Abstract
In this work, multifilament, continuous polyacrylonitrile (PAN) fiber tow was solution spun mimicking industrial processing at the small pilot scale (0.5 k tow), while carefully altering the composition of the coagulation bath, in order to determine the effect on the resulting fiber shape, density, orientation, and tensile properties at varying points in the spinning process. Novel here are the abnormally high coagulation bath solvent compositions investigated, which surpass those often reported in the literature. In addition, the coagulation bath was maintained at a slightly chilled temperature, contrary to reported methods to produce round fibers. Further, by altering the composition of the bath in a step-wise fashion during a single spinning run, variations in all other process parameters were minimized. We found that with increasing solvent composition in the coagulation bath, the fibers not only became round in cross section, but also became smaller in diameter, which persisted down the spin line. With this decrease in diameter, all else equal, came an accompanying increase in apparent fiber density via a reduction in microvoid content. In addition, molecular orientation and tensile properties also increased. Therefore, it was found that inadequate understanding of the coagulation bath effects, and spinning at low coagulation bath solvent compositions, can hinder the ability of the fiber to reach optimum properties.
1. Introduction
Carbon fiber properties are directly dependent on the quality of the precursor fiber [1,2,3]. Carbon fiber modulus has been shown to be directly proportional to the polyacrylonitrile (PAN) precursor modulus [4]. As a result, careful control of precursor properties is necessary. It has been shown that precursor fibers with non-round cross-section cannot withstand a high draw ratio during subsequent spinning, stabilization, and carbonization due to stress concentration [5]. Circular fibers, on the other hand, experience a homogeneous Poisson’s contraction when exposed to tensile forces [6]. Tsai found that the shape of the cross-section is one of the most important characteristics of fibers, with deviation from circularity affecting luster, mechanical, and other physical properties [6].
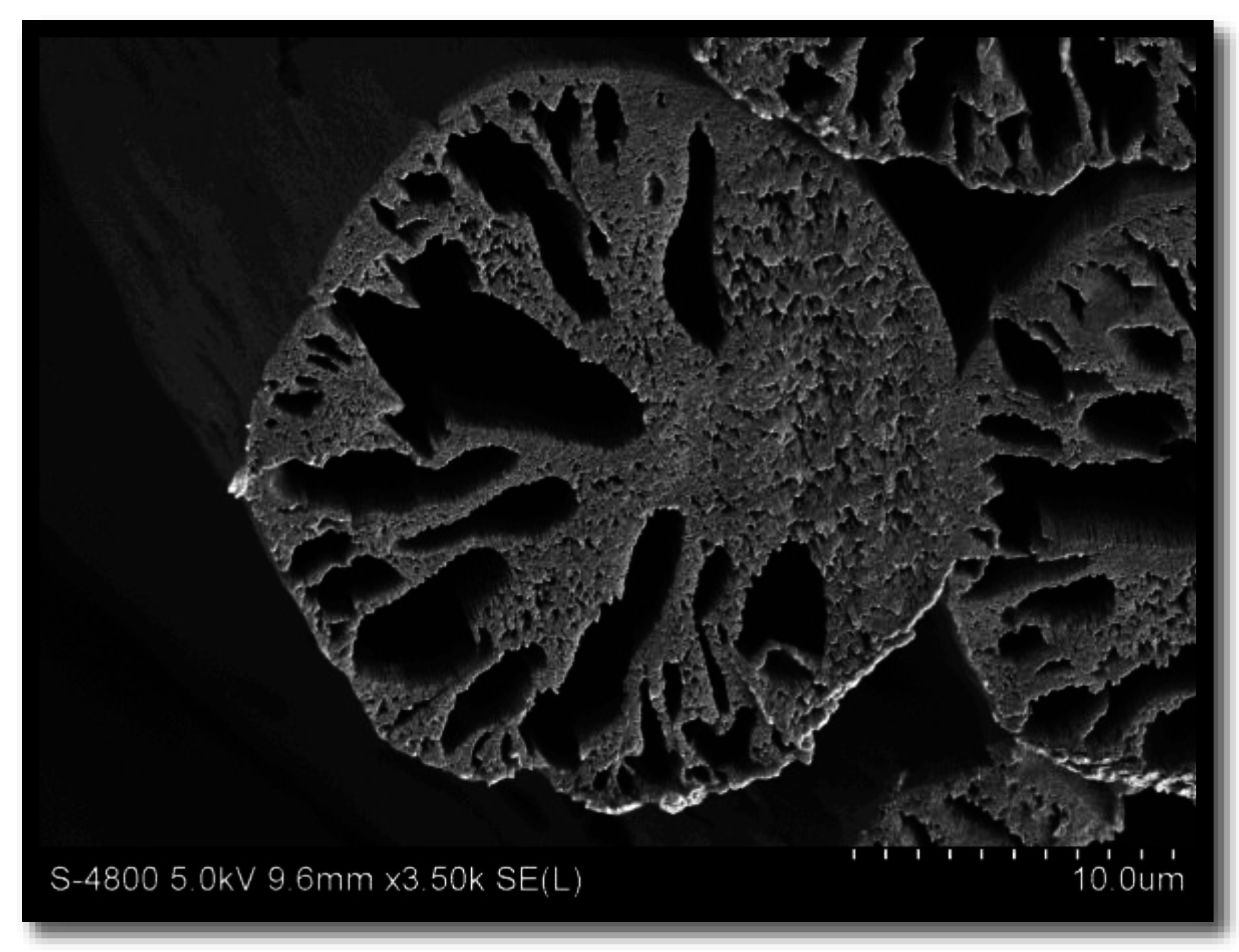
Achievement of a circular cross section is largely determined by controlling the relative diffusion of solvent out and non-solvent in the filaments during coagulation. When the flux of solvent outward is less than the inward flux, the filament swells and a circular cross-section can be expected. This swelling occurs at high coagulation bath temperatures and high solvent content in the coagulating bath [6]. For this reason, cross sections are characteristically round at 50 °C or above. However, high bath temperatures have also been attributed to an increase in void content and subsequent decrease in fiber density, resulting in poor fiber properties [2,3,7]. An example of a PAN fiber spun into a high temperature coagulation bath (60 °C) is shown in Figure 1. Although circular in cross sectional shape, large macrovoids are evident.
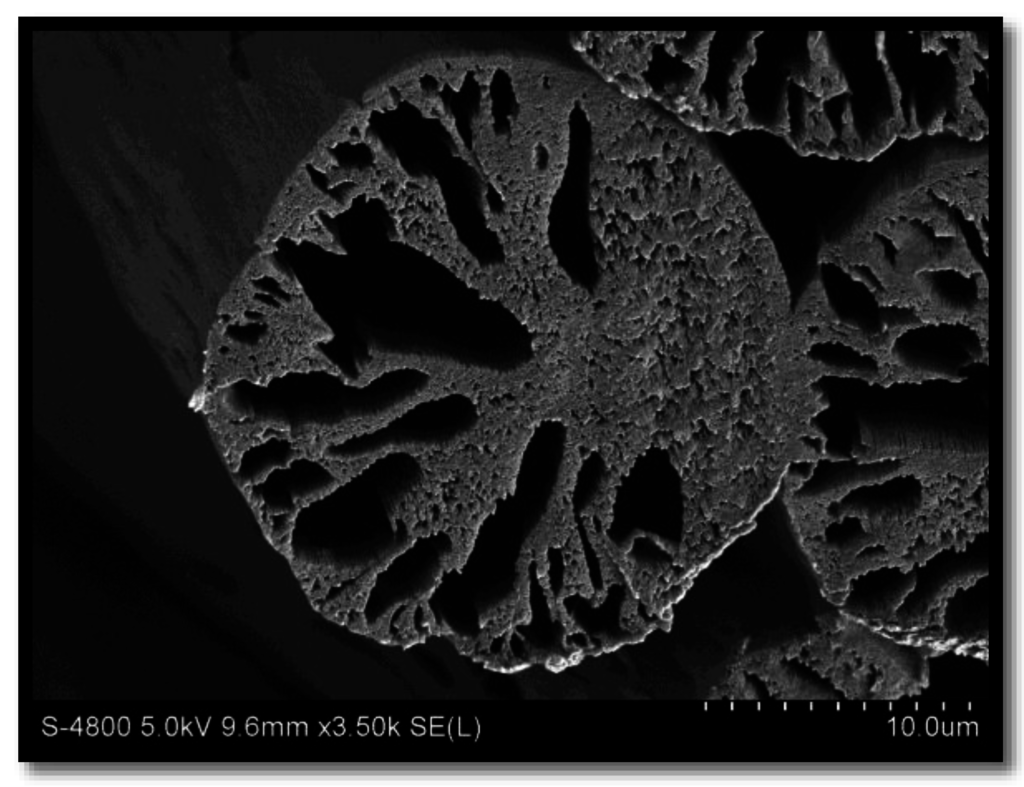
Figure 1.
