The Macromolecular Machines that Duplicate the Escherichia coli Chromosome as Targets for Drug Discovery

Abstract
1. History, and the Current State of Antibiotics in Medicine and the Food Industry
2. Experimental Approaches to Identify New Antibacterial Compounds
3. DNA Replication Proteins, of Which Many Are Members of the AAA+ Family of ATPases, as Targets for Drug Discovery
4. SSB
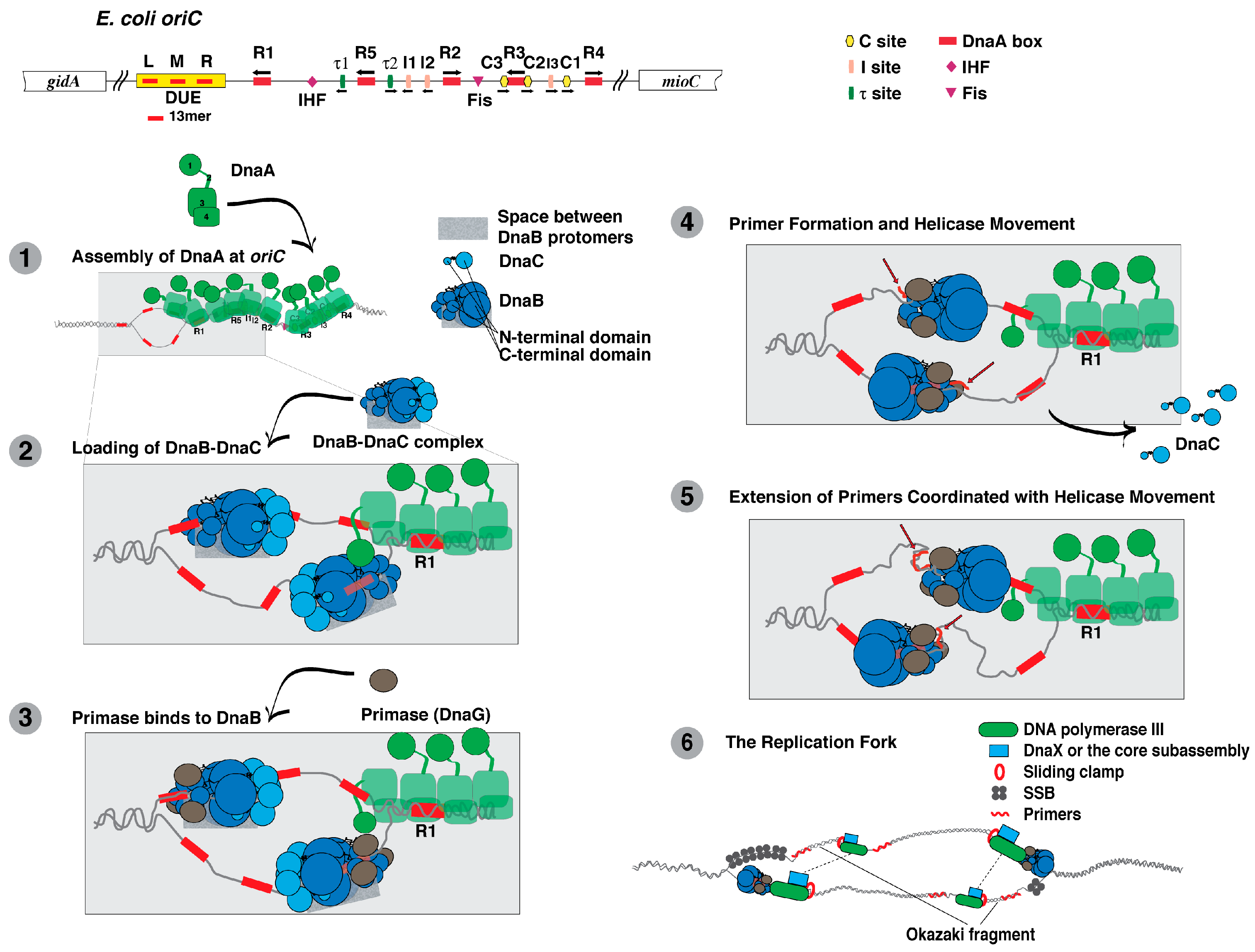
5. DnaA
6. DnaB
7. DnaC
8. Primase (DnaG)
9. DNA Polymerase I
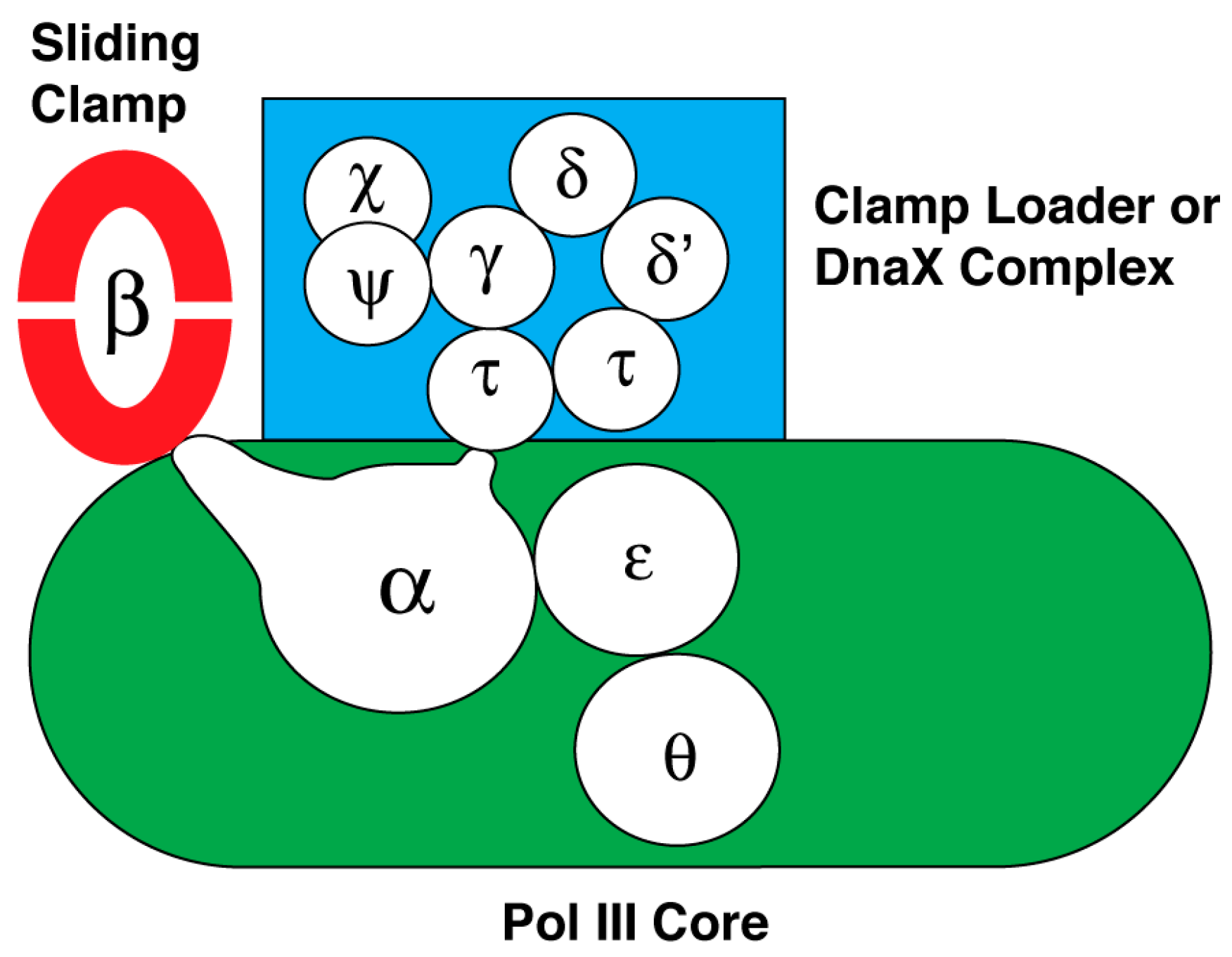
10. DNA Polymerase III Holoenzyme
11. Topoisomerases of E. coli
12. DNA Ligase
13. Conclusions
Acknowledgments
Conflicts of Interest
References
- Gates, G.A. Cost-effectiveness considerations in otitis media treatment. Otolaryngol. Head Neck Surg. 1996, 114, 525–530. [Google Scholar] [CrossRef]
- Ear Infections (Otitis Media) in Children (0–17): Use and Expenditures. 2006. Available online: https://meps.ahrq.gov/data_files/publications/st228/stat228.pdf (accessed on 15 December 2017).
- Teele, D.W.; Klein, J.O.; Rosner, B. Epidemiology of otitis media during the first seven years of life in children in greater Boston: A prospective, cohort study. J. Infect. Dis. 1989, 160, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Vergison, A. Microbiology of otitis media: A moving target. Vaccine 2008, 26, G5–G10. [Google Scholar] [CrossRef] [PubMed]
- Barnett, E.D.; Klein, J.O. The problem of resistant bacteria for the management of acute otitis media. Pediatr. Clin. N. Am. 1995, 42, 509–517. [Google Scholar] [CrossRef]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Chopra, I. The 2012 Garrod lecture: Discovery of antibacterial drugs in the 21st century. J. Antimicrob. Chemother. 2013, 68, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.J.; Miller, L.F.; Findlay, D.; Anderson, J.; Marks, L. Time for a change: Addressing R&D and commercialization challenges for antibacterials. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140086. [Google Scholar] [PubMed]
- Brown, E.D.; Wright, G.D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Benchmark Performance. Available online: https://amrbenchmark.org/industry-performance (accessed on 15 December 2017).
