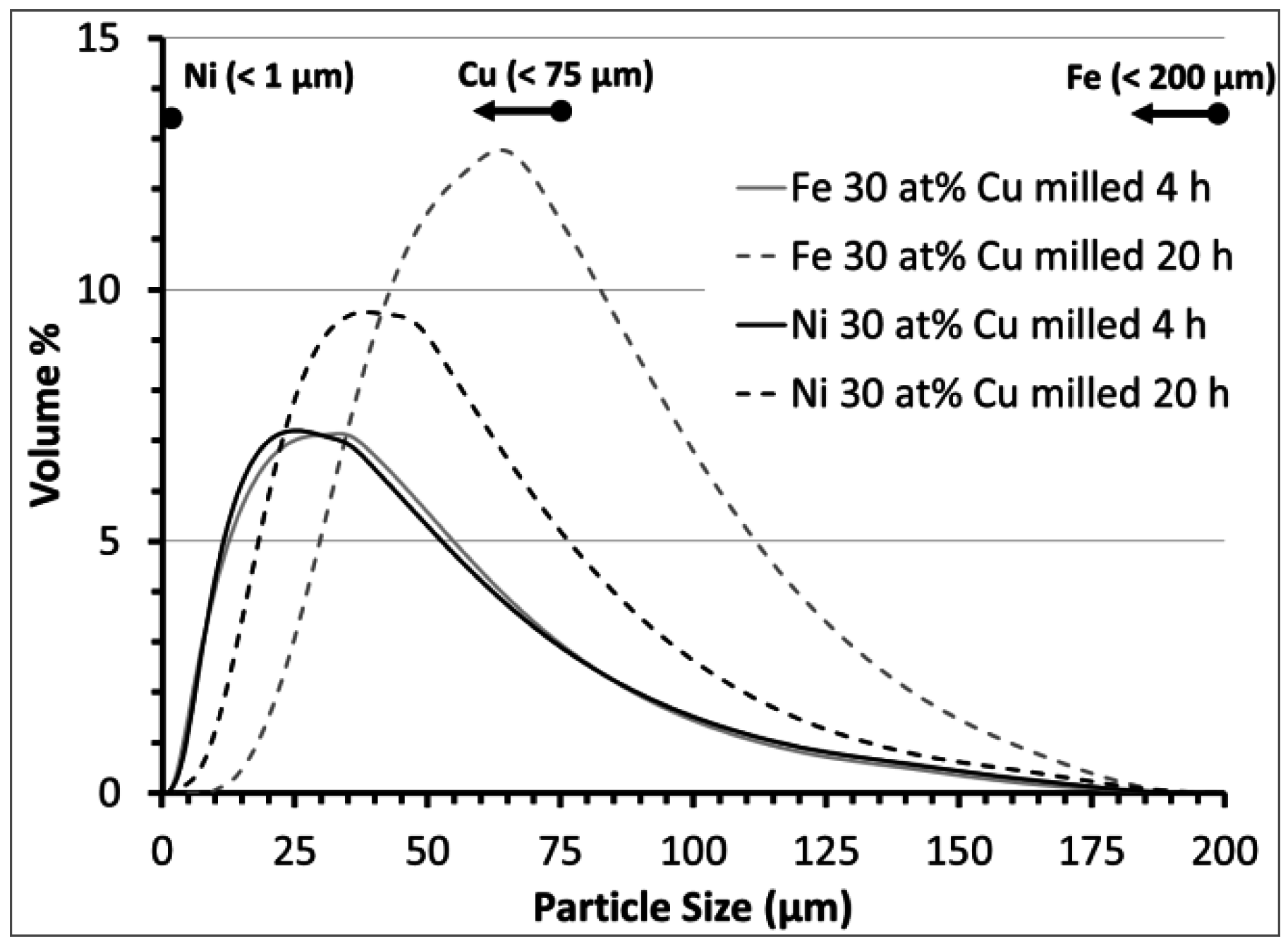
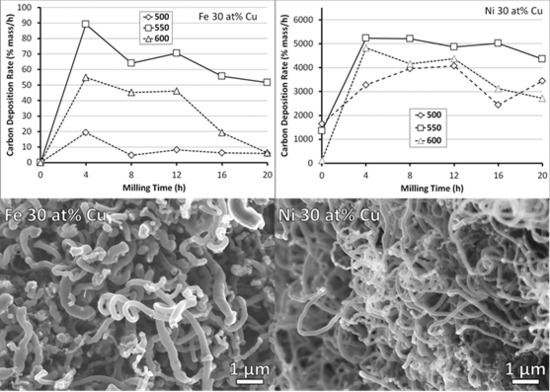
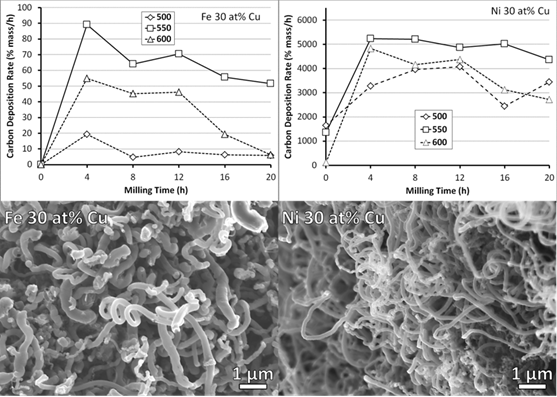
3.2. Carbon Deposition
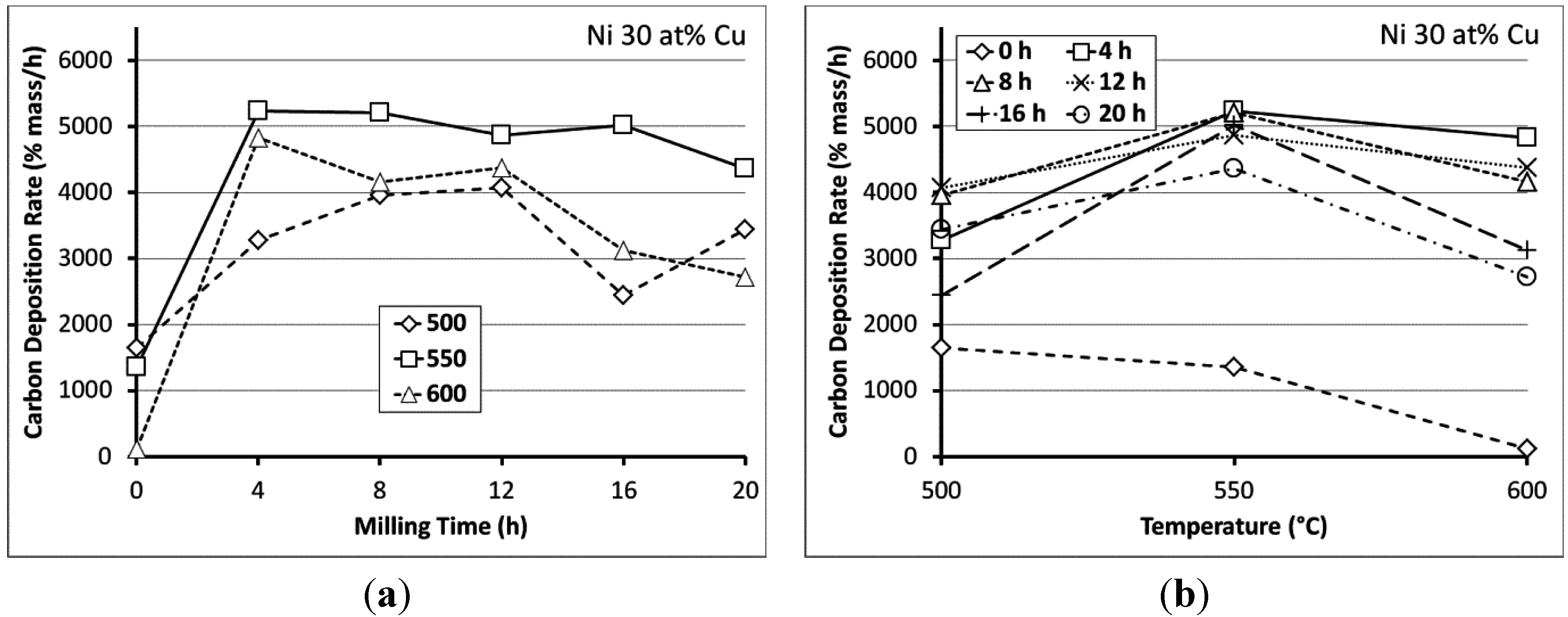
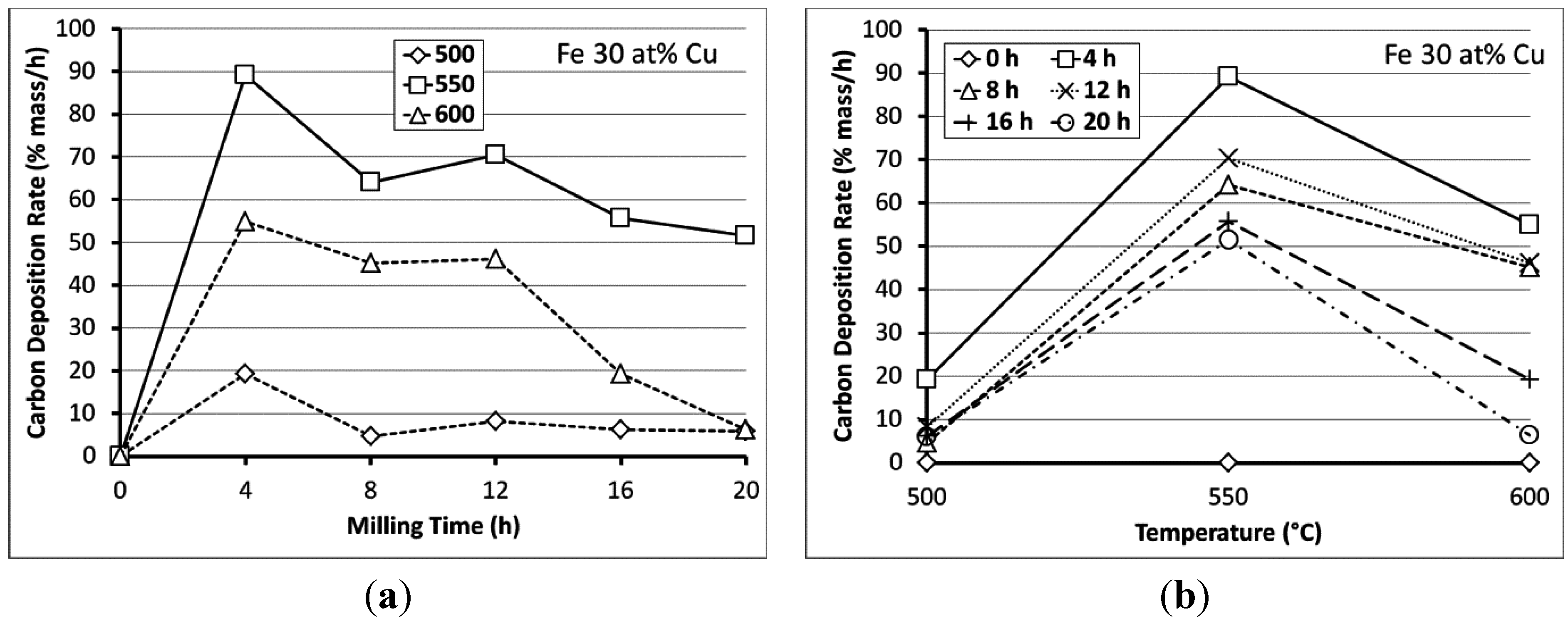
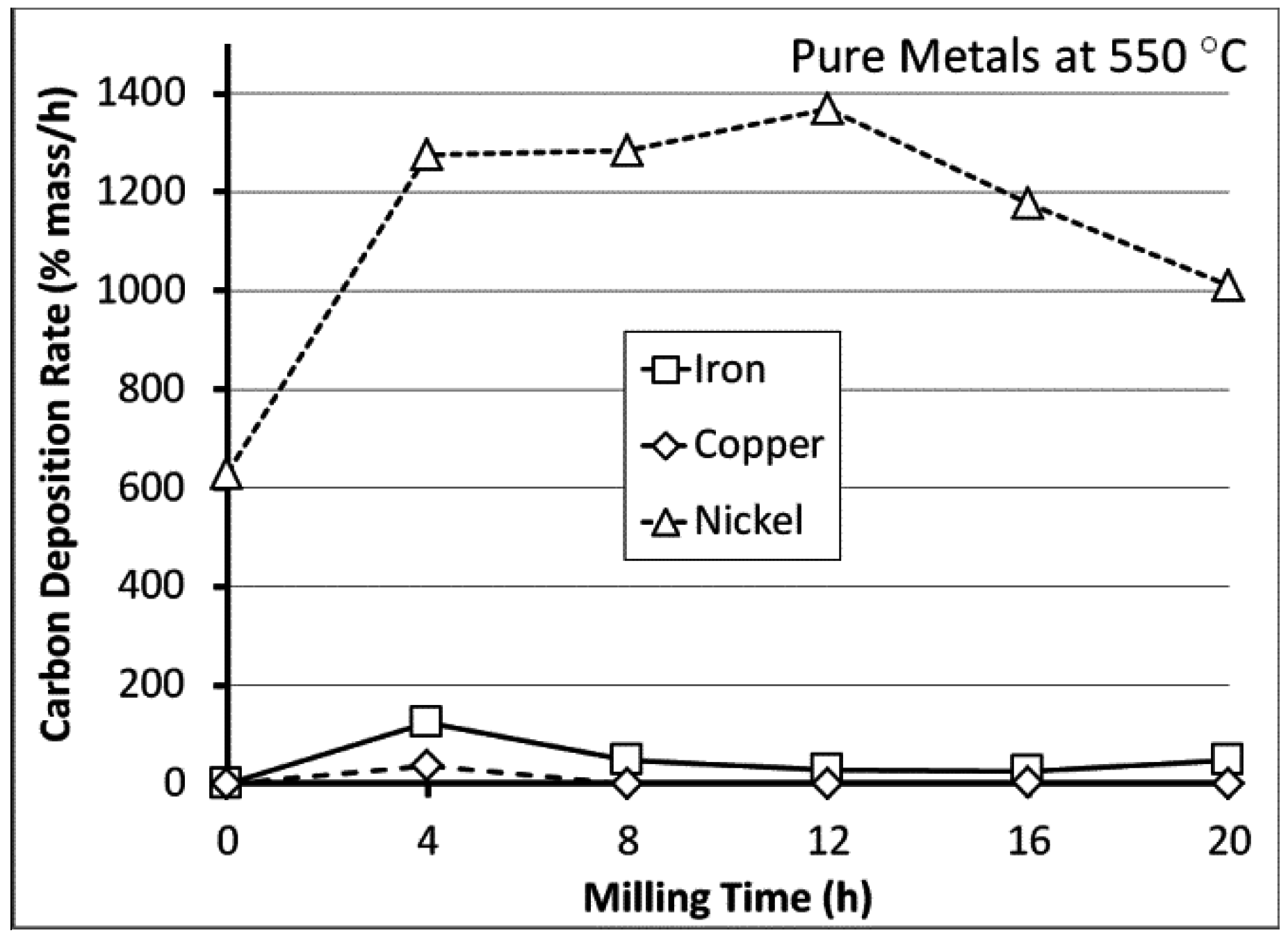

3.3. Processing Considerations
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Jong, K.P.; Geus, J.W. Carbon nanofiber: Synthesis and applications. Catal. Rev. 2000, 42, 481–510. [Google Scholar] [CrossRef]
- Takenaka, S.; Ishida, M.; Serizawa, M.; Tanabe, E.; Otsuka, K. Formation of carbon nanofibers and carbon nanotubes through methane decomposition over supported cobalt catalysts. J. Phys. Chem. B 2004, 108, 11464–11472. [Google Scholar] [CrossRef]
- Baird, T.; Fryer, J.R.; Grant, B. Structure of filamentous carbon. Nature 1971, 233, 329–330. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, N.M.; Kim, M.S.; Baker, R.T.K. Promotion effect of carbon monoxide on the deecomposition of ethylene over an iron catalyst. J. Catal. 1993, 144, 93–108. [Google Scholar] [CrossRef]
- Terrones, H.; Hayashi, T.; Munoz-Navia, M.; Terrones, M.; Kim, Y.A.; Grobert, N.; Kamalkaran, R.; Dorantes-Davila, J.; Escudero, R.; Dresselhaus, M.S.; et al. Graphitic cones in palladium catalysed carbon nanofibers. Chem. Phys. Lett. 2001, 343, 241–250. [Google Scholar] [CrossRef]
- Owens, W.T.; Rodriguez, N.M.; Baker, R.T.K. Carbon filament growth on platinum catalysts. J. Phys. Chem. 1992, 96, 5048–5053. [Google Scholar] [CrossRef]
- Baker, R.T.K.; Barber, M.A.; Harris, P.S.; Feates, F.S.; Waite, R.J. Nucleation and growth of carbon deposits from the nickel catalyzed decomposition of acetylene. J. Catal. 1972, 26, 51–62. [Google Scholar] [CrossRef]
- Borgna, A.; Moraweck, B.; Massardier, J.; Renouprez, A.J. New supported palladium-chromium catalysts: Characterization and catalytic properties. J. Catal. 1991, 128, 99–112. [Google Scholar] [CrossRef]