Example of a polyacrylonitrile (PAN) fiber spun at high coagulation bath temperature (60 °C), containing macrovoids.
Figure 1.
Example of a polyacrylonitrile (PAN) fiber spun at high coagulation bath temperature (60 °C), containing macrovoids.
In contrast, at lower coagulation temperatures and typical solvent concentrations (<70 wt%), the outward diffusion of solvent dominates, but the perimeter of the filament in contact with non-solvent is solidified early. As outward diffusion of solvent continues, the filament collapses, resulting in non-round (often bean-shaped) cross-sectional shape [3,5,8].
Due to the difficulties in achieving a round, dense cross-section fiber, numerous researchers have investigated the effect of coagulation bath composition on the resulting fiber. Few, if any, have focused on high solvent concentrations in the coagulation bath (>70 wt% solvent/non-solvent), with chilled coagulation bath temperature, and its effect on the resulting fiber. Multiple authors have studied bath concentrations up to 70 wt% [2,3,6,8,9,10,11]. Many of these authors were spinning at elevated bath temperatures (>20 °C). As a result, they were often unable to attain void-free round fibers.
Unique to this research effort, the results gathered here were produced on a small pilot scale (0.5 k filament, continuous tow), with industrially relevant filament diameters, and all samples were collected throughout the course of a single spin run. Sampling from a single run limited changes to spinning variables which may be present in data collected during various runs, with differing spinning solutions, environmental factors, etc. Here, a systematic study of coagulation bath composition, at high solvent composition and low bath temperature is reported. The resulting fiber shape, density, orientation, and tensile properties at varying points in the spinning process are discussed.
2. Experimental Section
2.1. Materials
Polyacrylonitrile-co-methyl acrylate (PAN-co-MA) polymer from Scientific Polymer Products (catalog number 665) and reagent grade N,N-Dimethylacetamide (DMAc) from Fisher Chemical (Pittsburg, PA, USA) were used. Deionized water was used as the non-solvent.
2.2. Fiber Spinning
All fibers were spun utilizing the multifilament solution spinning facility at the University of Kentucky Center for Applied Energy Research (UKY CAER) (Figure 2). A 23 wt% solution of PAN-co-MA/DMAc was prepared, deaerated, filtered, and solution spun into a DMAc/H2O coagulation bath using a 500 filament wet-jet spinnerette with 60 µm capillary diameters. All fibers presented in this study were spun during a single 5-hour spinning run. The only variable allowed was the concentration of solvent to water in the coagulation bath. The initial bath concentration, as measured by calibrated refractive index, was 79.0 wt% DMAc/H2O. Wet-jet solution spinning at >80 wt% solvent in the coagulation bath was previously found to be marginally effective at coagulating or demixing the polymer dope jets in a timely manner, and resulted in excessively fragile and viscous proto-filaments. Therefore, this was determined to be the upper limit in solvent concentration for the given system. After achieving spinning stability, samples were collected at various points along the spinline, shown in Figure 3.
Following sample collection at a given bath concentration, a calculated mass of solution was pumped from the coagulation bath into a collection container and replaced with a calculated mass of pure deionized water to dilute the bath, defining the bath concentration for the subsequent fiber sampling, etc. Each new coagulation bath solution was mixed thoroughly using an in-bath stirring system, until no further evidence of water-solvent dissolution was observed and the bath temperature was stable. For all samples, the coagulation bath temperature was held chilled to 19 ± 1.4 °C and fiber residence time within the coagulation bath was 87 s, with a calculated extrudate jet velocity of 1.3 m/min (not corrected for viscoelastic die swell), a take-up velocity of 0.9 m/min out of the coagulation bath, and a path length of 1.3 m. Excess heat of mixing from DMAc and water was dissipated using a circulating water/glycol external bath (heat sink). In addition, while five coagulation bath concentrations were studied, only three of the five coagulation conditions were studied down the entirety of the line, shown in Table 1.

Figure 2.
Solution spinning line at the University of Kentucky Center for Applied Energy Research (UKY CAER).
Figure 2.
Solution spinning line at the University of Kentucky Center for Applied Energy Research (UKY CAER).

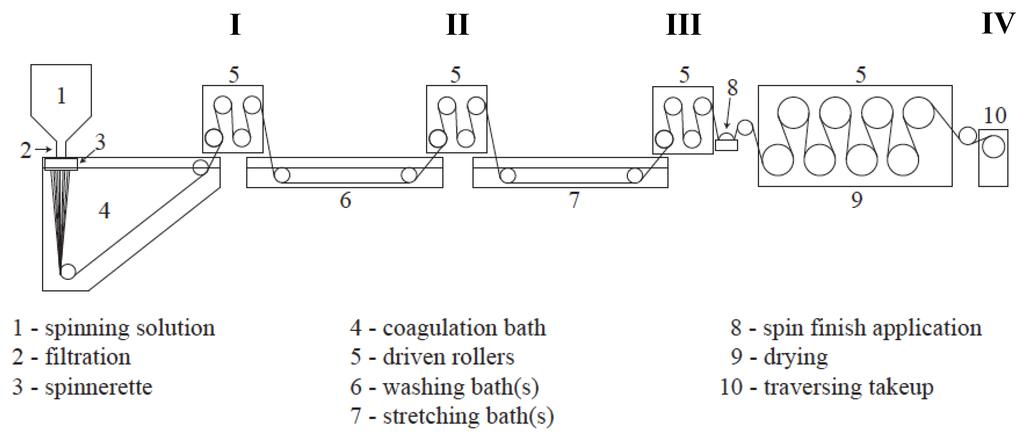
Figure 3.
Schematic of the solution spinning line at the UKY CAER, labeled with sampling points (I–IV). (I) After coagulation; (II) after washing; (III) after hot stretching; (IV) on the traversing takeup.
Figure 3.
Schematic of the solution spinning line at the UKY CAER, labeled with sampling points (I–IV). (I) After coagulation; (II) after washing; (III) after hot stretching; (IV) on the traversing takeup.
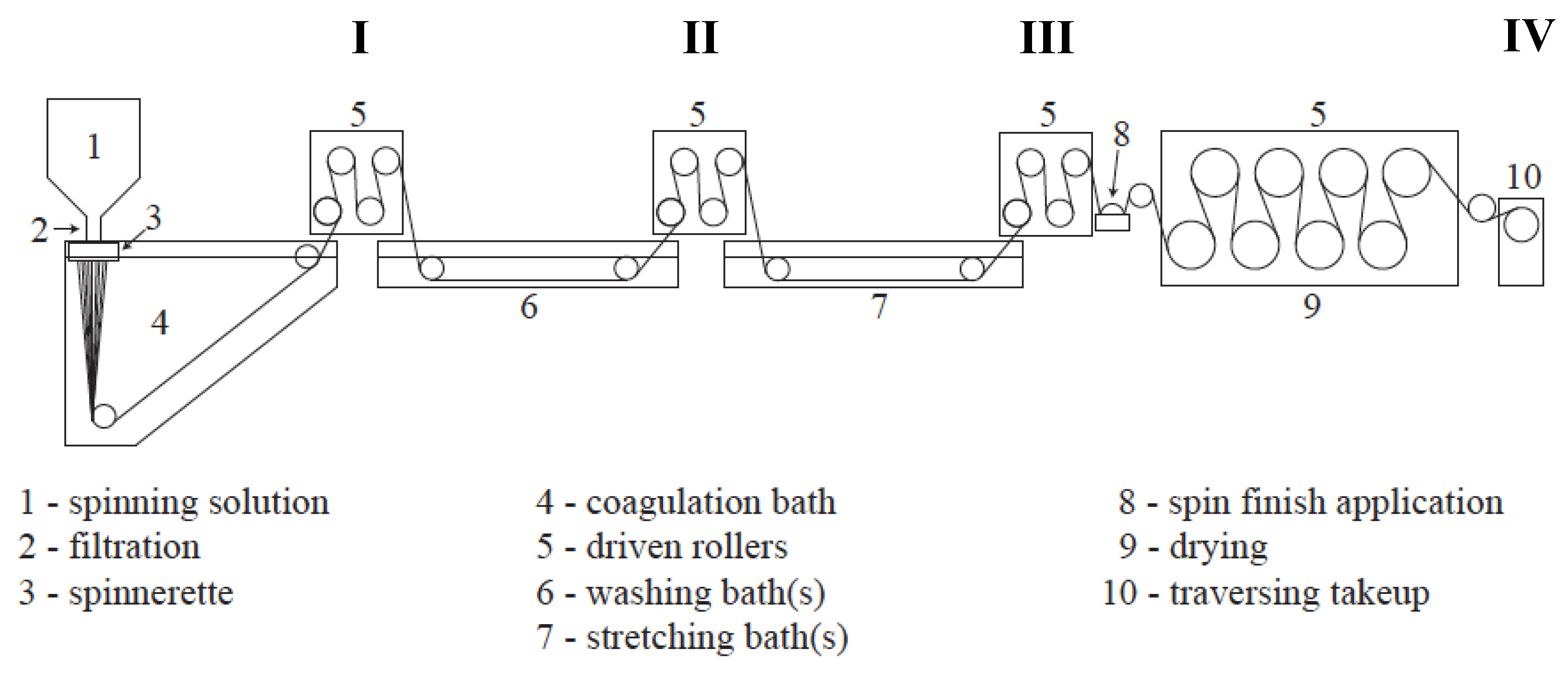

Table 1.
