
In Silico Modeling as a Perspective in Developing Potential Vaccine Candidates and Therapeutics for COVID-19



 ,
,
Abstract
:1. Introduction
2. Diverse Studies on Molecular Targets of SARS CoV
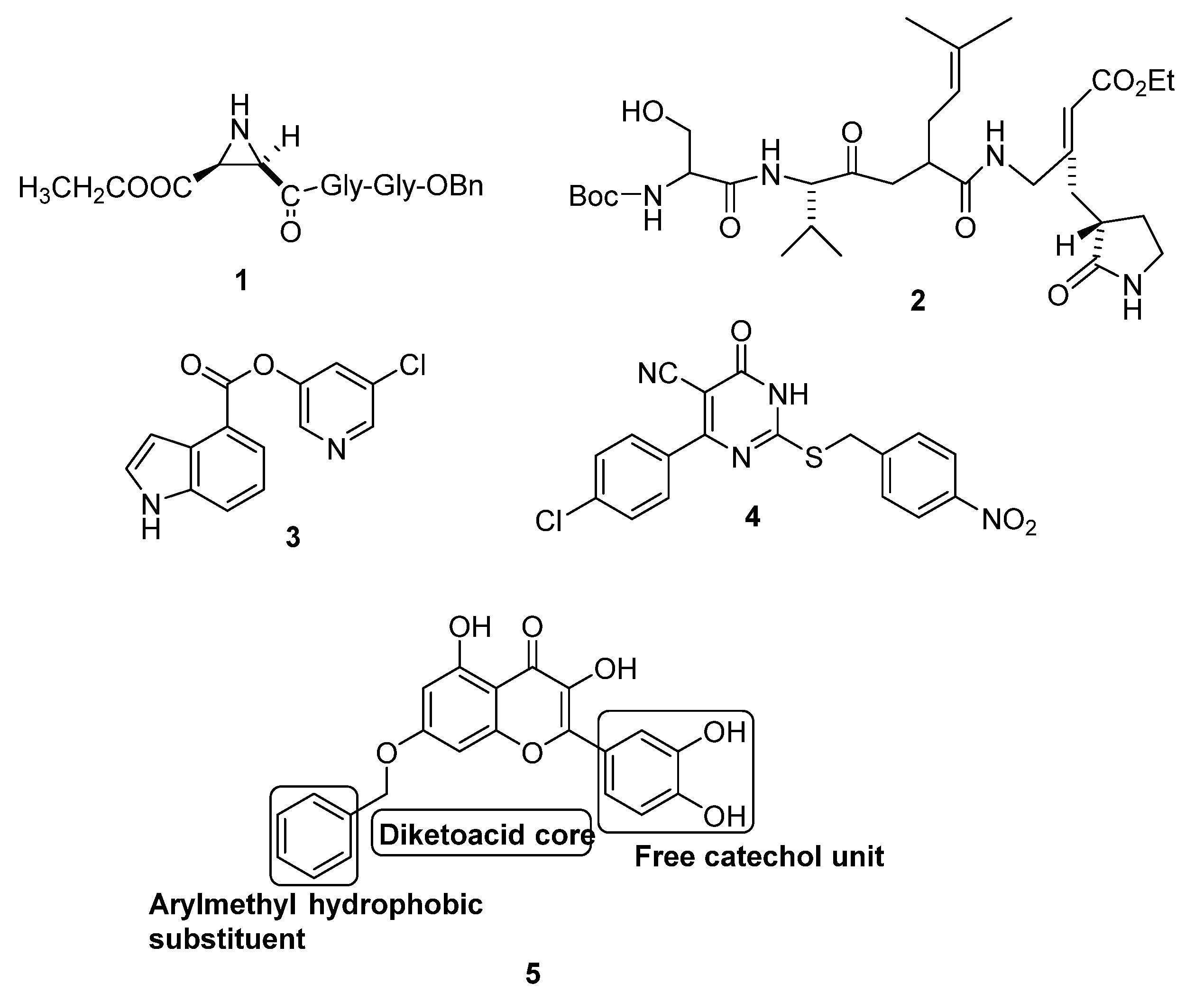
2.1. Molecular Targeting of the Main Protease of SARS CoV (Mpro)
2.2. Molecular Targeting of Chemotrypsin-Like Protease of SARS CoV (3CLpro)
2.3. Miscellaneous Targets of SARS CoV
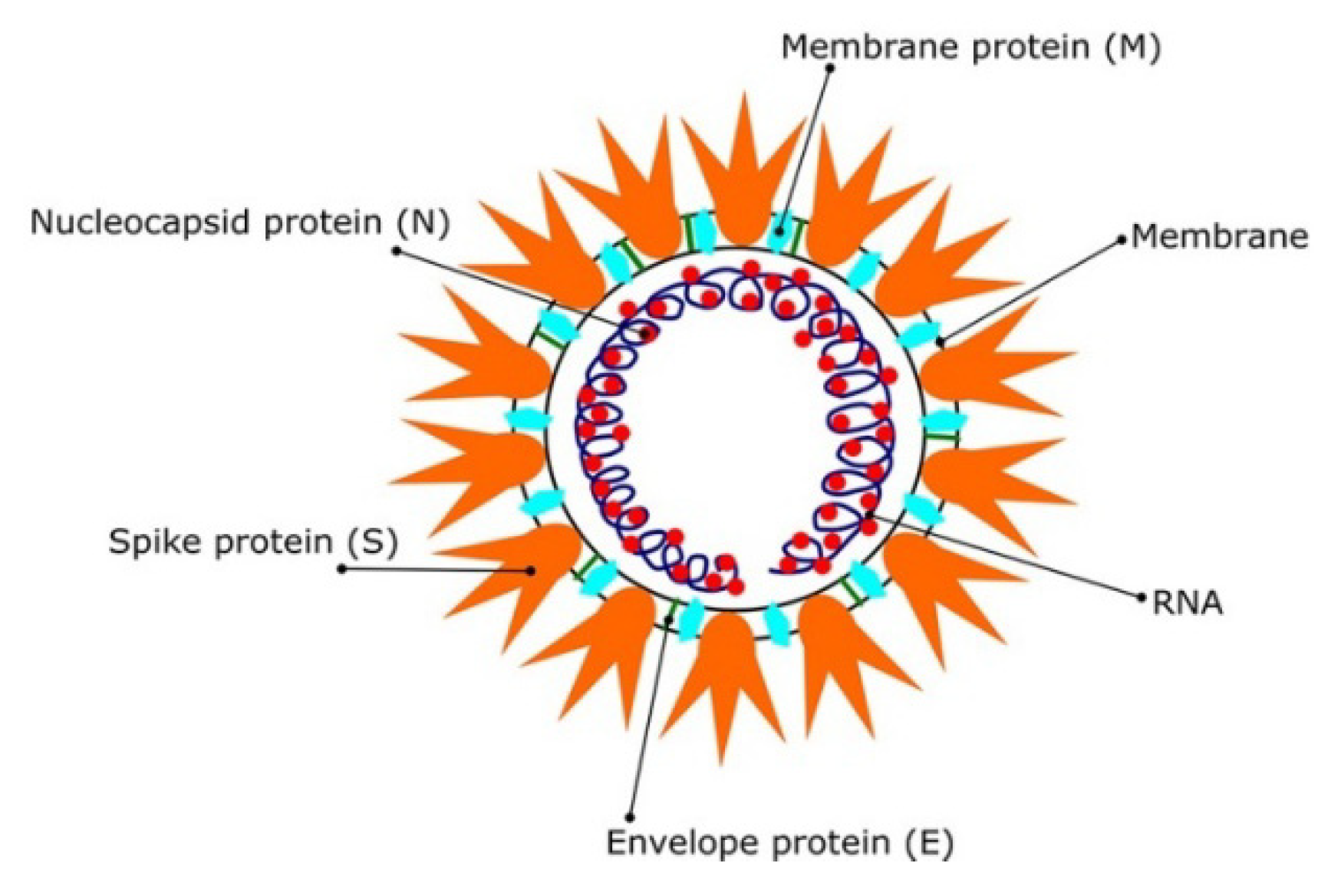
3. Molecular Modeling Studies for the SARS CoV-2Pandemic
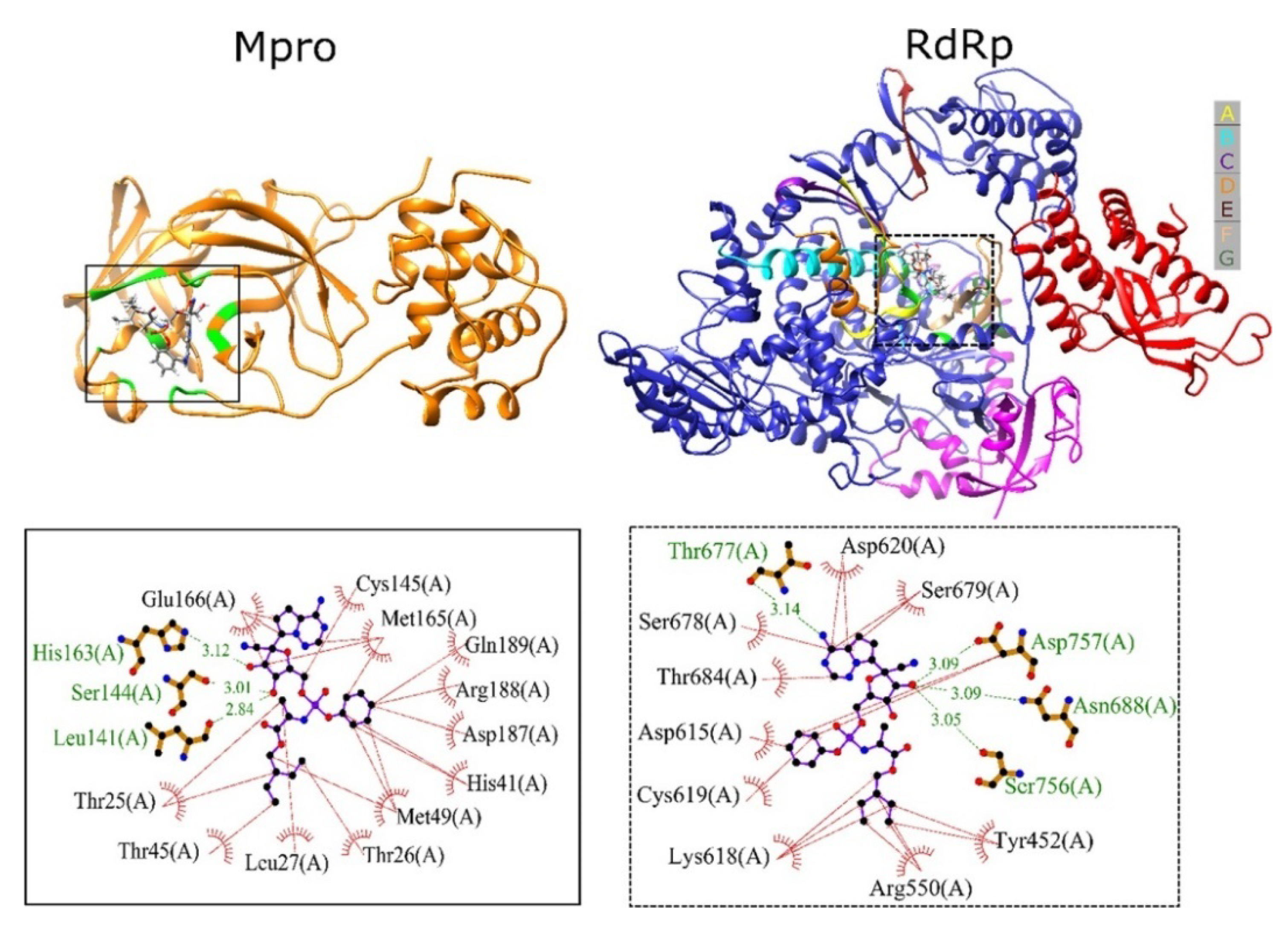
3.1. Molecular Modeling Studies on Drugs Acting on SARS CoV2 Mpro
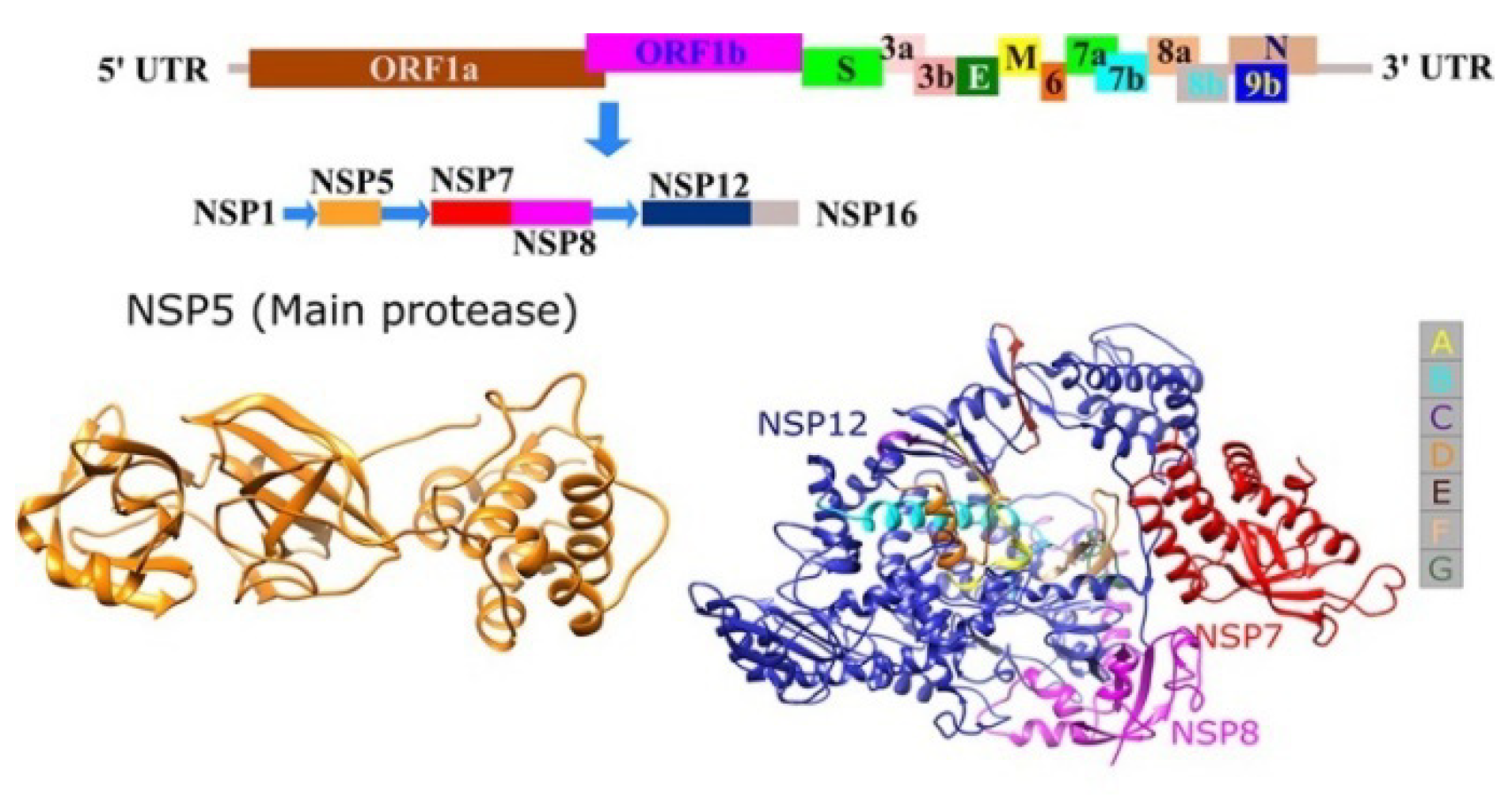
3.1.1. Sequence Analysis and Protease Homology Modeling of SARS CoV2 and SARS CoV
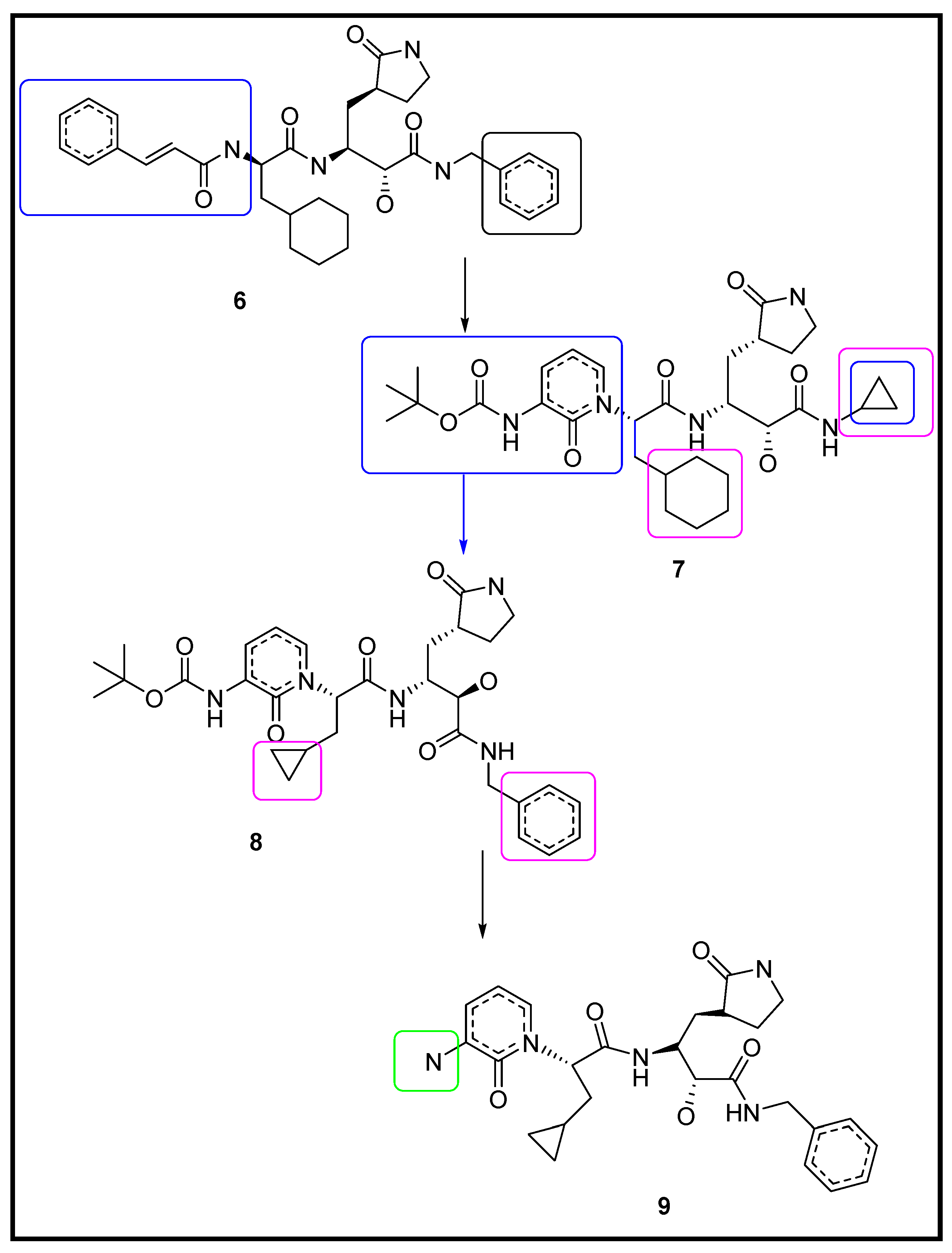
3.1.2. Designing the Improved Drugs for COVID-19: Targeting SARS CoV2 Main Protease Mpro
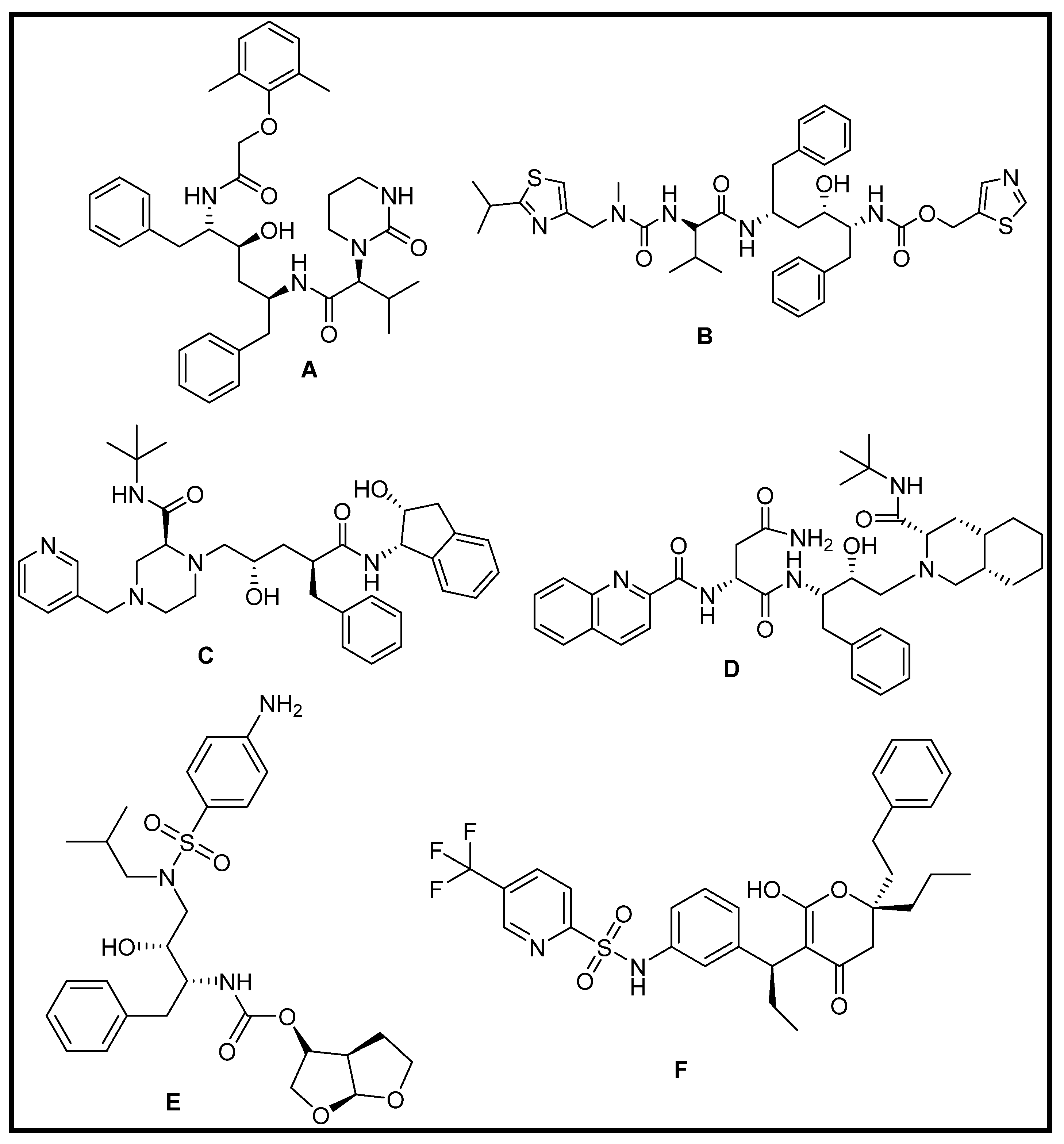
3.1.3. Repurposing of FDA-Approved Antiviral Drugs: Targeting the SARS CoV2 Main Protease Mpro
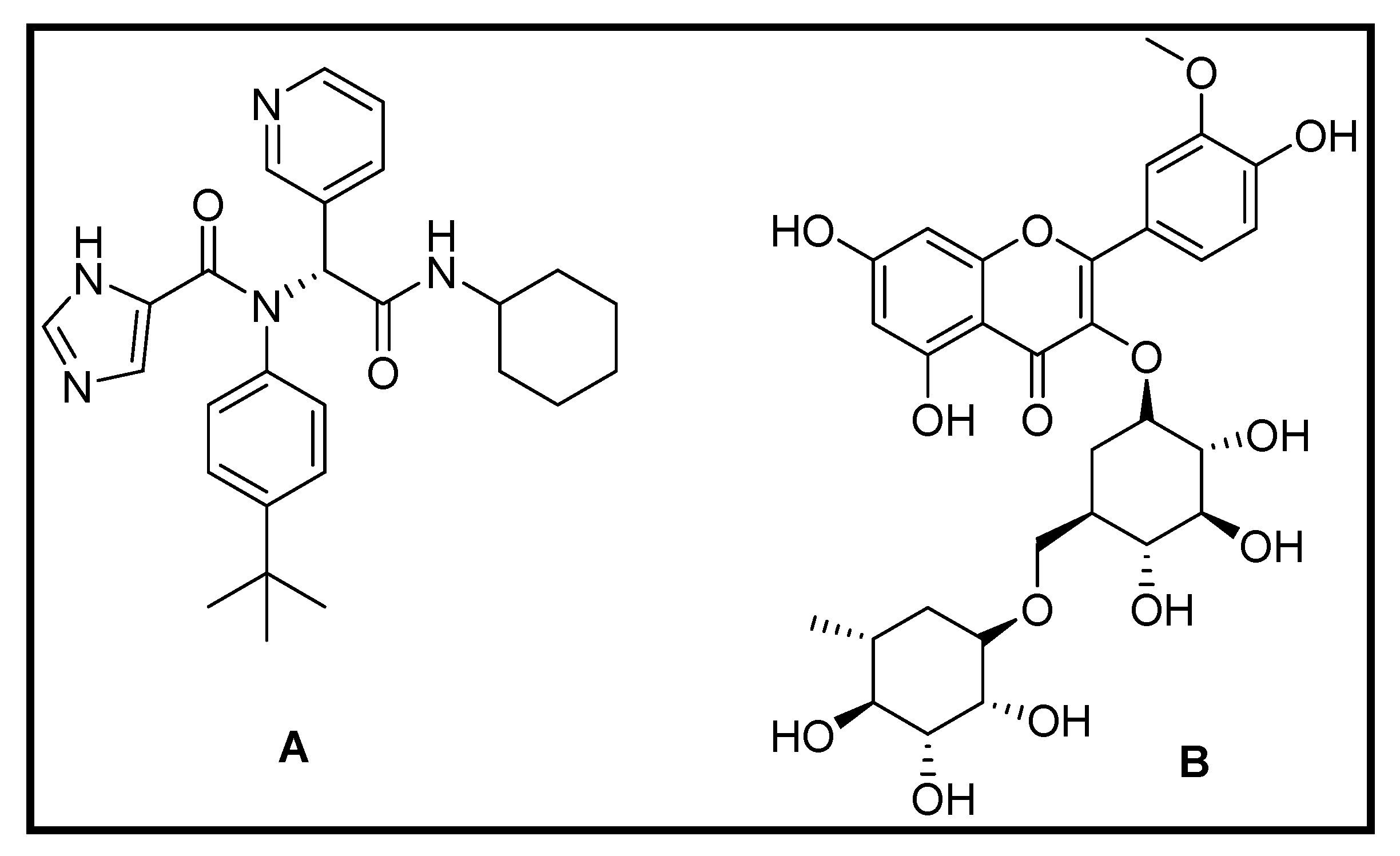
3.1.4. Repurposing of Natural Compound Drugs: Targeting the SARS CoV2 Main Protease Mpro
3.1.5. Virtual Screening Repurposing Studies: Targeting the SARS CoV2 Main Protease Mpro
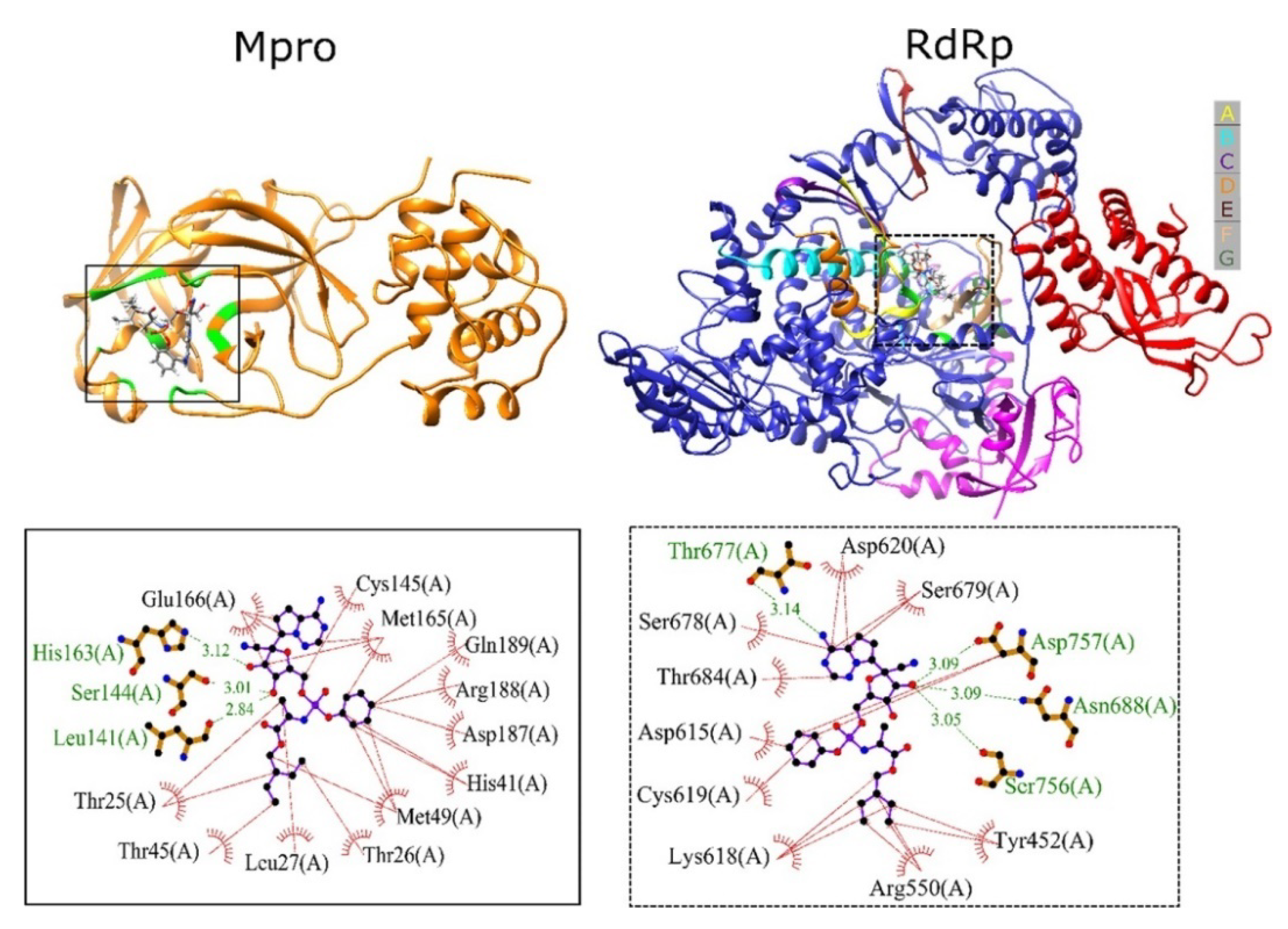