- Bax, B.D.; Chan, P.F.; Eggleston, D.S.; Fosberry, A.; Gentry, D.R.; Gorrec, F.; Giordano, I.; Hann, M.M.; Hennessy, A.; Hibbs, M.; et al. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 2010, 466, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2006. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, C.A.; Chuang, R.Y.; Noskov, V.N.; Assad-Garcia, N.; Deerinck, T.J.; Ellisman, M.H.; Gill, J.; Kannan, K.; Karas, B.J.; Ma, L.; et al. Design and synthesis of a minimal bacterial genome. Science 2016, 351. [Google Scholar] [CrossRef] [PubMed]
- Glass, J.I.; Merryman, C.; Wise, K.S.; Hutchison, C.A.; Smith, H.O. Minimal cells-real and imagined. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schaberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Duderstadt, K.E.; Berger, J.M. A structural framework for replication origin opening by AAA+ initiation factors. Curr. Opin. Struct. Biol. 2013, 23, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Leonard, A.C.; Grimwade, J.E. The orisome: Structure and function. Front. Microbiol. 2015, 6, 545. [Google Scholar] [CrossRef] [PubMed]
- Chodavarapu, S.; Kaguni, J.M. Replication initiation in bacteria. Enzymes 2016, 39, 1–30. [Google Scholar] [PubMed]
- Katayama, T.; Kasho, K.; Kawakami, H. The DnaA cycle in Escherichia coli: Activation, function and inactivation of the initiator rotein. Front. Microbiol. 2017, 8, 2496. [Google Scholar] [CrossRef] [PubMed]
- Dallmann, H.G.; Fackelmayer, O.J.; Tomer, G.; Chen, J.; Wiktor-Becker, A.; Ferrara, T.; Pope, C.; Oliveira, M.T.; Burgers, P.M.; Kaguni, L.S.; et al. Parallel multiplicative target screening against divergent bacterial replicases: Identification of specific inhibitors with broad spectrum potential. Biochemistry 2010, 49, 2551–2562. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Robinson, A.; Brzoska, A.J.; Turner, K.M.; Withers, R.; Harry, E.J.; Lewis, P.J.; Dixon, N.E. Essential biological processes of an emerging pathogen: DNA replication, transcription, and cell division in Acinetobacter spp. Microbiol. Mol. Biol. Rev. 2010, 74, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.; Causer, R.J.; Dixon, N.E. Architecture and conservation of the bacterial DNA replication machinery, an underexploited drug target. Curr. Drug Targets 2012, 13, 352–372. [Google Scholar] [CrossRef] [PubMed]
- Pucci, M.J.; Bush, K. Investigational antimicrobial agents of 2013. Clin. Microbiol. Rev. 2013, 26, 792–821. [Google Scholar] [CrossRef] [PubMed]
- Page, M.G.; Bush, K. Discovery and development of new antibacterial agents targeting Gram-negative bacteria in the era of pandrug resistance: Is the future promising? Curr. Opin. Pharmacol. 2014, 18, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Young, K.; Silver, L.L. What is an “ideal” antibiotic? Discovery challenges and path forward. Biochem. Pharmacol. 2017, 133, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Reiche, M.A.; Warner, D.F.; Mizrahi, V. Targeting DNA replication and repair for the development of novel therapeutics against tuberculosis. Front. Mol. Biosci. 2017, 4, 75. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Bernstein, D.A.; Satyshur, K.A.; Keck, J.L. Small-molecule tools for dissecting the roles of SSB/protein interactions in genome maintenance. Proc. Natl. Acad. Sci. USA 2010, 107, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Marceau, A.H.; Bernstein, D.A.; Walsh, B.W.; Shapiro, W.; Simmons, L.A.; Keck, J.L. Protein interactions in genome maintenance as novel antibacterial targets. PLoS ONE 2013, 8, e58765. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, T.; Sasaki, S.; Ohishi, H.; Kobayashi, M.; Katayama, T.; Miki, T.; Maeda, M.; Sekimizu, K. Molecular design of inhibitors of in vitro oriC DNA replication based on the potential to block the ATP binding of DnaA protein. J. Biol. Chem. 1996, 271, 25178–25183. [Google Scholar] [CrossRef] [PubMed]
- Griep, M.A.; Blood, S.; Larson, M.A.; Koepsell, S.A.; Hinrichs, S.H. Myricetin inhibits Escherichia coli DnaB helicase but not primase. Bioorg. Med. Chem. 2007, 15, 7203–7208. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.; Mierzwa, R.; Xu, L.; He, L.; Terracciano, J.; Patel, M.; Gullo, V.; Black, T.; Zhao, W.; Chan, T.M.; et al. Isolation and structure elucidation of Sch 642305, a novel bacterial DNA primase inhibitor produced by Penicillium verrucosum. J. Nat. Prod. 2003, 66, 1527–1530. [Google Scholar] [CrossRef] [PubMed]