- Chambers, A.; Rodriguez, N.M.; Baker, R.T.K. Modification of the catalytic behavior of cobalt by the addition of copper. J. Phys. Chem. 1995, 99, 10581–10589. [Google Scholar] [CrossRef]
- Jablonski, G.A.; Geurts, F.W.; A. Sacco, J. Carbon deposition over Fe, Ni, and Co foils from CO-H, -CH4-CO2-H2O, CO-CO2, CH4-H2, and CO-H2-H2O gas mixtures: II: Kinetics. Carbon 1992, 30, 99–106. [Google Scholar] [CrossRef]
- Kvande, I.; Chen, D.; Yu, Z.; Rønning, M.; Holmen, A. Optimization and scale-up of CNF production based on intrinsic kinetic data obtained from teom. J. Catal. 2008, 256, 204–214. [Google Scholar] [CrossRef]
- Park, C.; Baker, R.T.K. Carbon deposition on iron-nickel during interaction with ethylene-carbon monoxide-hydrogen mixtures. J. Catal. 2000, 190, 104–117. [Google Scholar] [CrossRef]
- Chambers, A.; Baker, R.T.K. Influence of the nature of the catalyst precursor on the carbon deposition characteristics during ethylene decomposition over copper–cobalt. J. Catal. 1996, 158, 356–360. [Google Scholar] [CrossRef]
- Rodriguez, N.M.; Kim, M.-S.; Baker, R.T.K. Carbon nanofibers: A unique catalyst support medium. J. Phys. Chem. 1994, 98, 13108–13111. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Mechanisms of catalyst deactivation. Appl. Catal. A Gen. 2001, 212, 17–60. [Google Scholar] [CrossRef]
- Wu, N.L.; Phillips, J. Catalytic etching of platinum during ethylene oxidation. J. Phys. Chem. 1985, 89, 591–600. [Google Scholar] [CrossRef]
- Wu, N.L.; Phillips, J. Carbon deposition on platinum during ethylene oxidation. J. Catal. 1988, 113, 383–397. [Google Scholar] [CrossRef]
- Phillips, J.; Shiina, T.; Nemer, M.; Lester, K. Graphitic structures by design. Langmuir 2006, 22, 9694–9703. [Google Scholar] [CrossRef] [PubMed]