3.2. Molecular Modeling Studies on Drugs Acting on RNA-Dependent RNA Polymerase of SARS CoV2
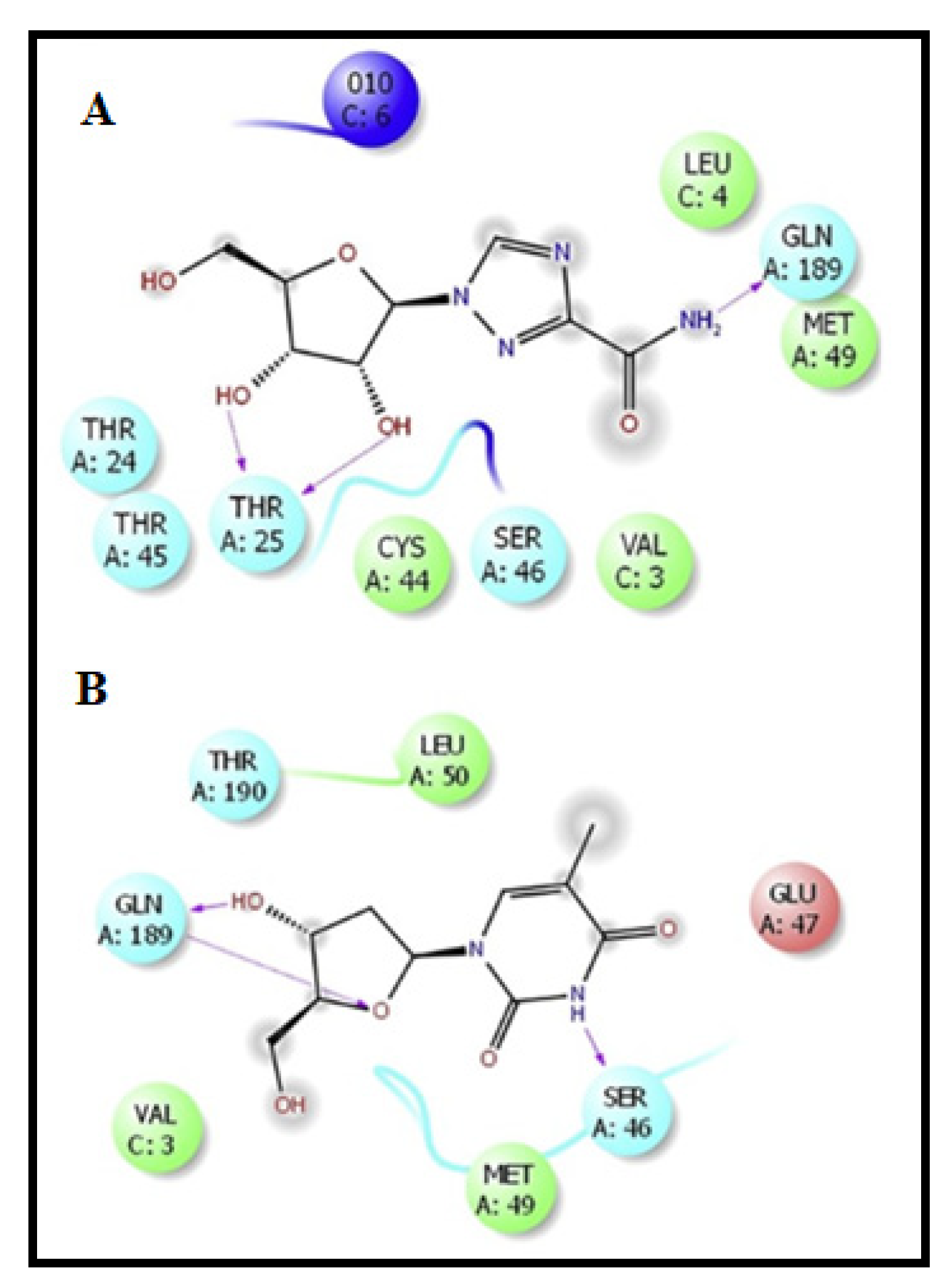
3.3. Molecular Modeling Studies on Drugs Acting on Endoribonuclease
Virtual Screening Repurposing Studies: Targeting Endoribonuclease
4. In Silico Modeling in Vaccine Development: A SARS CoV-2 Case Study
4.1. Molecular Modeling of the Designed Multi-Epitopic Vaccines
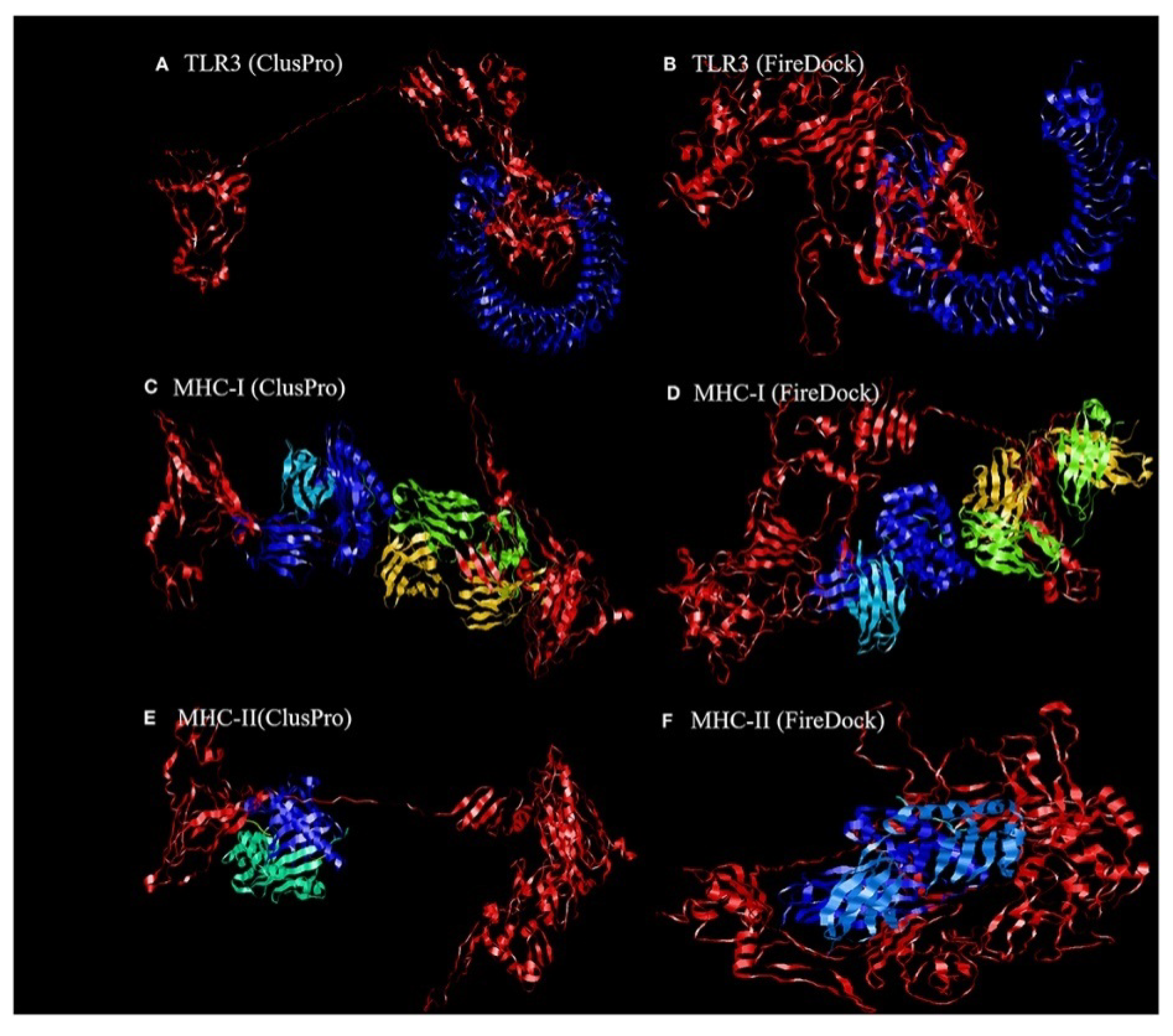
4.1.1. Molecular Docking of the Construct SARS CoV2 Vaccine with the Related Antigenic Recognition Receptors (TLR-3, MHC-I, and MHC-II)
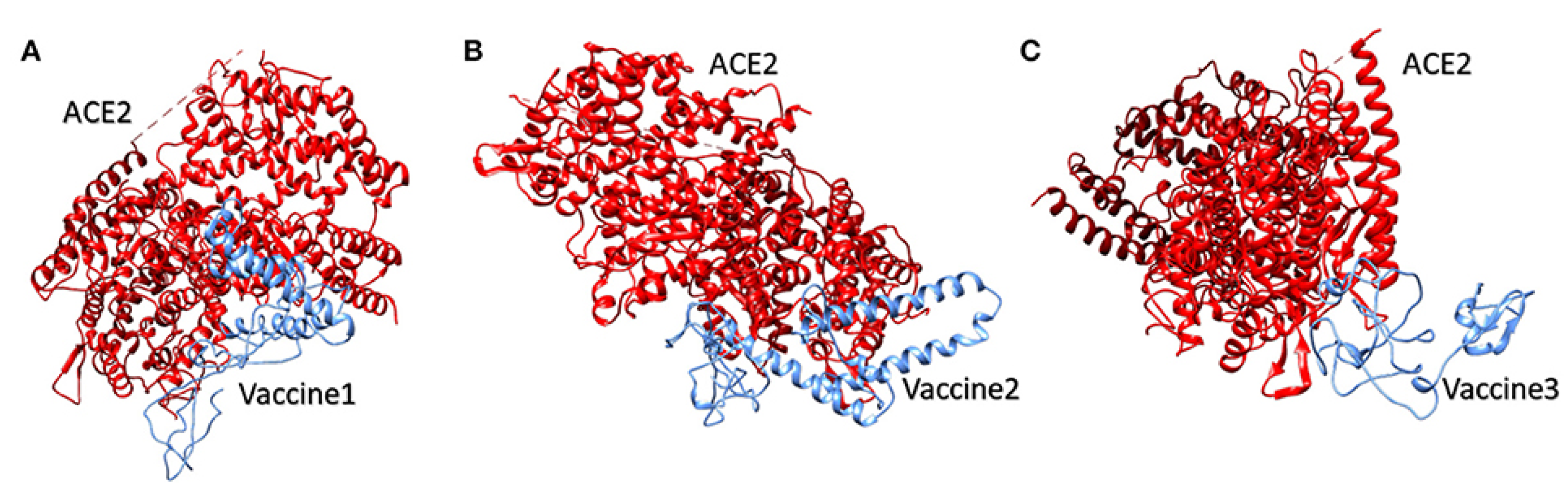
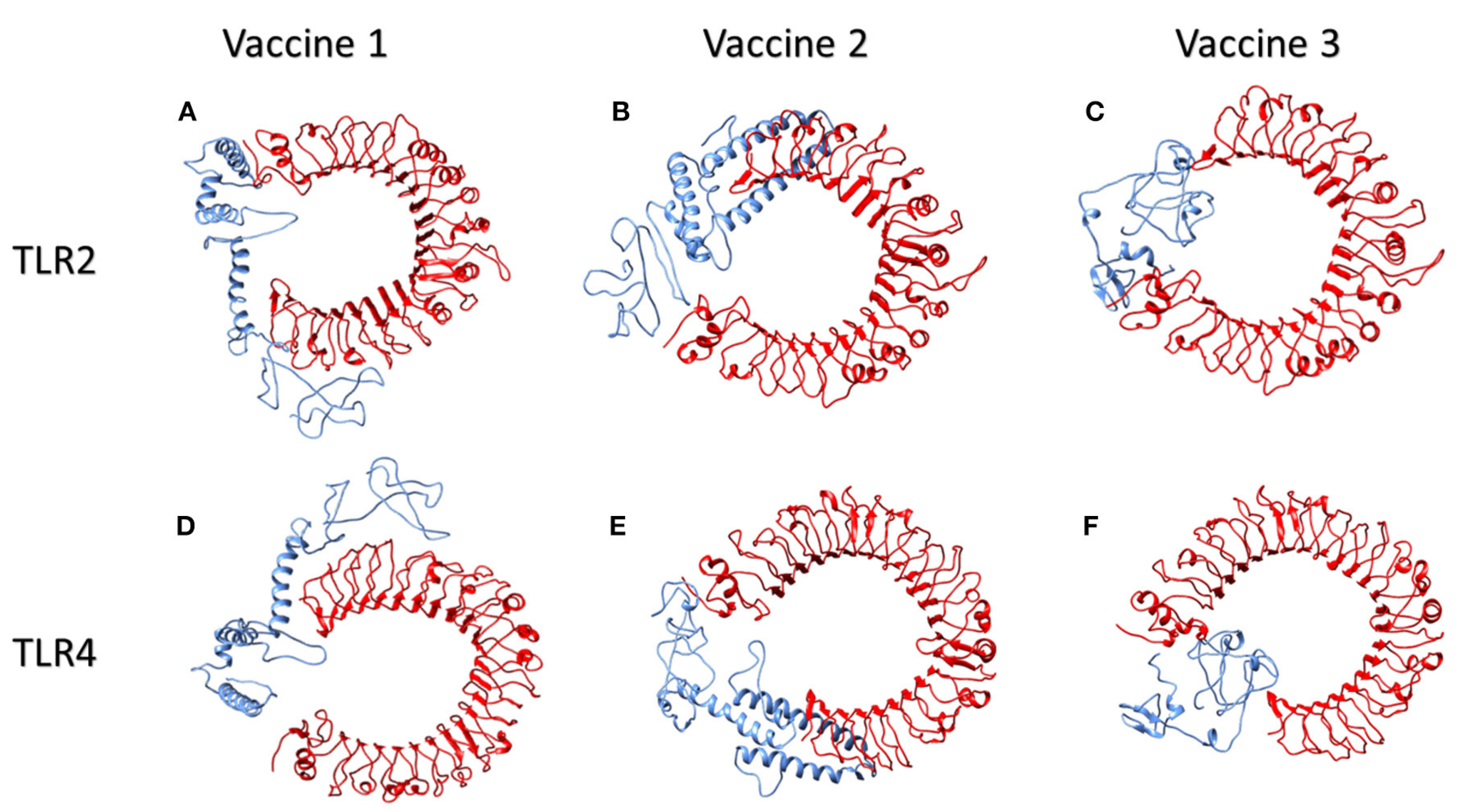
4.1.2. Molecular Docking of the SARS CoV2 Vaccine Construct with the Antigenic Recognition Receptors (ACE-2, TLR2, TLR4, HLA Alleles)
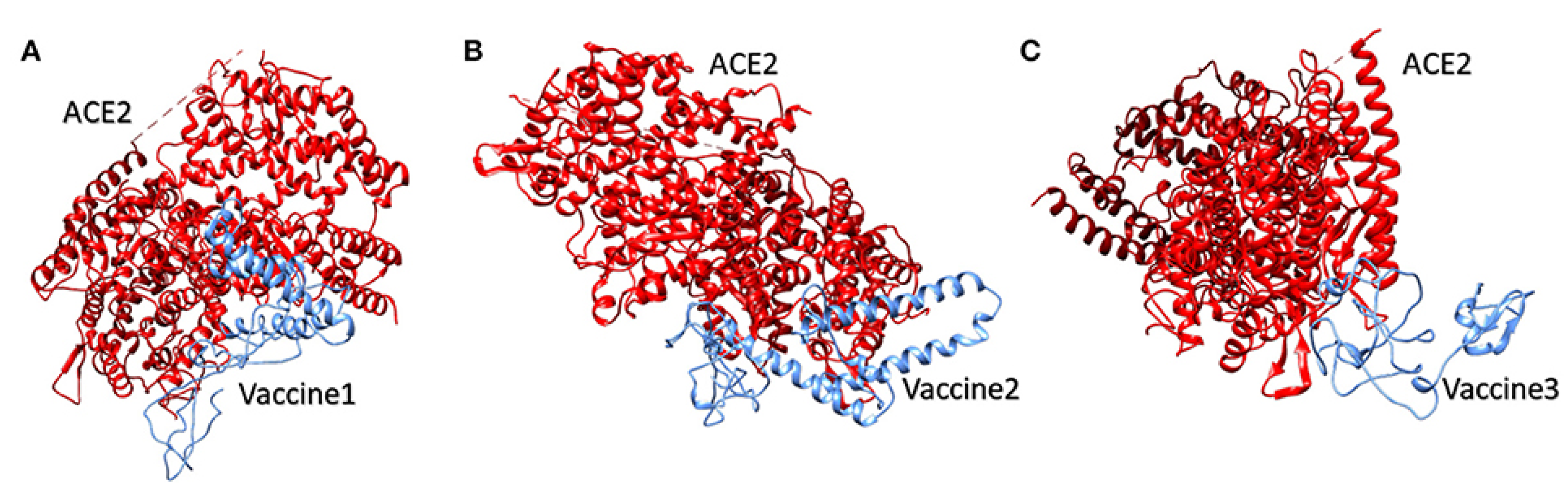
Molecular Docking of the Candidate Vaccines with ACE-2 Receptors
Molecular Docking of Vaccines with the Potential TLR2 and TLR4 Receptors
Molecular Modeling Interactions of the Suggested Vaccines with HLA Alleles
5. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, A.V.; Gemmati, D.; Vats, T.; Singh, A.; Zamboni, P. High throughput array technologies: Expanding applications from clinics to applied research. Front. Nanosci. Nanotechnol. 