- Hegde, V.R.; Pu, H.; Patel, M.; Black, T.; Soriano, A.; Zhao, W.; Gullo, V.P.; Chan, T.M. Two new bacterial DNA primase inhibitors from the plant Polygonum cuspidatum. Bioorg. Med. Chem. Lett. 2004, 14, 2275–2277. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Louise-May, S.; Thanassi, J.A.; Podos, S.D.; Cheng, J.; Thoma, C.; Liu, C.; Wiles, J.A.; Nelson, D.M.; Phadke, A.S.; et al. Small molecule inhibitors of E. coli primase, a novel bacterial target. Bioorg. Med. Chem. Lett. 2007, 17, 2807–2810. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, R.E.; Yurieva, O.; Kim, S.S.; Kuriyan, J.; Kong, X.P.; O’Donnell, M. Structure of a small-molecule inhibitor of a DNA polymerase sliding clamp. Proc. Natl. Acad. Sci. USA 2008, 105, 11116–11121. [Google Scholar] [CrossRef] [PubMed]
- Wijffels, G.; Johnson, W.M.; Oakley, A.J.; Turner, K.; Epa, V.C.; Briscoe, S.J.; Polley, M.; Liepa, A.J.; Hofmann, A.; Buchardt, J.; et al. Binding inhibitors of the bacterial sliding clamp by design. J. Med. Chem. 2011, 54, 4831–4838. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Wang, Y.; Whittell, L.R.; Jergic, S.; Liu, M.; Harry, E.; Dixon, N.E.; Kelso, M.J.; Beck, J.L.; Oakley, A.J. DNA replication is the target for the antibacterial effects of nonsteroidal anti-inflammatory drugs. Chem. Biol. 2014, 21, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Kjelstrup, S.; Hansen, P.M.; Thomsen, L.E.; Hansen, P.R.; Lobner-Olesen, A. Cyclic peptide inhibitors of the beta-sliding clamp in Staphylococcus aureus. PLoS ONE 2013, 8, e72273. [Google Scholar] [CrossRef] [PubMed]
- Kling, A.; Lukat, P.; Almeida, D.V.; Bauer, A.; Fontaine, E.; Sordello, S.; Zaburannyi, N.; Herrmann, J.; Wenzel, S.C.; Konig, C.; et al. Antibiotics. Targeting DnaN for tuberculosis therapy using novel griselimycins. Science 2015, 348, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.E.; Adam, G.C.; Arocho, M.; DiNunzio, E.; Donald, R.G.; Dorso, K.; Genilloud, O.; Gill, C.; Goetz, M.; Hairston, N.N.; et al. Elucidation of DnaE as the antibacterial target of the natural product, nargenicin. Chem. Biol. 2015, 22, 1362–1373. [Google Scholar] [CrossRef] [PubMed]
- Brotz-Oesterhelt, H.; Knezevic, I.; Bartel, S.; Lampe, T.; Warnecke-Eberz, U.; Ziegelbauer, K.; Habich, D.; Labischinski, H. Specific and potent inhibition of NAD+-dependent DNA ligase by pyridochromanones. J. Biol. Chem. 2003, 278, 39435–39442. [Google Scholar] [CrossRef] [PubMed]
- Miesel, L.; Kravec, C.; Xin, A.T.; McMonagle, P.; Ma, S.; Pichardo, J.; Feld, B.; Barrabee, E.; Palermo, R. A high-throughput assay for the adenylation reaction of bacterial DNA ligase. Anal. Biochem. 2007, 366, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.K.; Dube, D.; Tewari, N.; Dwivedi, N.; Tripathi, R.P.; Ramachandran, R. Mycobacterium tuberculosis NAD+-dependent DNA ligase is selectively inhibited by glycosylamines compared with human DNA ligase I. Nucleic Acids Res. 2005, 33, 7090–7101. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.K.; Dube, D.; Kukshal, V.; Jha, A.K.; Hajela, K.; Ramachandran, R. NAD+-dependent DNA ligase (Rv3014c) from Mycobacterium tuberculosis: Novel structure-function relationship and identification of a specific inhibitor. Proteins 2007, 69, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Meier, T.I.; Yan, D.; Peery, R.B.; McAllister, K.A.; Zook, C.; Peng, S.B.; Zhao, G. Identification and characterization of an inhibitor specific to bacterial NAD+-dependent DNA ligases. FEBS J. 2008, 275, 5258–5271. [Google Scholar] [CrossRef] [PubMed]
- Ciarrocchi, G.; MacPhee, D.G.; Deady, L.W.; Tilley, L. Specific inhibition of the eubacterial DNA ligase by arylamino compounds. Antimicrob. Agents Chemother. 1999, 43, 2766–2772. [Google Scholar] [PubMed]