- Atwater, M.A.; Phillips, J.; Doorn, S.K.; Luhrs, C.C.; Fernandez, Y.; Menendez, J.A.; Leseman, Z.C. The production of carbon nanofibers and thin films on palladium catalysts from ethylene–oxygen mixtures. Carbon 2009, 47, 2269–2280. [Google Scholar] [CrossRef]
- Atwater, M.A.; Phillips, J.; Leseman, Z.C. Formation of carbon nanofibers and thin films catalyzed by palladium in ethylene-hydrogen mixtures. J. Phys. Chem. C 2010, 114, 5804–5810. [Google Scholar] [CrossRef]
- Park, C.; Baker, R.T.K. Carbon deposition on iron-nickel during interaction with ethylene-hydrogen mixtures. J. Catal. 1998, 179, 361–374. [Google Scholar] [CrossRef]
- Rodriguez, N.M.; Kim, M.S.; Baker, R.T.K. Deactivation of copper-nickel catalysts due to changes in surface composition. J. Catal. 1993, 140, 16–29. [Google Scholar] [CrossRef]
- Klein, K.L.; Melechko, A.V.; Rack, P.D.; Fowlkes, J.D.; Meyer, H.M.; Simpson, M.L. Cu–Ni composition gradient for the catalytic synthesis of vertically aligned carbon nanofibers. Carbon 2005, 43, 1857–1863. [Google Scholar] [CrossRef]
- Rodriguez, N.M.; Chambers, A.; Baker, R.T.K. Catalytic engineering of carbon nanostructures. Langmuir 1995, 11, 3862–3866. [Google Scholar] [CrossRef]
- Chambers, A.; Rodriguez, N.M.; Baker, R.T.K. Influence of copper on the structural characteristics of carbon nanofibers produced from the cobalt-catalyzed decomposition of ethylene. J. Mater. Res. 1996, 11, 430–438. [Google Scholar] [CrossRef]
- Park, C.; Rodriguez, N.M.; Baker, R.T.K. Carbon deposition on iron-nickel during interaction with carbon monoxide-hydrogen mixtures. J. Catal. 1997, 169, 212–227. [Google Scholar] [CrossRef]
- Rodriguez, N.M.; Kim, M.S.; Baker, R.T.K. Deactivation of copper-nickel catalysts due to changes in surface composition. J. Catal. 2003, 140, 16–29. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, B.; Xie, X.; Liu, J.; Xu, Y.; Shen, W. Novel Ni catalysts for methane decomposition to hydrogen and carbon nanofibers. J. Catal. 2006, 238, 412–424. [Google Scholar] [CrossRef]