2019, 5, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.V.; Maharjan, R.-S.; Kanase, A.; Siewert, K.; Rosenkranz, D.; Singh, R.; Laux, P.; Luch, A. Machine-learning-based approach to decode the influence of nanomaterial properties on their interaction with cells. ACS Appl. Mater. Interf. 2021, 13, 1943–1955. [Google Scholar] [CrossRef] [PubMed]
- Muratov, E.N.; Amaro, R.; Andrade, C.H.; Brown, N.; Ekins, S.; Fourches, D.; Isayev, O.; Kozakov, D.; Medina-Franco, J.L.; Merz, K.M.; et al. A critical overview of computational approaches employed for COVID-19 drug discovery. Chem. Soc. Rev. 2021, 50, 9121–9152. [Google Scholar] [CrossRef]
- Abdellatiif, M.H.; Ali, A.; Ali, A.; Hussien, M.A. Computational studies by molecular docking of some antiviral drugs with COVID-19 receptors are an approach to medication for COVID-19. Open Chem. 2021, 19, 245–264. [Google Scholar] [CrossRef]
- Laws, M.; Surani, Y.M.; Hasan, M.; Chen, Y.; Jin, P.; Al-Adhami, T.; Chowdhury, M.; Imran, A.; Psaltis, I.; Jamshidi, S.; et al. Current trends and future approaches in small-molecule therapeutics for COVID-19. Curr. Med. Chem. 2018, 13, e0209034. [Google Scholar] [CrossRef]
- Kumar, R.; Harilal, S.; Al-Sehemi, A.G.; Mathew, G.E.; Carradori, S.; Mathew, B. The chronicle of COVID-19 and possible strategies to curb the pandemic. Curr. Med. Chem. 2020, 28, 2852–2886. [Google Scholar] [CrossRef] [PubMed]