- Mills, S.D.; Eakin, A.E.; Buurman, E.T.; Newman, J.V.; Gao, N.; Huynh, H.; Johnson, K.D.; Lahiri, S.; Shapiro, A.B.; Walkup, G.K.; et al. Novel bacterial NAD+-dependent DNA ligase inhibitors with broad-spectrum activity and antibacterial efficacy in vivo. Antimicrob. Agents Chemother. 2011, 55, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Stokes, S.S.; Huynh, H.; Gowravaram, M.; Albert, R.; Cavero-Tomas, M.; Chen, B.; Harang, J.; Loch, J.T.; Lu, M.; Mullen, G.B.; et al. Discovery of bacterial NAD+-dependent DNA ligase inhibitors: Optimization of antibacterial activity. Bioorg. Med. Chem. Lett. 2011, 21, 4556–4560. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Aster, S.D.; Graham, D.W.; Patel, G.F.; Taylor, G.E.; Tolman, R.L.; Painter, R.E.; Silver, L.L.; Young, K.; Ellsworth, K.; et al. Design and synthesis of novel antibacterial agents with inhibitory activity against DNA polymerase III. Bioorg. Med. Chem. Lett. 2001, 11, 2185–2188. [Google Scholar] [CrossRef]
- Tarantino, P.M., Jr.; Zhi, C.; Gambino, J.J.; Wright, G.E.; Brown, N.C. 6-Anilinouracil-based inhibitors of Bacillus subtilis DNA polymerase III: Antipolymerase and antimicrobial structure-activity relationships based on substitution at uracil N3. J. Med. Chem. 1999, 42, 2035–2040. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.S.; Giehl, T.J.; Brown, N.C.; Zhi, C.; Wright, G.E.; Ellison, R.T. In vitro antimicrobial activities of novel anilinouracils which selectively inhibit DNA polymerase III of gram-positive bacteria. Antimicrob. Agents Chemother. 2000, 44, 2217–2221. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhi, C.; Long, Z.Y.; Gambino, J.; Xu, W.C.; Brown, N.C.; Barnes, M.; Butler, M.; LaMarr, W.; Wright, G.E. Synthesis of substituted 6-anilinouracils and their inhibition of DNA polymerase IIIC and Gram-positive bacterial growth. J. Med. Chem. 2003, 46, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Guiles, J.; Sun, X.; Critchley, I.A.; Ochsner, U.; Tregay, M.; Stone, K.; Bertino, J.; Green, L.; Sabin, R.; Dean, F.; et al. Quinazolin-2-ylamino-quinazolin-4-ols as novel non-nucleoside inhibitors of bacterial DNA polymerase III. Bioorg. Med. Chem. Lett. 2009, 19, 800–802. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.E.; Brown, N.C.; Xu, W.C.; Long, Z.Y.; Zhi, C.; Gambino, J.J.; Barnes, M.H.; Butler, M.M. Active site directed inhibitors of replication-specific bacterial DNA polymerases. Bioorg. Med. Chem. Lett. 2005, 15, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.C.; Wright, G.E.; Brown, N.C.; Long, Z.Y.; Zhi, C.X.; Dvoskin, S.; Gambino, J.J.; Barnes, M.H.; Butler, M.M. 7-Alkyl-N(2)-substituted-3-deazaguanines. Synthesis, DNA polymerase III inhibition and antibacterial activity. Bioorg. Med. Chem. Lett. 2011, 21, 4197–4202. [Google Scholar] [CrossRef] [PubMed]
- Lohman, T.M.; Ferrari, M.E. Escherichia coli single-stranded DNA-binding protein: Multiple DNA-binding modes and cooperativities. Annu. Rev. Biochem. 1994, 63, 527–570. [Google Scholar] [CrossRef] [PubMed]
- Shereda, R.D.; Kozlov, A.G.; Lohman, T.M.; Cox, M.M.; Keck, J.L. SSB as an organizer/mobilizer of genome maintenance complexes. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 289–318. [Google Scholar] [CrossRef] [PubMed]
- Bianco, P.R. The tale of SSB. Prog. Biophys. Mol. Biol. 2017, 127, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Butland, G.; Peregrin-Alvarez, J.M.; Li, J.; Yang, W.; Yang, X.; Canadien, V.; Starostine, A.; Richards, D.; Beattie, B.; Krogan, N.; et al. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature 2005, 433, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Glover, B.P.; McHenry, C.S. The chi psi subunits of DNA polymerase III holoenzyme bind to single- stranded DNA-binding protein (SSB) and facilitate replication of an SSB- coated template. J. Biol. Chem. 1998, 273, 23476–23484. [Google Scholar] [CrossRef] [PubMed]
- Kelman, Z.; Yuzhakov, A.; Andjelkovic, J.; O’Donnell, M. Devoted to the lagging strand-the subunit of DNA polymerase III holoenzyme contacts SSB to promote processive elongation and sliding clamp assembly. EMBO J. 1998, 17, 2436–2449. [Google Scholar] [CrossRef] [PubMed]
- Yuzhakov, A.; Kelman, Z.; O’Donnell, M. Trading places on DNA—A three-point switch underlies primer handoff from primase to the replicative DNA polymerase. Cell 1999, 96, 153–163. [Google Scholar] [CrossRef]
- Witte, G.; Urbanke, C.; Curth, U. DNA polymerase III chi subunit ties single-stranded DNA binding protein to the bacterial replication machinery. Nucleic Acids Res. 2003, 31, 4434–4440. [Google Scholar] [CrossRef] [PubMed]