- Ngo, Q.; Cassell, A.M.; Radmilovic, V.; Li, J.; Krishnan, S.; Meyyappan, M.; Yang, C.Y. Palladium catalyzed formation of carbon nanofibers by plasma enhanced chemical vapor deposition. Carbon 2007, 45, 424–428. [Google Scholar] [CrossRef]
- Melechko, A.V.; Merkulov, V.I.; McKnight, T.E.; Guillorn, M.A.; Klein, K.L.; Lowndes, D.H.; Simpson, M.L. Vertically aligned carbon nanofibers and related structures: Controlled synthesis and directed assembly. J. Appl. Phys. 2005, 97. [Google Scholar] [CrossRef]
- Palumbo, M.; Curiotto, S.; Battezzatia, L. Thermodynamic analysis of the stable and metastable Co–Cu and Co–Cu–Fe phase diagrams. Calphad 2006, 30, 171–178. [Google Scholar] [CrossRef]
- Turchanin, M.A.; Agraval, P.G.; Nikolaenko, I.V. Thermodynamics of alloys and phase equilibria in the copper-iron system. J. Phase Equilib. 2003, 24, 307–319. [Google Scholar] [CrossRef]
- Suryanarayana, C.; Ivanob, E.; Boldyrev, V.V. The science and technology of mechanical alloying. Mater. Sci. Eng. A 2001, 304–306, 151–158. [Google Scholar] [CrossRef]
- Suryanarayana, C. Mechanical alloying and milling. Prog. Mater. Sci. 2001, 46, 1–184. [Google Scholar] [CrossRef]
- Suryanarayana, C.; Koch, C.C. Nanocrystalline materials—Current research and future directions. Hyperfine Interact. 2000, 130, 5–44. [Google Scholar] [CrossRef]
- Koch, C.C. Materials synthesis by mechanical alloying. Annu. Rev. Mater. Sci. 1989, 19, 121–143. [Google Scholar] [CrossRef]
- Koch, C.C. The synthesis and structure of nanocrystalline materials produced by mechanical attrition: A review. Nanostruct. Mater. 1993, 2, 109–129. [Google Scholar] [CrossRef]
- Koch, C.C.; Cho, Y.S. Nanocrystals by high energy ball milling. Nanostruct. Mater. 1992, 1, 207–212. [Google Scholar] [CrossRef]
- Lü, L.; Lai, M.O. Mechanical Alloying; Kluwer Academic Publishers: Boston, MA, USA, 1998; pp. 1–6. [Google Scholar]