- Ganser, L.R.; Lee, J.; Rangadurai, A.; Merriman, D.K.; Kelly, M.L.; Kansal, A.D.; Sathyamoorthy, B.; Al-Hashimi, H.M. High-performance virtual screening by targeting a high-resolution RNA dynamic ensemble. Nat. Struct. Mol. Biol. 2018, 25, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullock, J.; Luccioni, A.; Pham, K.H.; Lam, C.S.N.; Luengo-Oroz, M. Mapping the landscape of Artificial Intelligence applications against COVID-19. J. Artif. Intell. Res. 2020, 69, 807–845. [Google Scholar] [CrossRef]
- Al-Karmalawy, A.A.; Dahab, M.A.; Metwaly, A.M.; Elhady, S.S.; Elkaeed, E.B.; Eissa, I.H.; Darwish, K.M. molecular docking and dynamics simulation revealed the potential inhibitory activity of ACEIs against SARS-CoV-2 targeting the hACE2 receptor. Front. Chem. 2021, 9, 661230. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.L.; Salim, K.C.; Chu, J.J.H. Drug repurposing for COVID-19: Approaches, challenges and promising candidates. Pharmacol. Ther. 2021, 228, 107930–107944. [Google Scholar] [CrossRef]
- Kifle, Z.D.; Ayele, A.G.; Enyew, E.F. Drug repurposing approach, potential drugs, and novel drug targets for covid-19 treatment. J. Environ. Public Health 2021, 2021, 6631721. [Google Scholar] [CrossRef]