- Marceau, A.H.; Bahng, S.; Massoni, S.C.; George, N.P.; Sandler, S.J.; Marians, K.J.; Keck, J.L. Structure of the SSB-DNA polymerase III interface and its role in DNA replication. EMBO J. 2011, 30, 4236–4247. [Google Scholar] [CrossRef] [PubMed]
- Costes, A.; Lecointe, F.; McGovern, S.; Quevillon-Cheruel, S.; Polard, P. The C-terminal domain of the bacterial SSB protein acts as a DNA maintenance hub at active chromosome replication forks. PLoS Genet. 2010, 6, e1001238. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.G.; Jezewska, M.J.; Bujalowski, W.; Lohman, T.M. Binding specificity of Escherichia coli single-stranded DNA binding protein for the chi subunit of DNA pol III holoenzyme and PriA helicase. Biochemistry 2010, 49, 3555–3566. [Google Scholar] [CrossRef] [PubMed]
- Naue, N.; Fedorov, R.; Pich, A.; Manstein, D.J.; Curth, U. Site-directed mutagenesis of the chi subunit of DNA polymerase III and single-stranded DNA-binding protein of E. coli reveals key residues for their interaction. Nucleic Acids Res. 2011, 39, 1398–1407. [Google Scholar] [CrossRef] [PubMed]
- Molineux, I.J.; Gefter, M.L. Properties of the Escherichia coli in DNA binding (unwinding) protein: Interaction with DNA polymerase and DNA. Proc. Natl. Acad. Sci. USA 1974, 71, 3858–3862. [Google Scholar] [CrossRef] [PubMed]
- Furukohri, A.; Nishikawa, Y.; Akiyama, M.T.; Maki, H. Interaction between Escherichia coli DNA polymerase IV and single-stranded DNA-binding protein is required for DNA synthesis on SSB-coated DNA. Nucleic Acids Res. 2012, 40, 6039–6048. [Google Scholar] [CrossRef] [PubMed]
- Arad, G.; Hendel, A.; Urbanke, C.; Curth, U.; Livneh, Z. Single-stranded DNA-binding protein recruits DNA polymerase V to primer termini on RecA-coated DNA. J. Biol. Chem. 2008, 283, 8274–8282. [Google Scholar] [CrossRef] [PubMed]
- Cadman, C.J.; McGlynn, P. PriA helicase and SSB interact physically and functionally. Nucleic Acids Res. 2004, 32, 6378–6387. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Lin, M.J.; Huang, C.Y. Yeast two-hybrid analysis of PriB-interacting proteins in replication restart primosome: A proposed PriB-SSB interaction model. Protein J. 2013, 32, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Wessel, S.R.; Marceau, A.H.; Massoni, S.C.; Zhou, R.; Ha, T.; Sandler, S.J.; Keck, J.L. PriC-mediated DNA replication restart requires PriC complex formation with the single-stranded DNA-binding protein. J. Biol. Chem. 2013, 288, 17569–17578. [Google Scholar] [CrossRef] [PubMed]
- Naue, N.; Beerbaum, M.; Bogutzki, A.; Schmieder, P.; Curth, U. The helicase-binding domain of Escherichia coli DnaG primase interacts with the highly conserved C-terminal region of single-stranded DNA-binding protein. Nucleic Acids Res. 2013, 41, 4507–4517. [Google Scholar] [CrossRef] [PubMed]
- Zechner, E.L.; Wu, C.A.; Marians, K.J. Coordinated leading- and lagging-strand synthesis at the Escherichia coli DNA replication fork. III. A polymerase-primase interaction governs primer size. J. Biol. Chem. 1992, 267, 4054–4063. [Google Scholar] [PubMed]
- Tougu, K.; Peng, H.; Marians, K.J. Identification of a domain of Escherichia coli primase required for functional interaction with the DnaB helicase at the replication fork. J. Biol. Chem. 1994, 269, 4675–4682. [Google Scholar] [PubMed]
- Tougu, K.; Marians, K.J. The extreme C terminus of primase is required for interaction with DnaB at the replication fork. J. Biol. Chem. 1996, 271, 21391–21397. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.B.; Ratnakar, P.V.; Mohanty, B.K.; Bastia, D. Direct physical interaction between DnaG primase and DnaB helicase of Escherichia coli is necessary for optimal synthesis of primer RNA. Proc. Natl. Acad. Sci. USA 1996, 93, 12902–12907. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.; Marians, K.J. Identification of a region of Escherichia coli DnaB required for functional interaction with DnaG at the replication fork. J. Biol. Chem. 2000, 275, 26187–26195. [Google Scholar] [CrossRef] [PubMed]
- Thirlway, J.; Turner, I.J.; Gibson, C.T.; Gardiner, L.; Brady, K.; Allen, S.; Roberts, C.J.; Soultanas, P. DnaG interacts with a linker region that joins the N- and C-domains of DnaB and induces the formation of 3-fold symmetric rings. Nucleic Acids Res. 2004, 32, 2977–2986. [Google Scholar] [CrossRef] [PubMed]