- Yavari, A.R.; Desre, P.J.; Benameur, T. Mechanically driven alloying of immiscible elements. Phys. Rev. Lett. 1992, 68, 2235–2238. [Google Scholar] [CrossRef] [PubMed]
- Carsley, J.E.; Fisher, A.; Milligan, W.W.; Aifantis, E.C. Mechanical behavior of a bulk nanostructured iron alloy. Metall. Mater. Trans. A 1998, 29A, 2261–2271. [Google Scholar] [CrossRef]
- Benjamin, J.S. Mechanical alloying. Sci. Am. 1976, 234, 40–49. [Google Scholar] [CrossRef]
- Darling, K.A.; Roberts, A.J.; Mishin, Y.; Mathaudhu, S.N.; Kecskes, L.J. Grain size stabilization of nanocrystalline copper at high temperatures by alloying with tantalum. J. Alloys Compd. 2013, 573, 142–150. [Google Scholar] [CrossRef]
- Frolov, T.; Darling, K.A.; Kecskes, L.J.; Mishin, Y. Stabilization and strengthening of nanocrystalline copper by alloying with tantalum. Acta Mater. 2012, 60, 2158–2168. [Google Scholar] [CrossRef]
- Atwater, M.A.; Roy, D.; Darling, K.A.; Butler, B.G.; Scattergood, R.O.; Koch, C.C. The thermal stability of nanocrystalline copper cryogenically milled with tungsten. Mater. Sci. Eng. A 2012, 558, 226–233. [Google Scholar] [CrossRef]
- Raghu, T.; Sundaresan, R.; Ramakrishnan, P.; Mohan, T.R.R. Synthesis of nanocrystalline copper–tungsten alloys by mechanical alloying. Mater. Sci. Eng. A 2001, 304–306, 438–441. [Google Scholar] [CrossRef]
- Aboud, T.; Weiss, B.-Z.; Chaim, R. Mechanical alloying of the immiscible system W-Cu. Nanostruct. Mater. 1995, 6, 405. [Google Scholar] [CrossRef]
- Sahani, P.; Mula, S.; Roy, P.K.; Kang, P.C.; Koch, C.C. Structural investigation of vacuum sintered Cu–Cr and Cu–Cr–4% SiC nanocomposites prepared by mechanical alloying. Mater. Sci. Eng. A 2011, 528, 7781–7789. [Google Scholar] [CrossRef]
- Patel, A.N.; Diamond, S. The effects of non-equilibrium processing in the development of copper alloys. Mater. Sci. Eng. A 1988, 98, 329–334. [Google Scholar] [CrossRef]