- Boopathi, S.; Poma, A.B.; Kolandaivel, P. Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. J. Biomol. Struct. Dyn. 2020, 29, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sayed, A.M.; Khattab, A.R.; AboulMagd, A.M.; Hassan, H.M.; Rateb, M.E.; Zaid, H.; Abdelmohsen, U.R. Nature as a treasure trove of potential anti-SARS-CoV drug leads: A structural/mechanistic rationale. RSC Adv. 2020, 10, 19790–19802. [Google Scholar] [CrossRef]
- Corey, B.L.; Mascola, J.R.; Fauci, A.S.; Collins, F.S. A strategic approach to COVID-19 vaccine R&D. Science 2020, 368, 948–950. [Google Scholar]
- Graham, B.S. Rapid COVID-19 vaccine development. Science 2020, 368, 945–946. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. World Report COVID-19 vaccine development pipeline gears up. Lancet 2020, 395, 1751–1752. [Google Scholar] [CrossRef]
- Callaway, E. The race for coronavirus vaccines: A graphical guide. Nature 2020, 580, 576–577. [Google Scholar] [CrossRef]
- WHO. A Coordinated Global Research Roadmap—2019 Novel Coronavirus; World Health Organization: Geneva, Switzerland, 2020; pp. 1–96. Available online: https://www.who.int/blueprint/priority-diseases/key-action/Roadmap-version-FINAL-for-WEB.pdf?ua=1 (accessed on 7 October 2020).
- Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Molecular evolution of human coronavirus genomes. Trends Microbiol. 2017, 25, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, C.M.; Wang, N.; Yang, X.L.; Liu, H.Z.; Zhang, W.; Li, B.; Hu, B.; Peng, C.; Geng, Q.B.; Zhu, G.J.; et al. Discovery of novel bat coronaviruses in South China that use the same receptor as Middle East respiratory syndrome coronavirus. J. Virol. 2018, 92, 00116–00118. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.V.; Chandrasekar, V.; Janapareddy, P.; Mathews, D.E.; Laux, P.; Luch, A.; Yang, Y.; Garcia-Canibano, B.; Balakrishnan, S.; Abinahed, J.; et al. Emerging application of nanorobotics and artificial intelligence to cross the bbb: Advances in design, controlled maneuvering, and targeting of the barriers. ACS Chem. Neurosci. 2021, 12, 1835–1853. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.V.; Jahnke, A.V.; Wang, S.; Xiao, Y.; Alapan, Y.; Kharratian, S.; Onbasli, M.C.; Kozielski, K.; David, H.; Richter, G.; et al. Anisotropic gold nanostructures: Optimization via in silico modeling for hyperthermia. ACS Appl. Nano Mater. 2018, 1, 6205–6216. [Google Scholar] [CrossRef]
- Shanker, A.; Bhanu, D.; Alluri, A.; Gupta, S. Whole genome sequence analysis and homology modelling of a 3c like peptidase and a non-structural protein 3 of the sars-cov-2 shows protein ligand interaction with an aza-peptide and a noncovalent lead inhibitor with possible antiviral properties. New J. Chem. 2020, 44, 9202–9212. [Google Scholar] [CrossRef]
- Sharma, V.; Sharma, A.; Bharate, S.B. Natural products in mitigation of SARS CoV infections. Curr. Med. Chem. 2020, 28, 4454–4483. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.T.; Pandey, I.; Zamboni, P.; Gemmati, D.; Kanase, A.; Singh, A.V.; Singh, M.P. Traditional herbal remedies with a multifunctional therapeutic approach as an implication in covid-19 associated co-infections. Coatings 2020, 10, 761. [Google Scholar] [CrossRef]
- Ansari, M.H.D.; Lavhale, S.; Kalunke, R.M.; Srivastava, P.L.; Pandit, V.; Gade, S.; Yadav, S.; Laux, P.; Luch, A.; Gemmati, D.; et al. Recent advances in plant nanobionics and nanobiosensors for toxicology applications. Curr. Nanosci. 2020, 16, 27–41. [Google Scholar] [CrossRef]
- Pandeya, A.T.; Pandeyb, I.; Hachenberger, Y.; Krause, B.-C.; Haidar, R.; Laux, P.; Luch, A.; Singh, M.P.; Singh, A.V. Emerging paradigm against global antimicrobial resistance via bioprospecting of mushroom into novel nanotherapeutics development. Trends Food Sci. Technol. 2020, 106, 333–344. [Google Scholar] [CrossRef]
- Shen, Y.; Hao, T.; Ou, S.; Hu, C.; Chen, L. Applications and perspectives of nanomaterials in novel vaccine development. Med. Chem. Comm. 2018, 9, 226–238. [Google Scholar] [CrossRef]
- Gregory, A.E.; Titball, R.; Williamson, D. Vaccine delivery using nanoparticles. Front. Cell. Infect. Microbiol. 2013, 3, 13. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.K.; Lo, S.C.; Wang, Y.S.; Hou, M.H. Recent insights into the development of therapeutics against coronavirus diseases by targeting N protein. Drug Discov. 2016, 21, 562–572. [Google Scholar] [CrossRef]
- Singh, A.V.; Ansari, M.H.D.; Rosenkranz, D.; Maharjan, R.S.; Kriegel, F.L.; Gandhi, K.; Kanase, A.; Singh, R.; Laux, P.; Luch, A. Artificial intelligence and machine learning in computational nanotoxicology: Unlocking and empowering nanomedicine. Adv. Healthc. Mater. 2020, 9, 1901862. [Google Scholar] [CrossRef]
- Singh, A.V.; Rosenkranz, D.; Ansari, M.H.D.; Singh, R.; Kanase, A.; Singh, S.P.; Johnston, B.; Tentschert, J.; Laux, P.; Luch, A. Artificial intelligence and machine learning empower advanced biomedical material design to toxicity prediction. Adv. Intell. Syst. 2020, 2, 2000084. [Google Scholar] [CrossRef]
- Panyam, J.; Labhasetwar, V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv. Drug Deliv. Rev. 2003, 55, 329–347. [Google Scholar] [CrossRef]
- Sainz, V.; Conniot, J.; Matos, A.I.; Peres, C.; Zupanǒiǒ, E.; Moura, L.; Silva, L.C.; Florindo, H.F.; Gaspar, R.S. Regulatory aspects on nanomedicines. Biochem. Biophys. Res. Commun. 2015, 468, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Cao, D.; Zhang, Y.; Ma, J.; Qi, J.; Wang, Q.; Lu, G.; Wu, Y.; Yan, J.; Shi, Y.; et al. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat. Comm. 2017, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Martina, E.; Stiefl, N.; Degel, B.; Schulz, F.; Breuning, A.; Schiller, M.; Vicik, R.; Baumann, K.; Ziebuhr, J.; Schirmeister, T. Screening of electrophilic compounds yields an aziridinyl peptide as new active-site directed SARS-CoV main protease inhibitor. Bioorg. Med. Chem. Lett. 2005, 15, 5365–5369. [Google Scholar] [CrossRef]
- Niu, C.; Yin, J.; Zhang, J.; Vederas, J.C.; James, M.N. Molecular docking identifies the binding of 3-chloropyridine moieties specifically to the S1 pocket of SARS-CoV Mpro. Bioorg. Med. Chem. 2008, 16, 293–302. [Google Scholar] [CrossRef]
- Chen, L.; Li, J.; Luo, C.; Liu, H.; Xu, W.; Chen, G.; Liew, O.W.; Zhu, W.; Puah, C.M.; Shen, X.; et al. Binding interaction of quercetin-3-β-galactoside and its synthetic derivatives with SARS-CoV 3CLpro: Structure–activity relationship studies reveal salient pharmacophore features. Bioorg. Med. Chem. 2006, 14, 8295–8306. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Xi, K.; Grum-Tokars, V.; Xu, X.; Ratia, K.; Fu, W.; Houser, K.V.; Baker, S.C.; Johnson, M.E.; Mesecar, A.D. Structure-based design, synthesis, and biological evaluation of peptidomimetic SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 5876–5880. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Gong, G.; Grum-Tokars, V.; Mulhearn, D.C.; Baker, S.C.; Coughlin, M.; Prabhakar, B.S.; Sleeman, K.; Johnson, M.E.; Mesecar, A.D. Design, synthesis and antiviral efficacy of a series of potent chloropyridyl ester-derived SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 5684–5688. [Google Scholar] [CrossRef]
- Ramajayam, R.; Tan, K.P.; Liu, H.G.; Liang, P.H. Synthesis, docking studies, and evaluation of pyrimidines as inhibitors of SARS-CoV 3CL protease. Bioorg. Med. Chem. Lett. 2010, 20, 3569–3572. [Google Scholar] [CrossRef]
- Lee, V.S.; Wittayanarakul, K.; Remsungnen, T.; Parasuk, V.; Sompornpisut, P.; Chantratita, W.; Sangma, C.; Vannarat, S.; Srichaikul, P.; Hannongbua, S.; et al. Structure and dynamics of SARS coronavirus proteinase: The primary key to the designing and screening for anti-SARS drugs. Sci. Asia 2003, 29, 181–188. [Google Scholar]
- Yap, Y.; Zhang, X.; Andonov, A.; He, R. Structural analysis of inhibition mechanisms of aurintricarboxylic acid on SARS-CoV polymerase and other proteins. Comput. Biol. Chem. 2005, 29, 212–219. [Google Scholar] [CrossRef]
- Huang, Y.; Zhao, R.; Luo, J.; Xiong, S.; Shangguan, D.; Zhang, H.; Liu, G.H.; Chen, Y. Design, synthesis and screening of antisense peptide based combinatorial peptide libraries towards an aromatic region of SARS-CoV. J. Mol. Recognit. 2008, 2, 122–131. [Google Scholar] [CrossRef]
- Lee, C.; Lee, J.M.; Lee, N.R.; Kim, D.E.; Jeong, Y.J.; Chong, Y. Investigation of the pharmacophore space of Severe Acute Respiratory Syndrome coronavirus (SARS-CoV) NTPase/helicase by dihydroxychromone derivatives. Bioorg. Med. Chem. Lett. 2009, 19, 4538–4541. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. Coronaviridae Study Group of the International Committee on Taxonomy of viruses. The species severe acute respiratory syndromerelated coronavirus: Classifying 2019-nCoV and naming it SARS-CoV2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar]
- Zhou, P.; Yang, X.L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus−induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.L.; Lan, P.D.; Thai, N.Q.; Nissley, D.A.; O’Brien, E.P.; Li, M.S. Does SARS-CoV-2 bind to human ACE2 more strongly than does SARS-CoV? J. Phys. Chem. B 2020, 124, 7336–7347. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Chan, J.F.W.; Azhar, E.I.; Hui, D.S.C.; Yuen, K.-Y. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.L.; Thai, N.Q.; Truong, D.T.; Li, M.S. Remdesivir strongly binds to both RNA-dependent RNA polymerase and main protease of SARS-CoV-2: Evidence from molecular simulations. J. Phys. Chem. B 2020, 124, 11337–11348. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–313. [Google Scholar] [CrossRef] [Green Version]
- Anand, K.; Palm, G.J.; Mesters, J.R.; Siddell, S.G.; Ziebuhr, J.; Hilgenfeld, R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain. EMBO J. 2002, 21, 3213–3224. [Google Scholar] [CrossRef]
- Naik, V.R.; Munikumar, M.; Ramakrishna, U.; Srujana, M.; Goudar, G.; Naresh, P.; Kumar, B.N.; Hemalatha, R. Remdesivir (GS-5734) as a therapeutic option of 2019-nCOV main protease−in silico approach. J. Biomol. Struct. Dyn. 2020, 39, 4701–4714. [Google Scholar] [CrossRef]
- Mengist, H.M.; Fan, X.; Jin, T. Designing of improved drugs for COVID-19: Crystal structure of SARS-CoV-2 main protease M pro. Signal Transduct. Target Ther. 2020, 5, 1–2. [Google Scholar] [CrossRef]
- Sang, P.; Tian, S.H.; Meng, Z.H.; Yang, L.Q. Anti-HIV drug repurposing against SARS-CoV-2. RSC Adv. 2020, 10, 15775–15783. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Shen, R.; Guo, X. Molecular modeling evaluation of the binding abilities of ritonavir and lopinavir to Wuhan pneumonia coronavirus proteases. Biorxiv 2020. [Google Scholar] [CrossRef]
- Beck, B.R.; Shin, B.; Choi, Y.; Park, S.; Kang, K. Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput. Struct. Biotechnol. 2020, 18, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Khaerunnisa, S.; Kurniawan, H.; Awaluddin, R.; Suhartati, S.; Soetjipto, S. Potential inhibitor of COVID-19 main protease (Mpro) from several medicinal plant compounds by molecular docking study. Preprints 2020, 20944, 1–4. [Google Scholar]
- Ravichandran, S.; Singh, N.; Donnelly, D.; Migliore, M.; Johnson, P.; Fishwick, C.; Luke, B.T.; Martin, B.; Maudsley, S.; Fugmann, S.D.; et al. Pharmacophore model of the quercetin binding site of the SIRT6 protein. J. Mol. Graph. 2014, 49, 38–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubey, K.; Dubey, R. Computation screening of narcissoside a glycosyloxyflavone for potential novel coronavirus 2019 (COVID-19) inhibitor. Biomed. J. 2020, 43, 363–367. [Google Scholar] [CrossRef]
- Kandeel, M.; Al-Nazawi, M. Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 2020, 251, 117627. [Google Scholar] [CrossRef]
- Farag, A.; Wang, P.; Ahmed, M.; Sadek, H. Identification of FDA approved drugs targeting COVID-19 virus by structure-based drug repositioning. ChemRxiv. 2020. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Chen, C.; Tan, W.; Yang, K.; Yang, H. Structure of main protease from human coronavirus NL63: Insights for wide spectrum anti-coronavirus drug design. Sci. Rep. 2016, 6, 22677. [Google Scholar] [CrossRef] [Green Version]
- Marinho, E.M.; de Andrade Neto, J.B.; Silva, J.; da Silva, C.R.; Cavalcanti, B.C.; Marinho, E.S.; Júnior, H.V. Virtual screening based on molecular docking of possible inhibitors of Covid-19 main protease. Microb. Pathog. 2020, 148, 104365. [Google Scholar] [CrossRef]
- Mahase, E. Hydroxychloroquine for covid-19: The end of the line? BMJ 2020, 369, 2378. [Google Scholar] [CrossRef] [PubMed]
- Funnell, S.G.; Dowling, W.E.; Muñoz-Fontela, C.; Gsell, P.S.; Ingber, D.E.; Hamilton, G.A.; Delang, L.; Rocha-Pereira, J.; Kaptein, S.; Dallmeier, K.H.; et al. Emerging preclinical evidence does not support broad use of hydroxychloroquine in COVID-19 patients. Nat. Commun. 2020, 11, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, R. Binding mechanism of remdesivir to SARS-CoV-2 RNA dependent RNA polymerase. Preprints 2020, 124, 6955–6962. [Google Scholar]
- Koulgi, S.; Jani, V.; Uppuladinne, M.V.; Sonavane, U.; Joshi, R. Remdesivir-bound and ligand-free simulations reveal the probable mechanism of inhibiting the RNA dependent RNA polymerase of severe acute respiratory syndrome coronavirus 2. RSC Adv. 2020, 10, 26792–26803. [Google Scholar] [CrossRef]
- Joshi, G.; Poduri, R. Virtual screening enabled selection of antiviral agents against Covid-19 disease targeting coronavirus endoribonuclease NendoU: Plausible mechanistic interventions in the treatment of new virus strain. Chemrxiv 2020. [Google Scholar] [CrossRef]
- Krishnan, D.A.; Sangeetha, G.; Vajravijayan, S.; Nandhagopal, N.; Gunasekaran, K. Structure-based drug designing towards the identification of potential anti-viral for COVID-19 by targeting endoribonuclease NSP15. IMU 2020, 20, 100392. [Google Scholar] [CrossRef] [PubMed]
- Lurie, N.; Saville, M.; Hatchett, R.; Halton, J. Developing Covid-19 Vaccines at pandemic Speed. N. Engl. J. Med. 2020, 382, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Gavi. The COVID-19 Vaccine Race. 2020. Available online: https://www.gavi.org/vaccineswork/covid-19-vaccine-race (accessed on 10 October 2020).