Locations of fiber sample collections along the spinline at various coagulation bath concentrations.
| Coagulation Bath Concentration (wt% DMAc/H2O) | Locations of Fiber Sample Collection | |||
|---|---|---|---|---|
| I | II | III | IV | |
| 57.8 | ✓ | ✓ | ✓ | ✓ |
| 62.1 | ✓ | |||
| 66.6 | ✓ | ✓ | ✓ | ✓ |
| 72.2 | ✓ | |||
| 79.0 | ✓ | ✓ | ✓ | ✓ |
Note: ✓ indicates fiber sampled at that point.
Following the coagulation bath, the tow was passed through a series of ambient temperature wash baths and hot stretching baths prior to spin finish application, drying, and take-up using a traversing winder. The draw down ratio is defined as the ratio of the tow velocities, exiting-to-entering the zone of interest. The spin draw (sampling point I), or draw applied during coagulation, calculated from the apparent entering and exiting velocities, was less than 1, which has been observed by others [12,13,14,15,16,17]. The tow velocity entering coagulation was calculated using the known, constant volumetric flow rate of the dope, and the dimension of the spinneret, and was used, uncorrected for viscoelastic die swell of the extruded jets, for the velocity calculation. Therefore, spin draw less than one did not necessarily indicate a tow shrinkage in the coagulation bath. Throughout the spin run the volumetric flow rate of the dope and take-up velocity out of the coagulation bath remained constant. The draw down ratios (stretch) applied at points I, II, III, and IV were 0.67×, 2.20×, 3.06×, and 1.07×, respectively, totaling 4.83×. The tow experiences a “gel draw” of 2.20× in the first wash bath, as the tow still contains enough solvent to plasticize the fibers. No stretch is applied to the fibers in the remaining wash baths. The largest draw (3.06×) occurred in the stretching baths (III). Here, the solidified, washed fiber was heated above its glass transition to 160 °C, allowing for stretch and collapse of intra-filament porosity. Again, the total draw experienced by the as-spun fiber was 4.83×.
2.3. Refractive Index
The coagulation bath concentrations were verified using a digital bench-top refractometer (RX-5000, Atago, Bellevue, NE, USA). A calibration curve for DMAc/H2O solutions was determined, shown in Figure 4. For concentrations from 0 wt% to 65 wt% DMAc/H2O, there was a linear trend (R2 = 0.99). However, from 65 wt% to 100 wt% DMAc/H2O, the calibration curve produced a polynomial trend line (R2 = 0.98). These trends, and the measured refractive index from each of the bath concentrations, were used to determine the exact bath concentration at each of the fiber coagulation conditions.
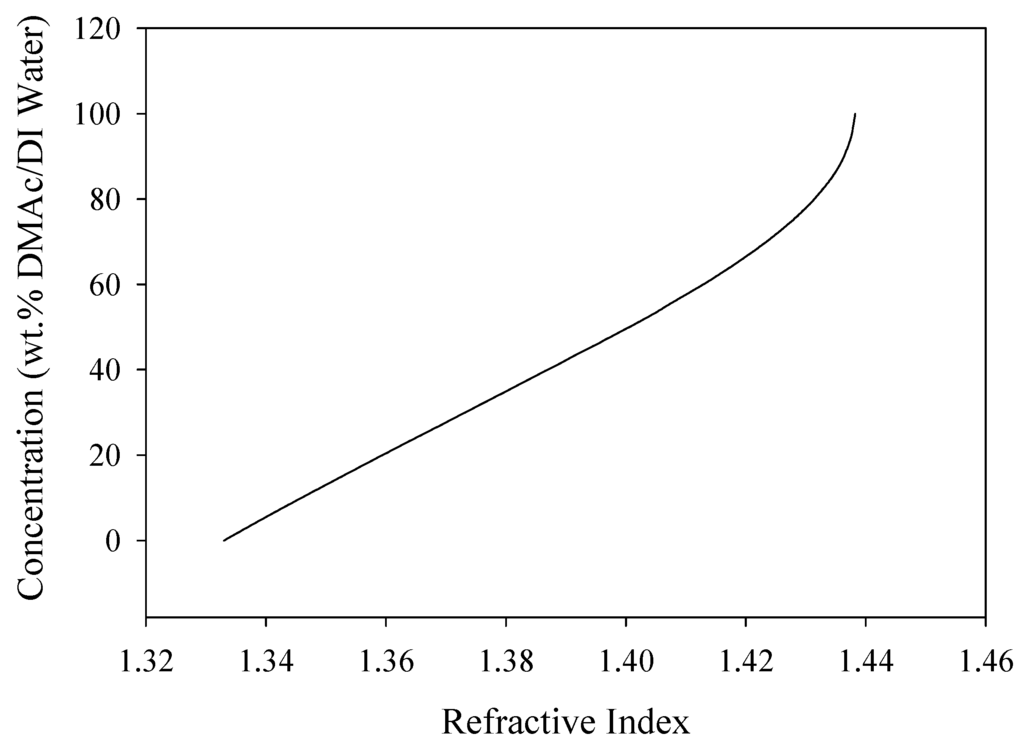
Figure 4.
Calibration curve developed using the refractive index for DMAc/H2O solutions.
Figure 4.
Calibration curve developed using the refractive index for DMAc/H2O solutions.
2.4. Freeze Drying
Fibers sampled from zones I–III were freeze-dried to preserve the uncollapsed fiber structure by first washing the collected, undried fiber in deionized water for the removal of excess solvent (as-spun fibers (zone IV) did not require freeze-drying, as the fibers at this point were collapsed and dried prior to takeup). The freeze-drying unit was chilled to −75 °C using liquid nitrogen. Following removal of solvent, the fibers were placed in the chilled freeze dryer, sealed, and subject to vacuum to reduce the pressure below the triple point of water (611.73 Pa, 273.16 K). While maintaining pressure below 611.73 Pa, as monitored with a thermocouple pressure gauge, the unit was allowed to slowly warm to room temperature over several hours following evaporation of the liquid nitrogen. Samples were removed when room temperature was achieved.
2.5. Fiber Sectioning
Following freeze-drying, fibers were sectioned to observe the cross section and structure using a Shandon Cryotome FSE (Themo Fisher Scientific, Kalamazoo, MI, USA). Fiber bundles were embedded in Tissue-Tek OCT compound (Sakura Finetek USA, Inc., Torrance, CA, USA) and cooled to −27 °C to harden the matrix compound. With the specimen at −27 °C and the chamber at −25 °C, the sample was trimmed with a microtome blade perpendicular to the fiber axis to achieve a clean cut, without pinching or altering the fiber cross-sections. The cut fibers were then washed in room temperature deionized water for removal of the matrix compound. Coag fibers, or fibers collected directly from the coagulation bath, were suitably brittle, following freeze-drying, to be pull-fractured under liquid nitrogen. Pull-fracturing coag fiber in this manner preserved the intra-filament fibrillar and void structure, which can be damaged by the microtome.