- Aguilara, C.; Ordonez, S.; Guzman, D.; Rojas, P.A. Mechanical alloying of Cu–xCr (x = 3, 5 and 8 wt.%) alloys. J. Alloys Compd. 2010, 504, 102–109. [Google Scholar] [CrossRef]
- Miettinen, J. Thermodynamic description of Cu–Mg–Ni and Cu–Mg–Zn systems. Calphad 2008, 32, 389–398. [Google Scholar] [CrossRef]
- Atwater, M.A.; Phillips, J.; Leseman, Z.C. Accelerated growth of carbon nanofibers using physical mixtures and alloys of Pd and Co in an ethylene–hydrogen environment. Carbon 2011, 49, 1058–1066. [Google Scholar] [CrossRef]
- Black, D.R.; Windover, D.; Henins, A.; Gil, D.; Filliben, J.; Cline, J.P. Certification of nist standard reference material 640d. Powder Diffr. 2010, 25, 187–190. [Google Scholar] [CrossRef]
- Koch, C.C.; Scattergood, R.O.; Darling, K.A.; Semones, J.E. Stabilization of nanocrystalline grain sizes by solute additions. J. Mater. Sci. 2008, 43, 7264–7272. [Google Scholar] [CrossRef]
- Krill, C.E.; Ehrhardt, H.; Birringer, R. Thermodynamic stabilization of nanocrystallinity. Z. Metallkd. 2005, 96, 1134–1141. [Google Scholar] [CrossRef]
- Millett, P.C.; Selvam, R.P.; Saxena, A. Molecular dynamics simulations of grain size stabilization in nanocrystalline materials by addition of dopants. Acta Mater. 2006, 54, 297–303. [Google Scholar] [CrossRef]
- Chookajorn, T.; Murdoch, H.A.; Schuh, C.A. Design of stable nanocrystalline alloys. Science 2012, 337, 951–954. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, H.A.; Schuh, C.A. Stability of binary nanocrystalline alloys against grain growth and phase separation. Acta Mater. 2013, 61, 2121–2132. [Google Scholar] [CrossRef]
- Darling, K.A.; Tschopp, M.A.; VanLeeuwen, B.K.; Atwater, M.A.; Liu, Z.K. Mitigating grain growth in binary nanocrystalline alloys through solute selection based on thermodynamic stability maps. Comp. Mater. Sci. 2014, 84, 255–266. [Google Scholar] [CrossRef]