- Le, T.T.; Andreadakis, Z.; Kumar, A.; Román, R.G.; Tollefsen, S.; Saville, M.; Mayhew, S. The COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 2020, 19, 305–306. [Google Scholar] [CrossRef] [PubMed]
- WHO Draft Landscape of Covid-19 Candidate Vaccines. 2020. Available online: https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines?utm_source=Human+Vaccines+Project+COVID+Report&utm_campaign=089a88336d-EMAIL_CAMPAIGN_2018_08_21_06_59_COPY_01&utm_medium=email&utm_term=0_89e0163bb8-089a88336d-180996954 (accessed on 10 October 2020).
- Reed, S.G.; Orr, M.T.; Fox, C.B. Key roles of adjuvants in modern vaccines. Nat. Med. 2013, 19, 1597–1608. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Liao, F.; Wang, H.; Tang, H.; Yang, Z.; Hou, W. Evaluation of antibody-dependent enhancement of SARS-CoV infection in rhesus macaques immunized with an inactivated SARS-CoV vaccine. Virol. Sin. 2018, 33, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Funk, C.D.; Laferrière, C.; Ardakani, A. A Snapshot of the Global Race for Vaccines Targeting SARS-CoV-2 and the COVID-19 Pandemic. Front. Pharmacol. 2020, 11, 937. [Google Scholar] [CrossRef]
- Wang, N.; Shang, J.; Jiang, S.; Du, L. Subunit vaccines against emerging pathogenic human coronaviruses. Front. Microbiol. 2020, 11, 298. [Google Scholar] [CrossRef]
- Yong, C.Y.; Ong, H.K.; Yeap, S.K.; Ho, K.L.; Tan, W.S. Recent advances in the vaccine development against Middle East respiratory syndrome-coronavirus. Front. Microbiol. 2019, 10, 1781. [Google Scholar] [CrossRef] [Green Version]
- Shang, W.; Yang, Y.; Rao, Y.; Rao, X. The outbreak of SARS-CoV-2 pneumonia calls for viral vaccines. NPJ Vaccines 2020, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Anon. COVID-19 Treatment and Vaccine Tracker. Available online: https://airtable.com/shrSAi6t5WFwqo3GM/tblEzPQS5fnc0FHYR/viweyymxOAtNvo7yH?blocks=bipZFzhJ7wHPv7x9z (accessed on 30 October 2020).
- Anon. Draft Landscape of COVID-19 Candidate Vaccines. Available online: https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines (accessed on 30 October 2020).
- Anon. HKU Joins Global Partnership to Develop COVID-19 Vaccine. Available online: https://fightcovid19.hku.hk/hku-state-key-laboratory-for-emerging-infectious-diseases-joins-global-effort-to-develop-covid-19-vaccine/ (accessed on 11 December 2020).
- Voltron Therapeutics, Inc. Enters into Sponsored Research Agreement with The Vaccine & Immunotherapy Center at the Massachusetts General Hospital to Develop Potential COVID-19 Vaccine. Available online: https://www.prnewswire.com/news-releases/voltron-therapeutics-inc-enters-into-sponsored-research-agreement-with-the-vaccine--immunotherapy-center-at-the-massachusetts-general-hospital-to-develop-potential-covid-19-vaccine-301034225.html (accessed on 24 December 2020).
- Zhang, N.; Tang, J.; Lu, L.; Jiang, S.; Du, L. Receptor-binding domain-based subunit vaccines against MERS-CoV. Virus Res. 2015, 202, 151–159. [Google Scholar] [CrossRef]
- Lee, N.H.; Lee, J.A.; Park, S.Y.; Song, C.S.; Choi, I.S.; Lee, J.B. A review of vaccine development and research for industry animals in Korea. Clin. Exp. Vaccine Res. 2012, 1, 18–34. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Zhu, X.; Hossen, M.N.; Kakar, P.; Zhao, Y.; Chen, X. Augmentation of vaccine-induced humoral and cellular immunity by a physical radiofrequency adjuvant. Nat. Commun. 2018, 9, 3695. [Google Scholar] [CrossRef]
- Tu, Y.-F.; Chien, C.-S.; Yarmishyn, A.A.; Lin, Y.-Y.; Luo, Y.-H.; Lin, Y.-Y.; Lai, W.-Y.; Yang, D.-M.; Chou, S.-J.; Yang, Y.-P.; et al. A Review of SARS-CoV-2 and the Ongoing Clinical Trials. Int. J. Mol. Sci. 2020, 21, 2657. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Erdos, G.; Huang, S.; Kenniston, T.W.; Balmert, S.C.; Carey, C.D.; Raj, V.S.; Epperly, M.W.; Klimstr, W.B.; Haagmans, B.L.; et al. Microneedle array delivered recombinant coronavirus vaccines: Immunogenicity and rapid translational development. EBioMedicine 2020, 55, 102743. [Google Scholar] [CrossRef]
- Arora, K.; Rastogi, R.; Arora, N.M.; Parashar, D.; Paliwal, J.; Naqvi, A.; Srivastava, A.; Singh, S.K.; Kalyanaraman, S.; Potdar, S.; et al. Multi-Antigenic Virus-like Particle of SARS-CoV-2 produced in Saccharomyces cerevisiae as a vaccine candidate. bioRxiv 2020, preprint. [Google Scholar] [CrossRef]
- Thanh, L.T.; Zacharias, A.; Arun, K.; G’omez, R.R.; Stig, T.; Melanie, S.; Stephen, M. The COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 2020, 19, 305–306. [Google Scholar] [CrossRef]
- Anon. Countries where COVID-19 Has Spread. Available online: https://www.worldometers.info/coronavirus/countries-where-coronavirus-has-spread (accessed on 24 December 2020).
- Anon. UW–Madison, FluGen, Bharat Biotech to Develop CoroFlu, a Coronavirus Vaccine. 2020. Available online: https://www.businesswire.com/news/home/20200402005666/en/UW%E2%80%93Madison-FluGen-Bharat-Biotech-develop-CoroFlu-coronavirus (accessed on 24 December 2020).