2.6. Scanning Electron Microscopy (SEM)
Imaging of the filament surface and cross sectional morphology was performed using a Hitachi S-4800 field emission SEM (Hitachi High-Technologies Corporation, Tokyo, Japan). Samples were sputter-coated with gold for 120 s using a Hummer 6.2 Sputter System (Anatech USA, Union City, CA, USA) using a sputtering current of 20 mA. For cross section observation, the previously microtomed fiber bundles were positioned vertically in a thin specimen split mount SEM sample holder (Ted Pella, Redding, CA, USA). Fibers were imaged with a 5 kV accelerating voltage and a 10 μA beam current, using the secondary electron detector (Hitachi High-Technologies Corporation, Tokyo, Japan). SEM digital image analysis, including cross sectional area and perimeter (for fiber diameter calculation), was done using Adobe Photoshop CS6 (Adobe, San Jose, CA, USA).
2.7. Wide-Angle X-Ray Diffraction (WAXD)
A Rigaku Smartlab 1 kW X-ray diffraction system (Rigaku Corporation, Tokyo, Japan) providing Ni-filtered CuKα radiation (λ = 1.54 Å) as the source and an operational voltage of 40 kV and current of 44 mA was used to measure the crystalline-related properties and orientation of the fibers. A Rigaku αβ-stage attachment (Rigaku Corporation, Tokyo, Japan) was used to investigate the 360-degree azimuthal circle, allowing the fiber axis to be rotated 360 degrees around the vertical. A step width of 0.2° at 5°/min was used for the azimuthal scan from −90 to 270°. Multi-filament tows were aligned perpendicular to the incident X-ray beam as carefully as possible, although some overlap of filaments could not be avoided (causing slight asymmetry in azimuthal peaks). The diffracted X-ray intensity data was analyzed using PDXL 2.0 software (Rigaku Corporation, Tokyo, Japan), and peaks were fit using a split pseudo-Voigt function. Data was smoothed using B-spline smoothing (chi = 1.50). The Herman’s orientation factor, fc, was determined from the corrected azimuthal intensity distribution from the (100) reflection (2θ = 16.9°), using Equations (1) and (2):
where is the azimuthal angle and is the scattered intensity along the angle . The Herman’s orientation factor has the value of unity when the normal of the reflection plane is parallel to the reference direction and a value of −0.5 when the normal is perpendicular to the reference direction. A value of zero occurs when there is completely random orientation. The crystal size () and d-spacing were calculated using Equation (3) (the Scherrer equation) and Equation (4) (the Bragg equation), respectively:
where is the apparatus constant (0.94) and is the FWHM. Corrections for instrumental broadening were neglected in the calculation.
2.8. Tensile Property Analysis
Tensile properties of fiber samples were measured using ≥20 specimens per sample at a crosshead speed of 5 mm/min. The specimens were prepared by bonding a single filament to a 20 mm gauge length aperture card using Easypoxy K-20 (Cytec, Woodland Park, CO, USA). Following curing of the epoxy, the aperture card was mounted in miniature tensile grips of an MTS Systems Q10 machine (MTS, Eden Prairie, MN, USA) fitted with a 150 g load cell. The sides of the aperture card were then cut, leaving the filament intact.
3. Results and Discussion
3.1. Cross Sectional Shape
Freeze dried then cryo-fractured fibers obtained from sampling point I, directly after the coagulation bath, are shown in Figure 5. The fibers clearly show a change in fiber cross section from bean shaped to circular with increasing solvent concentration in the coagulation bath. This has been shown by numerous authors [5,9,10,11,18] and is due to the formation of a skin on the fiber surface when spun into water-rich conditions, which encourage instantaneous demixing of the polymer dope jets. This skin remains as the filament core collapses during diffusion, resulting in a bean shaped fiber. At higher coagulation bath solvent concentrations, which encourage delayed demixing of the polymer dope jets, little to no skin is formed, and the fiber is allowed to contract uniformly. One unique observation is the decrease in equivalent circular fiber diameter with increasing bath solvent content at sampling point I, as shown in Figure 6. From 57.8 wt% to 79.0 wt% DMAc/H2O, the fiber diameter decreases from 65 to 44 μm. This will be addressed in the following section.

Figure 5.
SEM images of freeze-dried, cryo-fractured fiber from the coagulation bath (sampling point I) at varying coagulation bath concentrations (DMAc/H2O). (a) 57.8 wt%; (b) 62.1 wt%; (c) 66.6 wt%; (d) 72.2 wt%; (e) 79.0 wt%.
Figure 5.
SEM images of freeze-dried, cryo-fractured fiber from the coagulation bath (sampling point I) at varying coagulation bath concentrations (DMAc/H2O). (a) 57.8 wt%; (b) 62.1 wt%; (c) 66.6 wt%; (d) 72.2 wt%; (e) 79.0 wt%.

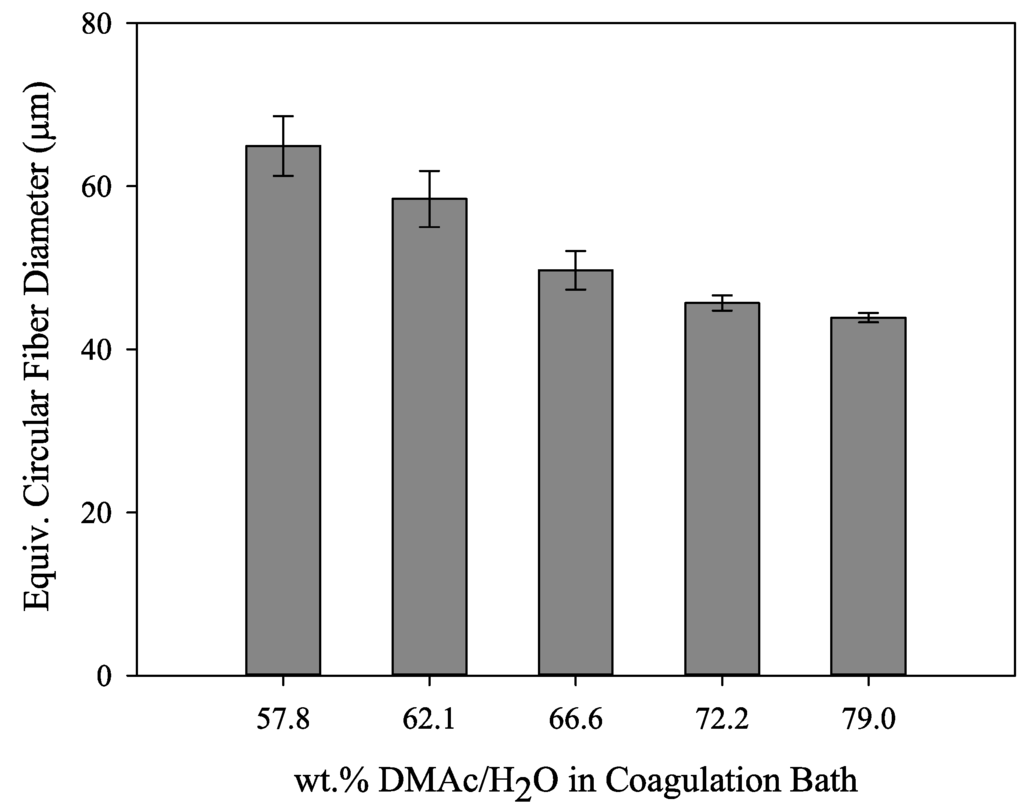
Figure 6.
Equivalent circular fiber diameter for the fibers shown in Figure 5 (sampling point 1).
Figure 6.
Equivalent circular fiber diameter for the fibers shown in Figure 5 (sampling point 1).
3.2. Fiber Development down the Spinning Line
Following analysis of the fibers collected at sampling point I, fibers from the 57.8, 66.6, and 79.0 wt% DMAc/H2O coagulation baths were spun down the line under identical conditions and sampled at points II–IV. Figure 7 shows the SEM cross-sections of the resulting fibers at 1000× magnification. Changes in fiber diameter are evident as the fibers progress from I to IV, due to the stretches applied as discussed in Section 2.2, while cross-sectional shape is retained.