- Andrievski, R.A. Review of thermal stability of nanomaterials. J. Mater. Sci. 2014, 49, 1449–1460. [Google Scholar] [CrossRef]
- Cullity, B.D.; Stock, S.R. Elements of X-ray Diffraction, 3rd ed.; Prentice Hall: Upper Saddle River, NJ, USA, 2001; p. 170. [Google Scholar]
- Kleiner, S.; Bertocco, F.; Khalid, F.A.; Beffort, O. Decomposition of process control agent during mechanical milling and its influence on displacement reactions in the Al–TiO2 system. Mater. Chem. Phys. 2005, 89, 362–366. [Google Scholar] [CrossRef]
- Zhang, Y.F.; Lu, L.; Yap, S.M. Prediction of the amount of pca for mechanical milling. J. Mater. Process Tech. 1999, 89–90, 260–265. [Google Scholar] [CrossRef]
- Jarrah, N.A.; Ommen, J.G.V.; Lefferts, L. Mechanistic aspects of the formation of carbon-nanofibers on the surface of Ni foam: A new microstructured catalyst support. J. Catal. 2006, 239, 460–469. [Google Scholar] [CrossRef]
- Martin-Gullon, I.; Vera, J.; Conesa, J.A.; Gonzalez, J.L.; Merino, C. Differences between carbon nanofibers produced using Fe and Ni catalysts in a floating catalyst reactor. Carbon 2006, 44, 1572–1580. [Google Scholar] [CrossRef]
- Anderson, P.E.; Rodriguez, N.M. Influence of the support on the structural characteristics of carbon nanofibers produced from the metal-catalyzed decomposition of ethylene. Chem. Mater. 2000, 12, 823–830. [Google Scholar] [CrossRef]
- Boellaard, E.; Bokx, P.K.D.; Kock, A.J.H.M.; Geus, J.W. The formation of filamentous carbon on iron and nickel catalysts—III morphology. J. Catal. 1985, 96, 481–490. [Google Scholar] [CrossRef]
- Bokx, P.K.D.; Kock, A.J.H.M.; Boellaard, E.; Klop, W.; Geus, J.W. The formation of filamentous carbon on iron and nickel catalysts: I. thermodynamics. J. Catal. 1985, 96, 454–467. [Google Scholar]
- Kock, A.J.H.M.; Bokx, P.K.D.; Boellaard, E.; Klop, W.; Geus, J.W. The formation of filamentous carbon on iron and nickel catalysts. J. Catal. 1985, 96, 468–480. [Google Scholar] [CrossRef]