- Lauring, A.S.; Jones, J.O.; Andino, R. Rationalizing the development of live attenuated virus vaccines. Nat Biotechnol. 2010, 28, 573–579. [Google Scholar] [CrossRef]
- Yang, Z.Y.; Kong, W.P.; Huang, Y.; Roberts, A.; Murphy, B.R.; Subbarao, K.; Nabel, G.J. A DNA vaccine induces SARS coronavirus neutralization and protective immunity in mice. Nature 2004, 428, 561–564. [Google Scholar] [CrossRef] [Green Version]
- Modjarrad, K.; Roberts, C.C.; Mills, K.T.; Castellano, A.R.; Paolino, K.; Muthumani, K.; Reuschel, E.L.; Robb, M.L.; Racine, T.; Oh, M.; et al. Safety and immunogenicity of an anti-Middle East respiratory syndrome coronavirus DNA vaccine: A phase 1, open-label, single-arm, dose-escalation trial. Lancet Infect. Dis. 2019, 19, 1013–1022. [Google Scholar] [CrossRef] [Green Version]
- Ura, T.; Okuda, K.; Shimada, M. Developments in viral vector-based vaccines. Vaccines 2014, 624–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulligan, M.J.; Lyke, K.E.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Raabe, V.; Bailey, R.; Swanson, K.A.; et al. Phase 1/2 study to describe the safety and immunogenicity of a COVID-19 RNA vaccine candidate (BNT162b1) in adults 18 to 55 years of age: Interim report. MedRxiv 2020, preprint. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobernik, D.; Bros, M. DNA vaccines-how far from clinical use? Int. J. Mol. Sci. 2018, 19, 3605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomar, N.; De, R.K. Immunoinformatics: A brief review. Methods Mol. Biol. 2014, 1184, 23–55. [Google Scholar] [PubMed]
- Oli, A.N.; Obialor, W.O.; Ifeanyichukwu, M.O.; Odimegwu, D.C.; Okoyeh, J.N.; Emechebe, G.O.; Adejumo, S.A.; Ibeanu, G.C. Immunoinformatics and vaccine development: An overview. Immunotargets Ther. 2020, 26, 13–30. [Google Scholar] [CrossRef] [Green Version]
- Lia, L.; Suna, T.; He, Y.; Lia, W.; Fana, Y.; Zhanga, J. Epitope-based peptide vaccines predicted against novel coronavirus disease caused by SARS-CoV-2. Virus Res. 2020, 288, 198082. [Google Scholar]
- Bency, J.; Helen, M. Novel epitope based peptides for vaccine against SARS-CoV-2 virus: Immunoinformatics with docking approach. Int. J. Res. Med. Sci. 2020, 8, 2385–2394. [Google Scholar]
- Qamar, M.T.; Rehman, A.; Tusleem, K.; Ashfaq, U.A.; Qasim, M.; Zhu, X.; Fatima, I.; Shahid, F.; Chen, L.-L. Designing of a next generation multiepitope based vaccine (MEV) against SARS-COV-2: Immunoinformatics and in silico approaches. PLoS ONE 2020, 15, e0244176. [Google Scholar]
- Sanami, S.; Zandi, M.; Pourhossein, B.; Mobini, G.-R.; Safaei, M.; Abed, A.; Arvejeh, P.M.; Chermahini, F.A.; Alizadeh, M. Design of a multi-epitope vaccine against SARS-CoV-2 using immunoinformatics approach. Int. J. Biol. Macromol. 2020, 164, 871–883. [Google Scholar] [CrossRef]
- Jyotisha Singh, S.; Qureshi, I.A. Multi-epitope vaccine against SARS-CoV-2 applying immunoinformatics and molecular dynamics simulation approaches. J. Biomol. Struct. Dyn. 2021. [Google Scholar] [CrossRef]
- Chen, H.-Z.; Tang, L.-L.; Yu, X.-L.; Zhou, J.; Chang, Y.-F.; Wu, X. Bioinformatics analysis of epitope-based vaccine design against the novel SARS-CoV-2. Infect. Dis. Poverty 2020, 9, 88. [Google Scholar] [CrossRef]
- Sirohi, P.R.; Gupta, J.; Somvanshi, P.; Prajapati, V.K.; Grover, A. Multiple epitope-based vaccine prediction againstSARS-CoV-2 spike glycoprotein. J. Biomol. Struct. Dyn. 2020. [Google Scholar] [CrossRef] [PubMed]
- Dar, H.A.; Waheed, Y.; Najmi, M.H.; Ismail, S.; Hetta, H.F.; Ali, A.; Muhammad, K. Multiepitope subunit vaccine design against COVID-19 based on the spike protein of SARS-CoV-2: An in silico analysis. J. Immunol. Res. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Baruah, V.; Bose, S. Immunoinformatics-aided identification of T cell and B cell epitopes in the surface glycoprotein of 2019-now. J. Med. Virol. 2020, 2, 5. [Google Scholar]
- Abraham, P.K.; Srihansa, T.; Krupanidhi, S.; Vijaya, S.A.; Venkateswarulu, T.C. Design of multi-epitope vaccine candidate against SARS-CoV-2: An in-silico study. J. Biomol. Struct. Dyn. 2020, 1, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, R.K.; Ojha, R.; Aathmanathan, V.S.; Krishnan, M.; Prajapati, V.K. Immunoinformatics approaches to design a novel multiepitope subunit vaccine against HIV infection. Vaccine 2018, 36, 2262–2272. [Google Scholar] [CrossRef]
- Nezafat, N.; Ghasemi, Y.; Javadi, G.; Khoshnoud, M.J.; Omidinia, E. A novel multiepitope peptide vaccine against cancer: An in silico approach. Biology 2014, 349, 121–134. [Google Scholar]
- Barh, D.; Misra, A.N.; Kumar, A.; Azevedo, V. A novel strategy of epitope design in Neisseria gonorrhoeae. Bioinformation 2010, 5, 77–82. [Google Scholar] [CrossRef]
- Dong, R.; Chu, Z.; Yu, F.; Zha, Y. Contriving multi-epitope subunit of vaccine for covid-19: Immunoinformatics approaches. Front. Immunol. 2020, 11, 1784. [Google Scholar] [CrossRef]
- Vajda, S.; Yueh, C.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Kozakov, D. New additions to the ClusPro server motivated by CAPRI. Proteins 2017, 85, 435–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naz, A.; Shahid, F.; Butt, T.T.; Awan, F.M.; Ali, A.; Malik, A. Designing multi-epitope vaccines to combat emerging coronavirus disease 2019 (COVID-19) by employing immuno-informatics approach. Front. Immunol. 2020, 11, 1663. [Google Scholar] [CrossRef]
- Guo, J.; Yang, Y.; Xiao, W.; Sun, W.; Yu, H.; Du, L.; Lustigman, S.; Jiang, S.; Koua, Z.; Zhou, Y. A truncated fragment of Ov-ASP-1 consisting of the core pathogenesis-related-1 (PR-1) domain maintains adjuvanticity as the full-length protein. Vaccine 2015, 33, 1974–1980. [Google Scholar] [CrossRef] [PubMed]
- Dowling, J.K.; Mansell, A. Toll-like receptors: The swiss army knife of immunity and vaccine development. Clin. Transl. Immunol. 2016, 5, e85. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. HADDOCK: A protein–protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [Green Version]
- Lester, S.N.; Li, K. Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar] [CrossRef]
- Plikusiene, I.; Maciulis, V.; Ramanaviciene, A.; Balevicius, Z.; Buzavaite-Verteliene, E.; Ciplys, E.; Slibinskas, R.; Simanavicius, M.; Zvirbliene, A.; Ramanavicius, A. Evaluation of kinetics and thermodynamics of interaction between immobilized SARS-CoV-2 nucleoprotein and specific antibodies by total internal reflection ellipsometry. J. Colloid Interface Sci. 2021, 594, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Drobysh, M.; Ramanaviciene, A.; Viter, R.; Ramanavicius, A. Affinity sensors for the diagnosis of COVID-19. Micromachines 2021, 12, 390. [Google Scholar] [CrossRef]
- Dronina, J.; Bubniene, U.S.; Ramanavicius, A. The application of DNA polymerases and Cas9 as representative of DNA-modifying enzymes group in DNA sensor design (Review). Biosens. Bioelectron. 2021, 175, 112867. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.V.; Romeo, A.; Scott, K.; Wagener, S.; Leibrock, L.; Laux, P.; Luch, A.; Kerkar, P.; Balakrishnan, S.; Dakua, S.P.; et al. Emerging technologies for in vitro inhalation toxicology. Adv. Healthc. Mater. 2021, 10, 2100633. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.V.A.; Maharjan, R.S.; Kromer, C.; Laux, P.; Luch, A.; Vats, T.; Chandrasekar, V.; Dakua, S.P.; Park, B.-W. Advances in smoking related in vitro inhalation toxicology: A perspective case of challenges and opportunities from progresses in lung-on-chip technologies. Chem. Res. Toxicol. 2021, 34. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | TLR3 | MHC-I | MHC-II |
|---|---|---|---|
| lowest binding energy score (kcal/mol) | −1156.2 | −1346.8 | −1309 |
| global energy | −38.40 | −22.97 | −27.52 |
| attractive van der Waals energy (VdW) | −26.02 | −26.84 | −26.86 |
| repulsive van der Waals energy (VdW) | 8.62 | 12.82 | 10.93 |
| atomic contact energy | −11.06 | −1.79 | 0.77 |
| Parameter | ACE-2/Vaccine 1 | ACE-2/Vaccine 2 | ACE-2/Vaccine 3 |
|---|---|---|---|
| HADDOCK score (kcal/mol) | 39.8 +/− 29.1 | 0.3 +/− 9.8 | 147.5 +/− 15.0 |
| Z-score | 1.6 | 1.3 | 1.2 |
| water-refined models | 18% | 9.0% | 11% |
| Parameter | Vaccine 1 | Vaccine 2 | Vaccine 3 | |||
|---|---|---|---|---|---|---|
| TLR2 | TLR4 | TLR2 | TLR4 | TLR2 | TLR4 | |
| HADDOCK score (kcal/mol) | 4. 2 +/− 20.8 | 37.9 +/− 7.8 | 23.7 +/− 12.1 | 16.8 +/− 23.4 | 16.7 +/− 14.0 | 23.3 +/− 5.7 |
| Z-score | 1.2 | 2.2 | 1.3 | 1.6 | 1.8 | 1.3 |
| water-refined models | 20.0% | 78.5 | 40.0% | 23.5 | 68.0% | 46.5 |
| Parameter | Vaccine 1 | Vaccine 2 | Vaccine 3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| HLA A Allele | HLA B Allele | HLA DRB1 Allele | HLA A Alleles | HLA B Alleles | HLA DRB1 allele | HLA A Alleles | HLA B Alleles | HLA DRB1 Allele | |
| HADDOCK score (kcal/mol) | 26.5 +/− 2.7 | 57.5 +/− 12.8 | 27.8 +/− 6.0 | 57.5 +/− 12.8 | 18.7 +/− 8.7 | 24.8 +/− 25.6 | 34.7 +/− 1.9 | 41.2 +/− 18.7 | 37.1 +/− 11. 8 |
| Z-score | 2.5 | 2.3 | 2.3 | 2.3 | 1.6 | 1.7 | 1.1 | 2.1 | −1.5 |
| water-refined models | 59.0% | 57.5 | 33.5 | 48.5 | 42 | 32 | 93.5 | 84 | 46.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barghash, R.F.; Fawzy, I.M.; Chandrasekar, V.; Singh, A.V.; Katha, U.; Mandour, A.A. In Silico Modeling as a Perspective in Developing Potential Vaccine Candidates and Therapeutics for COVID-19. Coatings 2021, 11, 1273. https://doi.org/10.3390/coatings11111273
Barghash RF, Fawzy IM, Chandrasekar V, Singh AV, Katha U, Mandour AA. In Silico Modeling as a Perspective in Developing Potential Vaccine Candidates and Therapeutics for COVID-19. Coatings. 2021; 11(11):1273. https://doi.org/10.3390/coatings11111273
Chicago/Turabian StyleBarghash, Reham F., Iten M. Fawzy, Vaisali Chandrasekar, Ajay Vikram Singh, Uma Katha, and Asmaa A. Mandour. 2021. "In Silico Modeling as a Perspective in Developing Potential Vaccine Candidates and Therapeutics for COVID-19" Coatings 11, no. 11: 1273. https://doi.org/10.3390/coatings11111273
APA StyleBarghash, R. F., Fawzy, I. M., Chandrasekar, V., Singh, A. V., Katha, U., & Mandour, A. A. (2021). In Silico Modeling as a Perspective in Developing Potential Vaccine Candidates and Therapeutics for COVID-19. Coatings, 11(11), 1273. https://doi.org/10.3390/coatings11111273