- Oakley, A.J.; Loscha, K.V.; Schaeffer, P.M.; Liepinsh, E.; Pintacuda, G.; Wilce, M.C.; Otting, G.; Dixon, N.E. Crystal and solution structures of the helicase-binding domain of Escherichia coli primase. J. Biol. Chem. 2005, 280, 11495–11504. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.; Eliason, W.K.; Steitz, T.A. Structure of hexameric DnaB helicase and its complex with a domain of DnaG primase. Science 2007, 318, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Soultanas, P. The bacterial helicase-primase interaction: A common structural/functional module. Structure 2005, 13, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Soultanas, P.; Bolt, E. Replicative DNA helicases and primases. In Molecular Life Sciences; Wells, R.D., Bond, J.S., Klinman, J., Masters, B.S.S., Bell, E., Eds.; Springer: New York, NY, USA, 2014. [Google Scholar]
- Mott, M.L.; Berger, J.M. DNA replication initiation: Mechanisms and regulation in bacteria. Nat. Rev. Microbiol. 2007, 5, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, H.; Katayama, T. DnaA, ORC, and Cdc6: Similarity beyond the domains of life and diversity. Biochem. Cell Biol. 2010, 88, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Kaguni, J.M. Replication initiation at the Escherichia coli chromosomal origin. Curr. Opin. Chem. Biol. 2011, 15, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Secondary Structure Predictions for 104 DnaA Protein Sequences. Available online: https://owww.molgen.mpg.de/~messer/DnaA%20comparison%202002.pdf (accessed on 7 March 2002).
- Erzberger, J.P.; Pirruccello, M.M.; Berger, J.M. The structure of bacterial DnaA: Implications for general mechanisms underlying DNA replication initiation. EMBO J. 2002, 21, 4763–4773. [Google Scholar] [CrossRef] [PubMed]
- Erzberger, J.P.; Mott, M.L.; Berger, J.M. Structural basis for ATP-dependent DnaA assembly and replication-origin remodeling. Nat. Struct. Mol. Biol. 2006, 13, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, S.; Noguchi, Y.; Hayashi, Y.; Miyazaki, E.; Katayama, T. Differentiation of the DnaA-oriC sub-complex for DNA unwinding in a replication initiation complex. J. Biol. Chem. 2012, 287, 37458–37471. [Google Scholar] [CrossRef] [PubMed]
- Rozgaja, T.A.; Grimwade, J.E.; Iqbal, M.; Czerwonka, C.; Vora, M.; Leonard, A.C. Two oppositely oriented arrays of low-affinity recognition sites in oriC guide progressive binding of DnaA during Escherichia coli pre-RC assembly. Mol. Microbiol. 2011, 82, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Zawilak-Pawlik, A.; Nowaczyk, M.; Zakrzewska-Czerwinska, J. The role of the N-terminal domains of bacterial initiator DnaA in the assembly and regulation of the bacterial replication initiation complex. Genes 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Jo, T.; Matsuda, Y.; Matsunaga, C.; Katayama, T.; Ueda, T. Structure and function of DnaA N-terminal domains: Specific sites and mechanisms in inter-DnaA interaction and in DnaB helicase loading on oriC. J. Biol. Chem. 2007, 282, 17816–17827. [Google Scholar] [CrossRef] [PubMed]
- Marszalek, J.; Kaguni, J.M. DnaA protein directs the binding of DnaB protein in initiation of DNA replication in Escherichia coli. J. Biol. Chem. 1994, 269, 4883–4890. [Google Scholar] [PubMed]
- Sutton, M.D.; Carr, K.M.; Vicente, M.; Kaguni, J.M. E. coli DnaA protein: the N-terminal domain and loading of DnaB helicase at the E. coli chromosomal origin. J. Biol. Chem. 1998, 273, 34255–34262. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.; Weigel, C.; Messer, W. The interaction domains of the DnaA and DnaB replication proteins of Escherichia coli. Mol. Microbiol. 2000, 37, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Weigel, C.; Schmidt, A.; Seitz, H.; Tungler, D.; Welzeck, M.; Messer, W. The N-terminus promotes oligomerization of the Escherichia coli initiator protein DnaA. Mol. Microbiol. 1999, 34, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Simmons, L.A.; Felczak, M.; Kaguni, J.M. DnaA Protein of Escherichia coli: Oligomerization at the E. coli chromosomal origin is required for initiation and involves specific N-terminal amino acids. Mol. Microbiol. 2003, 49, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Felczak, M.M.; Simmons, L.A.; Kaguni, J.M. An essential tryptophan of Escherichia coli DnaA protein functions in oligomerization at the E. coli replication origin. J. Biol. Chem. 2005, 280, 24627–24633. [Google Scholar] [CrossRef] [PubMed]