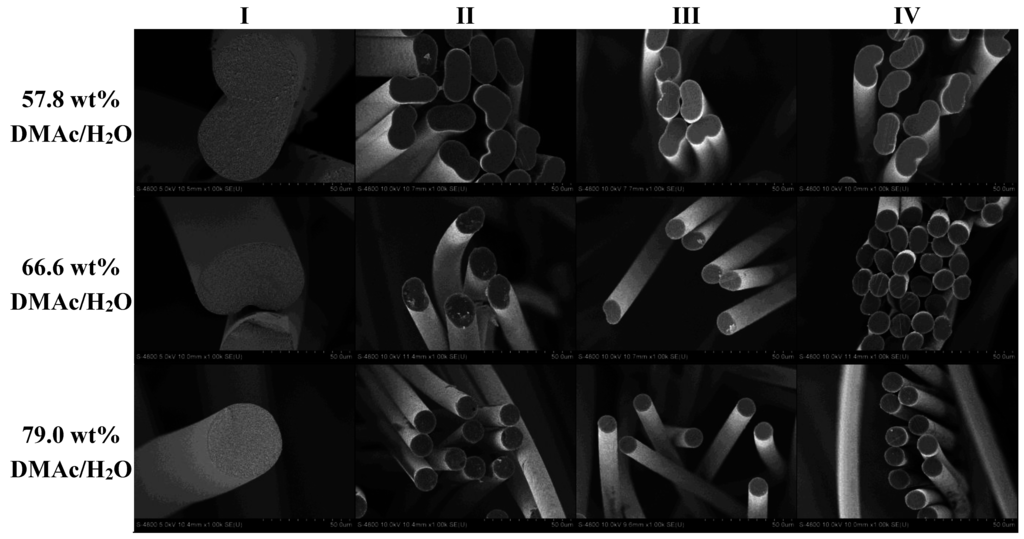
Figure 7.
Effect of coagulation bath composition on fiber shape and size as the fiber progresses down the spinning line.
Figure 7.
Effect of coagulation bath composition on fiber shape and size as the fiber progresses down the spinning line.
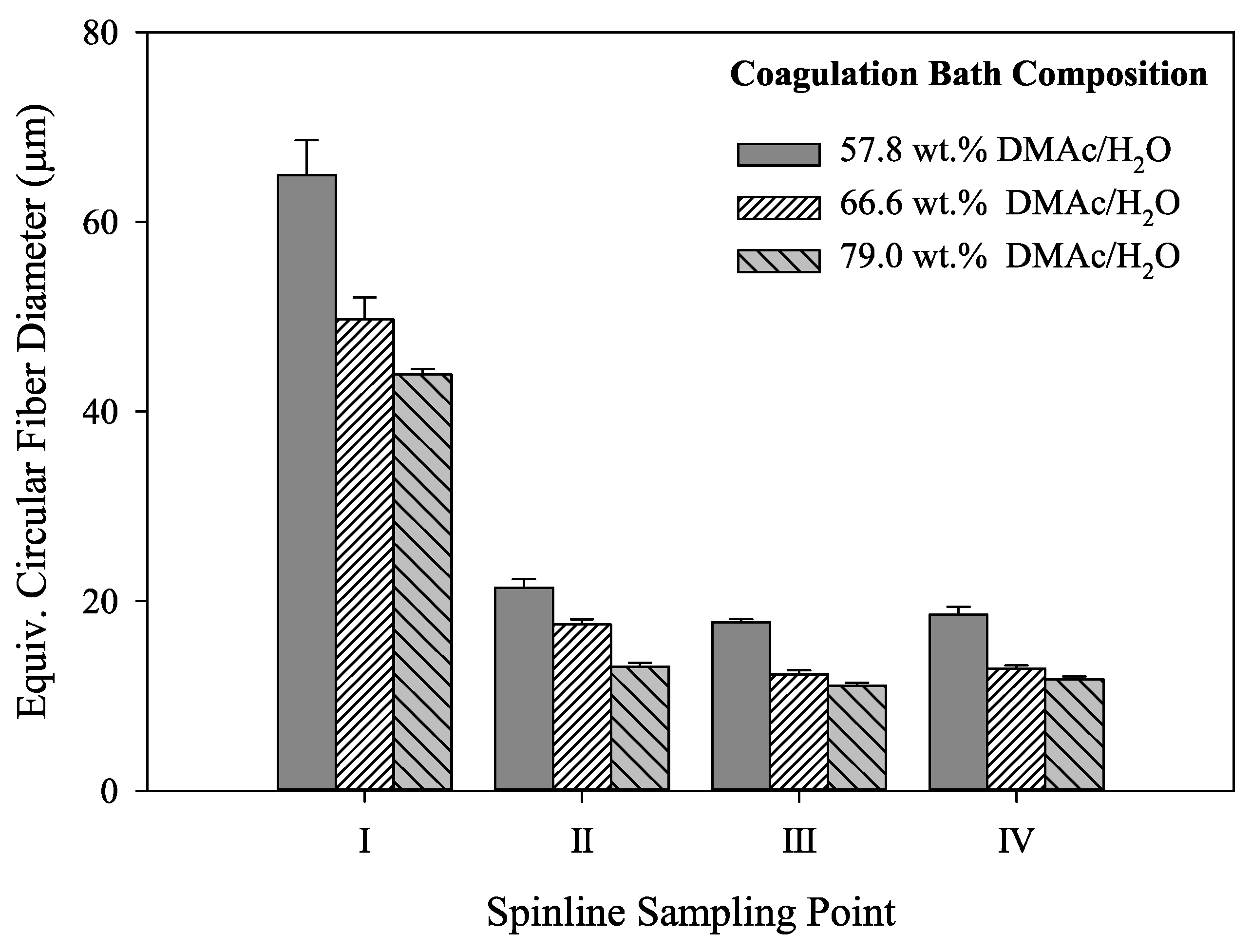
In addition, the calculated equivalent circular cross section diameters are shown in Figure 8. Of specific interest is the observation that although the fibers undergo the same stretching process, the fibers coagulated in 57.8 wt% DMAc/H2O never reach the same final diameter as those coagulated at higher solvent concentrations. Therefore, bath concentration has a strong influence on the final fiber diameter. Compared to smaller diameter fibers, larger diameter fibers have increased internal voids, reducing fiber density. Under the same spinning conditions, a smaller diameter fiber will therefore have increased density. In addition, the fiber shape set in the coagulation bath was retained along the spinning process and present in the final fiber. It is important to note that final fiber diameter, density and fiber shape affect fiber tensile properties.
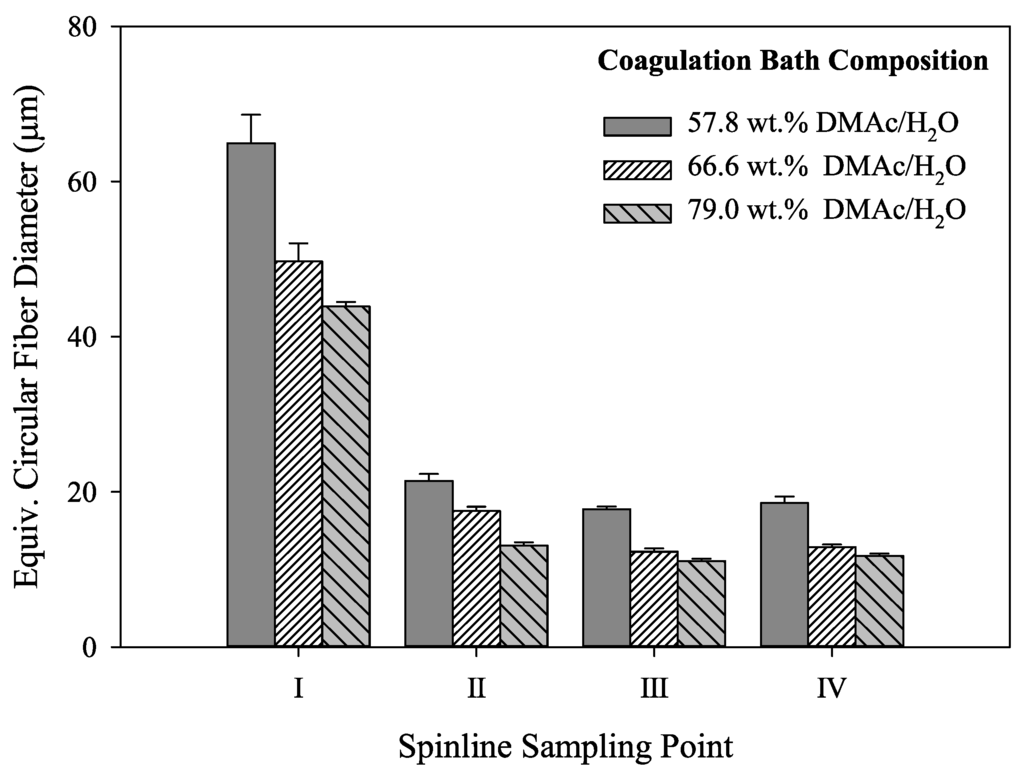
Figure 8.