- Kim, M.S.; Rodriguez, N.M.; Baker, R.T.K. The interaction of hydrocarbons with copper-nickel and nickel in the formation of carbon filaments. J. Catal. 1991, 131, 60–73. [Google Scholar] [CrossRef]
- Carneiro, O.C.; Rodriguez, N.M.; Baker, R.T.K. Growth of carbon nanofibers from the iron–copper catalyzed decomposition of CO/C2H4/H2 mixtures. Carbon 2005, 43, 2389–2396. [Google Scholar] [CrossRef]
- Rodriguez, N.M. A review of catlytically grown carbon nanofibers. J. Mater. Res. 1993, 8, 3233–3250. [Google Scholar] [CrossRef]
- Jablonski, G.A.; Geurts, F.W.; Sacco, J.A.; Biederman, R.R. Carbon deposition over Fe, Ni, and Co foils from CO-H, -CH4-CO2-H2O, CO-CO2, CH4-H2, and CO-H2-H2O gas mixtures: I. Morphology. Carbon 1992, 30, 87–98. [Google Scholar] [CrossRef]
- Yang, X.F.; Lu, J. A new way to synthesize carbon nanofiber film on bulk titanium via hybrid surface mechanical attrition treatment. Appl. Surf. Sci. 2013, 264, 191–196. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guevara, L.; Wanner, C.; Welsh, R.; Atwater, M.A. Using Mechanical Alloying to Create Bimetallic Catalysts for Vapor-Phase Carbon Nanofiber Synthesis. Fibers 2015, 3, 394-410. https://doi.org/10.3390/fib3040394
Guevara L, Wanner C, Welsh R, Atwater MA. Using Mechanical Alloying to Create Bimetallic Catalysts for Vapor-Phase Carbon Nanofiber Synthesis. Fibers. 2015; 3(4):394-410. https://doi.org/10.3390/fib3040394
Chicago/Turabian StyleGuevara, Laura, Crystal Wanner, Roger Welsh, and Mark A. Atwater. 2015. "Using Mechanical Alloying to Create Bimetallic Catalysts for Vapor-Phase Carbon Nanofiber Synthesis" Fibers 3, no. 4: 394-410. https://doi.org/10.3390/fib3040394
APA StyleGuevara, L., Wanner, C., Welsh, R., & Atwater, M. A. (2015). Using Mechanical Alloying to Create Bimetallic Catalysts for Vapor-Phase Carbon Nanofiber Synthesis. Fibers, 3(4), 394-410. https://doi.org/10.3390/fib3040394