Graphical representation of equivalent circular diameters for the fibers shown in Figure 7 (N ≥ 15 for each column).
Figure 8.
Graphical representation of equivalent circular diameters for the fibers shown in Figure 7 (N ≥ 15 for each column).
3.3. Cross-Sectional Morphology
Cross-sectional morphology showed denser fibril packing at high coagulation bath compositions, shown in Figure 9. Large pockets and voids are seen in the fibers from a 57.8 wt% DMAc/H2O coagulation bath, while the 79.0 wt% DMAc/H2O bath produced a much more homogeneous fibril distribution, indicating higher overall density. It is known that the fibrillar structure formed during coagulation is carried through the remaining steps [2,8] and that improvement in the fiber density resulted in improvements in the fiber tensile strength [10].

Figure 9.
High resolution SEM images of freeze dried, cryo-fractured fiber from the coagulation bath (sampling point I) at varying bath concentrations. (a) 57.8 wt% DMAc/H2O; (b) 79.0 wt% DMAc/H2O.
Figure 9.
High resolution SEM images of freeze dried, cryo-fractured fiber from the coagulation bath (sampling point I) at varying bath concentrations. (a) 57.8 wt% DMAc/H2O; (b) 79.0 wt% DMAc/H2O.

3.4. Fiber Orientation
Crystalline polymer chain alignment within the fiber samples, or fiber orientation, was studied using wide-angle X-ray diffraction (WAXD). Azimuthal scans of as-spun fibers produced using different composition coagulation baths are shown in Figure 10. Sharp azimuthal peaks for a given plane indicate higher orientation. Figure 10 shows increasing peak intensity and sharpness for fibers spun using increasingly higher coagulation bath compositions. This is supported further by the data in Table 2, Table 3 and Table 4, which show the fiber orientation and crystallite size at each sampling point of the spinning line, when spun into three different coagulation bath compositions.
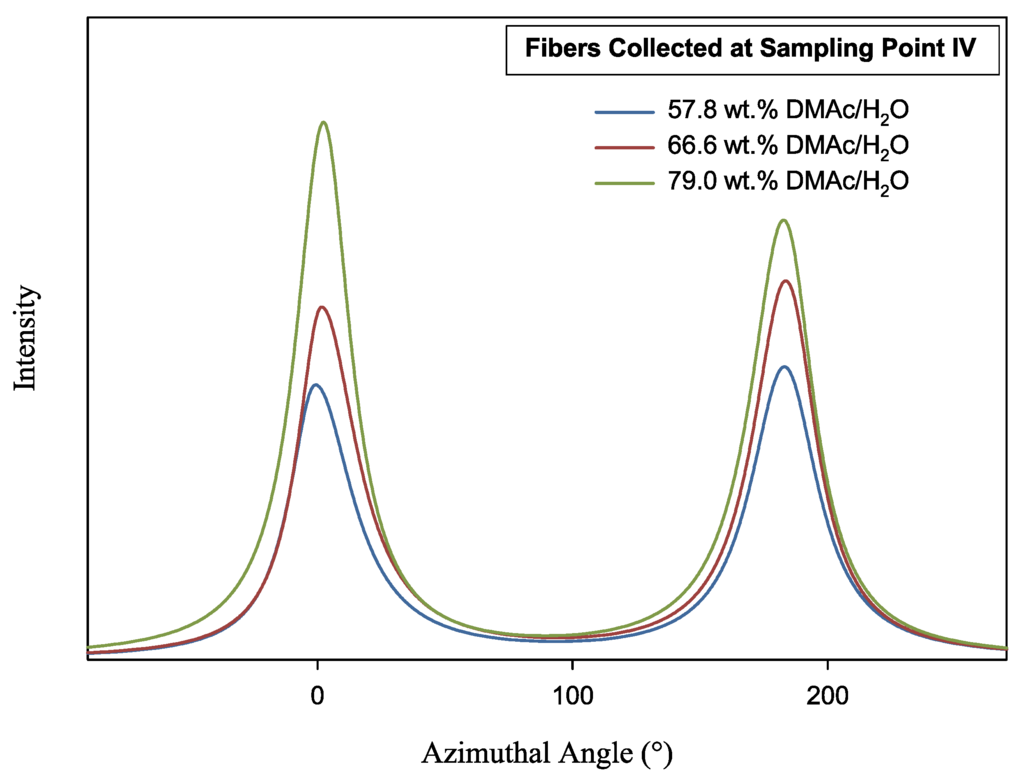
Figure 10.
Azimuthal scan of as-spun fiber at 2θ = 16.9°. Fibers were spun into varying composition coagulation baths.
Figure 10.
Azimuthal scan of as-spun fiber at 2θ = 16.9°. Fibers were spun into varying composition coagulation baths.
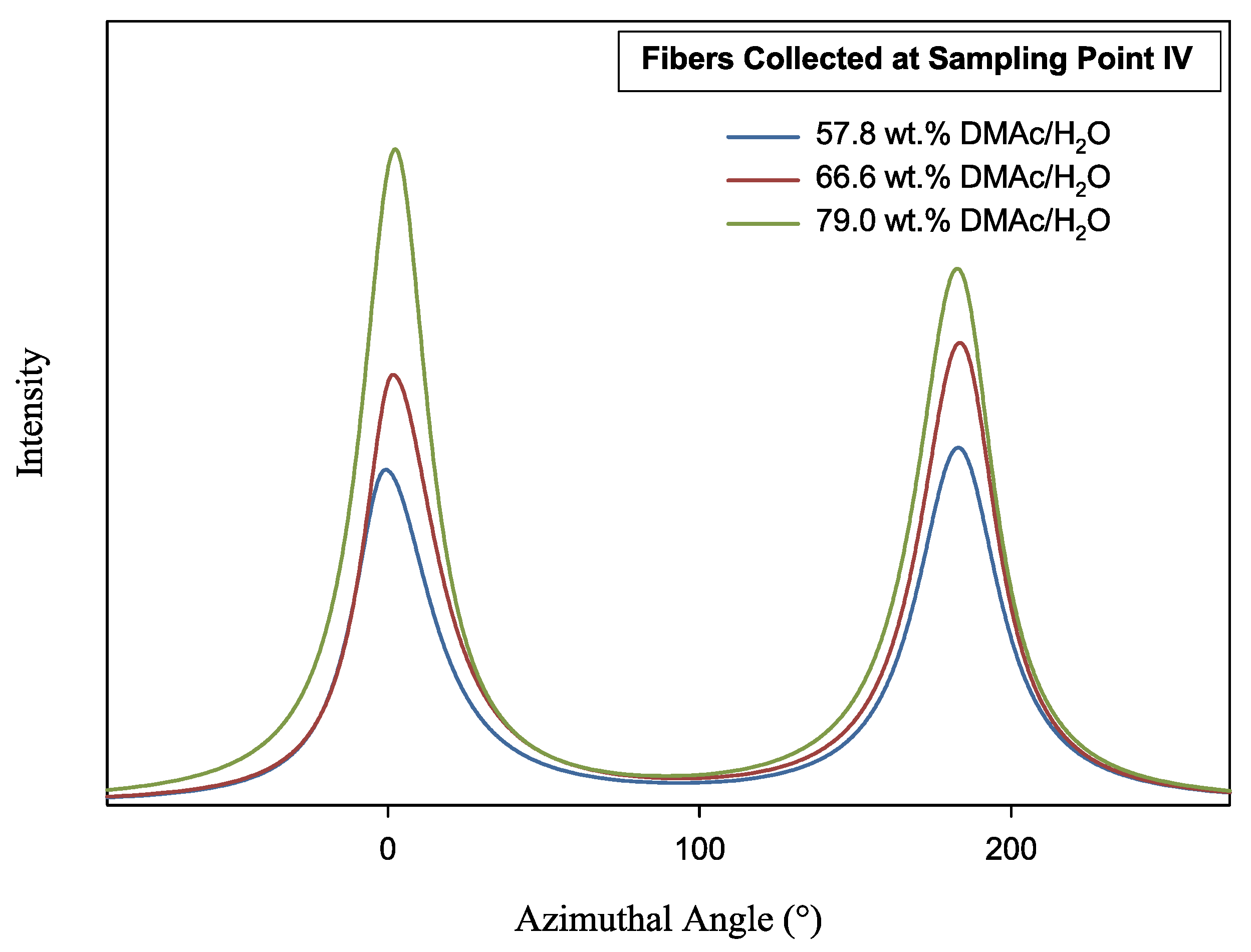
Table 2.
Orientation and crystallite size for fibers spun into a 57.8 wt% DMAc/H2O coagulation bath obtained from WAXD.
| Sampling Point | fc | FWHM | Crystal Size (Lc) |
|---|---|---|---|
| I | 0.186 | 81.40 | 1.06 |
| II | 0.356 | 37.06 | 2.40 |
| III | 0.398 | 30.70 | 2.79 |
| IV | 0.396 | 30.55 | 2.76 |
Table 3.
Orientation and crystallite size for fibers spun into a 66.6 wt% DMAc/H2O coagulation bath obtained from WAXD.
| Sampling Point | fc | FWHM | Crystal Size (Lc) |
|---|---|---|---|
| I | 0.198 | 72.32 | 1.18 |
| II | 0.338 | 41.51 | 2.00 |
| III | 0.516 | 16.37 | 5.35 |
| IV | 0.408 | 28.47 | 2.91 |
Table 4.
Orientation and crystallite size for fibers spun into a 79.0 wt% DMAc/H2O coagulation bath obtained from WAXD.
| Sampling Point | fc | FWHM | Crystal Size (Lc) |
|---|---|---|---|
| I | 0.145 | 86.12 | 1.00 |
| II | 0.362 | 37.14 | 2.39 |
| III | 0.533 | 15.10 | 5.44 |
| IV | 0.423 | 27.73 | 3.18 |
Fiber orientation at sampling point I was difficult to measure, due to the highly random and amorphous quality of the fiber at this stage in the spinning process. Indeed, WAXD resulted in very low intensities. Trends in fiber orientation began to appear at points II–IV. At sampling point II, fc for all fibers reached ≥0.3. While fibers spun into the 66.6 and 79.0 wt% coagulation baths continued to increase in orientation (fc ≥ 0.5), the orientation of fibers spun into the 57.8 wt% baths remained stunted, never achieving higher fiber orientation.
Of further interest is the decrease in fiber orientation from sampling point III to IV for all samples. However, this can be explained by heat application during fiber drying prior to take up (Figure 3), and a decrease in fiber tow tension prior to takeup. This allows for polymer chains to relax slightly prior to winding. Overall, the highest fiber orientation, determined by the fc and FWHM was achieved by the fibers spun into the 79.0 wt% DMAc/H2O coagulation bath, indicating the higher solvent content of the bath would allow the production of higher modulus fibers.
3.5. Tensile Properties
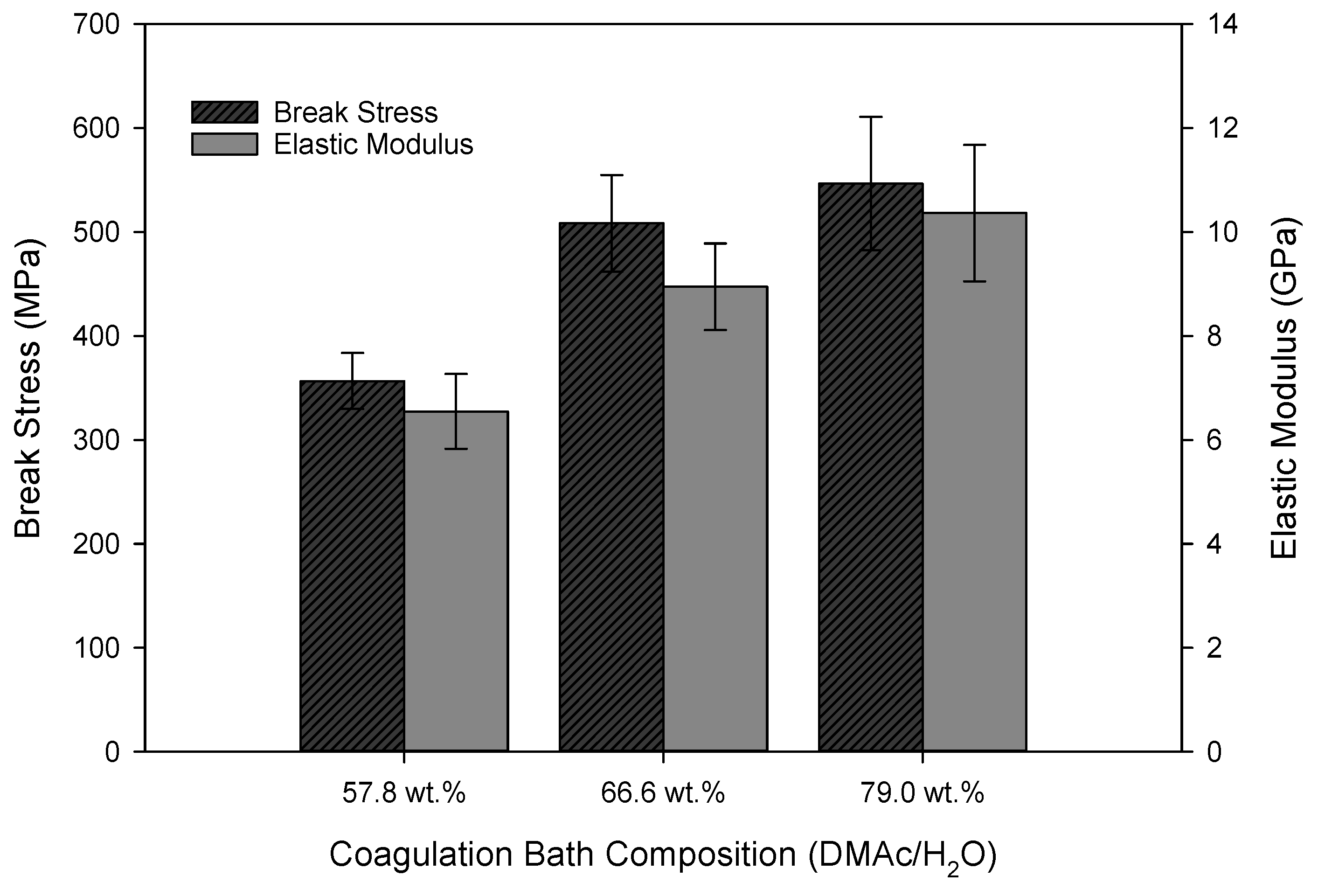
Molecular alignment has been well documented to impact the elastic modulus of the resulting PAN fibers [8,19,20]. Here, this is further supported. When considering the azimuthal scan in Figure 10, as well as the results listed in Table 2, Table 3 and Table 4, it is expected that the elastic modulus of the as-spun fibers will also increase with increasing coagulation bath composition. This, in fact, was observed as shown in Figure 11. Break stress also increased accordingly, as the fiber diameter decreased with an increase in the coagulation bath composition (Figure 8). The strength of PAN precursor fibers has been shown to drastically increase with decreasing fiber diameter [21].
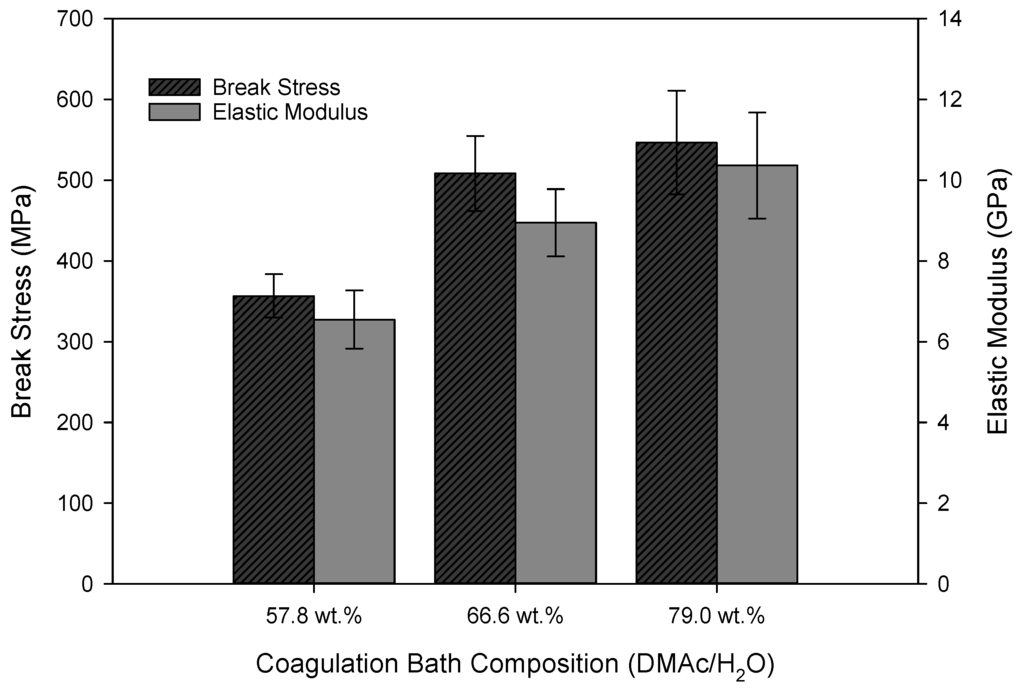
Figure 11.
Tensile properties of as-spun fiber (sampling point IV) (N ≥ 20 for each bath composition).
Figure 11.
Tensile properties of as-spun fiber (sampling point IV) (N ≥ 20 for each bath composition).
4. Conclusions
A systematic study of the effects of coagulation bath composition, at relatively high solvent-to-water composition and low bath temperature, is reported using a small pilot scale solution spinning process (0.5 k filament, continuous tow), with industrially relevant filament diameters, for PAN precursor fibers. The results indicated numerous advantages of using a high solvent content (79.0 wt% DMAc/H2O) coagulation bath to achieve optimum fiber properties, when compared to fibers produced using a lower solvent composition bath. Higher coagulation bath solvent composition produced fibers that were more circular in cross section, smaller in diameter, higher in density, achieved an overall higher orientation (fc = 0.423, FWHM = 27.73), and produced fibers with higher break stress and modulus. In addition, it was observed that the fiber shape and cross sectional area set by the coagulation bath composition persisted down the entirety of the spinline. Larger bean shaped filaments spun using a lower (57.8 wt%) solvent concentration bath never reached the smaller diameter of those spun into the 79.0 wt% concentration bath, in spite of both experiencing the exact same draw ratios. Due to all of these observations, the importance of understanding the coagulation bath composition effect on the final fiber cannot be overstated, and is necessary to achieve optimum fiber performance.
Acknowledgments
The authors would like to gratefully thank Terry Rantell for his assistance with the experiments that enabled this work.
Author Contributions
Elizabeth Ashley Morris assisted in experimental design, fiber spinning, and sample collection, completed all sample characterization, data acquisition, and analysis, and led manuscript preparation. Gregory Wilson Rice designed and constructed the apparatus for real-time modification of the coagulation bath composition and was instrumental in designing the experiment and carrying out spinning and sample collection. Matthew Collins Weisenberger co-designed the experiments, and advised the work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Barnet, F.R.; Norr, M.K. A three-dimensional structural model for a high modulus pan-based carbon fibre. Composites 1976, 7, 93–99. [Google Scholar] [CrossRef]
- Craig, J.P.; Knudsen, J.P.; Holland, V.F. Characterization of acrylic fiber structure. Text. Res. J. 1962, 32, 435–448. [Google Scholar] [CrossRef]
- Bell, J.P.; Dumbleton, J.H. Changes in the structure of wet-spun acrylic fibers during processing. Text. Res. J. 1971, 41, 196–203. [Google Scholar] [CrossRef]
- Chari, S.S.; Bahl, O.P.; Mathur, R.B. Characterisation of acrylic fibres used for making carbon fibres. Fibre Sci. Technol. 1981, 15, 153–160. [Google Scholar] [CrossRef]
- Chen, J.; Wang, C.; Ge, H.; Bai, Y.; Wang, Y. Effect of coagulation temperature on the properties of poly(acrylonitrile-itaconic acd) fibers in wet spinning. J. Polym. Res. 2007, 14, 223–228. [Google Scholar] [CrossRef]
- Tsai, J.S.; Su, W.C. Control of cross-section shape for polyacrylonitrile fibre during wet-spinning. J. Mater. Sci. Lett. 1991, 10, 1253–1256. [Google Scholar] [CrossRef]
- Law, S.J.; Mukhopadhyay, S.K. Investigation of wet-spun acrylic fiber morphology by membrane technology techniques. J. Appl. Polym. Sci. 1996, 62, 32–47. [Google Scholar] [CrossRef]
- Knudsen, J. The influence of coagulation variables on the structure and physical properties of an acrylic fiber. Text. Res. J. 1963, 33, 13–20. [Google Scholar] [CrossRef]
- Ji, B. Effect of coagulation bath concentration on the structure and properties of polyacrylonitrile as-spun fibers during wet-spinning. Adv. Mater. Res. 2011, 287–290, 1832–1836. [Google Scholar] [CrossRef]
- Wang, Y.X.; Wang, C.G.; Yu, M.J. Effects of different coagulation conditions on polyacrylonitrile fibers wet spun in a system of dimethylsulphoxide and water. J. Appl. Polym. Sci. 2006, 104, 3723–3729. [Google Scholar] [CrossRef]
- Chen, J.; Ge, H.; Liu, H.; Li, G.; Wang, C. The coagulation process of nascent fibers in pan wet-spinning. J. Wuhan Univ. Technol. Mater. Sci. Ed. 2010, 25, 200–205. [Google Scholar] [CrossRef]
- Han, C.D.; Segal, L. A study of fiber extrusion in wet spinning. Ii. Effects of spinning conditions on fiber formation. J. Appl. Polym. Sci. 1970, 14, 2999–3019. [Google Scholar] [CrossRef]
- Sen, K.; Bahrami, S.H.; Bajaj, P. High-performance acrylic fibers. J. Macromol. Sci. B Polym. Rev. 1996, 36, 1–76. [Google Scholar] [CrossRef]
- Capone, G.J. Wet-spinning technology. In Acrylic Fiber Technology and Applications; Masson, J.C., Ed.; Marcel Dekker, Inc.: New York, NY, USA, 1995; pp. 69–103. [Google Scholar]
- Ji, B.H.; Wang, C.G.; Wang, Y.X. Effect of jet stretch on polyacrylonitrile as-spun fiber formation. J. Appl. Polym. Sci. 2007, 103, 3348–3352. [Google Scholar] [CrossRef]
- Zeng, X.; Hu, J.; Zhao, J.; Zhang, Y.; Pan, D. Investigating the jet stretch in the wet spinning of pan fiber. J. Appl. Polym. Sci. 2007, 106, 2267–2273. [Google Scholar] [CrossRef]
- Perepelkin, K.E.; Pugach, B.M. The jet stretch as a factor in wet spinning. Fibre Chem. 1974, 6, 69–72. [Google Scholar] [CrossRef]
- Takahashi, M.; Nukushina, Y.; Kosugi, S. Effect of fiber-forming conditions on the microstructure of acrylic fiber. Text. Res. J. 1964, 34, 87–97. [Google Scholar] [CrossRef]
- Gupta, A.K.; Paliwal, D.K.; Bajaj, P. Acrylic precursors for carbon fibers. J. Macromol. Sci. B Polym. Rev. 1991, 31, 1–89. [Google Scholar] [CrossRef]
- Ferguson, J.; Doulgeris, C.; McKay, G.R. Rheological and coagulation features in the wet spinning process. J. Non Newton. Fluid Mech. 1980, 6, 333–338. [Google Scholar] [CrossRef]
- Morris, E.A.; Weisenberger, M.C.; Bradley, S.B.; Abdallah, M.G.; Mecham, S.J.; Pisipati, P.; McGrath, J.E. Synthesis, spinning, and properties of very high molecular weight poly(acrylonitrile-co-methyl acrylate) for high performance precursors for carbon fiber. Polymer 2014, 55, 6471–6482. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).