- Felczak, M.M. Escherichia coli DnaA: Self-Oligomerization in Initiation at the Bacterial Chromosomal Origin; Polish Academy of Sciences: East Lansing, MI, USA; Warsaw, Poland, 2007. [Google Scholar]
- Bramhill, D.; Kornberg, A. Duplex opening by dnaA protein at novel sequences in initiation of replication at the origin of the E. coli chromosome. Cell 1988, 52, 743–755. [Google Scholar] [CrossRef]
- Speck, C.; Messer, W. Mechanism of origin unwinding: Sequential binding of DnaA to double- and single-stranded DNA. EMBO J. 2001, 20, 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Duderstadt, K.E.; Mott, M.L.; Crisona, N.J.; Chuang, K.; Yang, H.; Berger, J.M. Origin remodeling and opening in bacteria rely on distinct assembly states of the DnaA initiator. J. Biol. Chem. 2010, 285, 28229–28239. [Google Scholar] [CrossRef] [PubMed]
- Duderstadt, K.E.; Chuang, K.; Berger, J.M. DNA stretching by bacterial initiators promotes replication origin opening. Nature 2011, 478, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, S.; Katayama, T. Highly organized DnaA-oriC complexes recruit the single-stranded DNA for replication initiation. Nucleic Acids Res. 2012, 40, 1648–1665. [Google Scholar] [CrossRef] [PubMed]
- Sakiyama, Y.; Kasho, K.; Noguchi, Y.; Kawakami, H.; Katayama, T. Regulatory dynamics in the ternary DnaA complex for initiation of chromosomal replication in Escherichia coli. Nucleic Acids Res. 2017, 45, 12354–12373. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Akimitsu, N.; Kashioka, T.; Hatano, M.; Kubota, T.; Ogata, Y.; Sekimizu, K.; Katayama, T. DiaA, a novel DnaA-binding protein, ensures the timely initiation of Escherichia coli chromosome replication. J. Biol. Chem. 2004, 279, 45546–45555. [Google Scholar] [CrossRef] [PubMed]
- Keyamura, K.; Abe, Y.; Higashi, M.; Ueda, T.; Katayama, T. DiaA dynamics are coupled with changes in initial origin complexes leading to helicase loading. J. Biol. Chem. 2009, 284, 25038–25050. [Google Scholar] [CrossRef] [PubMed]
- Natrajan, G.; Noirot-Gros, M.F.; Zawilak-Pawlik, A.; Kapp, U.; Terradot, L. The structure of a DnaA/HobA complex from Helicobacter pylori provides insight into regulation of DNA replication in bacteria. Proc. Natl. Acad. Sci. USA 2009, 106, 21115–21120. [Google Scholar] [CrossRef] [PubMed]
- Chodavarapu, S.; Felczak, M.M.; Yaniv, J.R.; Kaguni, J.M. Escherichia coli DnaA interacts with HU in initiation at the E. coli replication origin. Mol. Microbiol. 2008, 67, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Drlica, K.; Rouviere-Yaniv, J. Histonelike proteins of bacteria. Microbiol. Rev. 1987, 51, 301–319. [Google Scholar] [PubMed]
- Browning, D.F.; Grainger, D.C.; Busby, S.J. Effects of nucleoid-associated proteins on bacterial chromosome structure and gene expression. Curr. Opin. Microbiol. 2010, 13, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Rimsky, S.; Travers, A. Pervasive regulation of nucleoid structure and function by nucleoid-associated proteins. Curr. Opin. Microbiol. 2011, 14, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Macvanin, M.; Adhya, S. Architectural organization in E. coli nucleoid. Biochim. Biophys. Acta 2012, 1819, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Keyamura, K.; Fujikawa, N.; Ishida, T.; Ozaki, S.; Su’etsugu, M.; Fujimitsu, K.; Kagawa, W.; Yokoyama, S.; Kurumizaka, H.; Katayama, T. The interaction of DiaA and DnaA regulates the replication cycle in E. coli by directly promoting ATP DnaA-specific initiation complexes. Genes Dev. 2007, 21, 2083–2099. [Google Scholar] [CrossRef] [PubMed]
- Zawilak-Pawlik, A.; Donczew, R.; Szafranski, S.; Mackiewicz, P.; Terradot, L.; Zakrzewska-Czerwinska, J. DiaA/HobA and DnaA: A pair of proteins co-evolved to cooperate during bacterial orisome assembly. J. Mol. Biol. 2011, 408, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Chodavarapu, S.; Felczak, M.M.; Kaguni, J.M. Two forms of ribosomal protein L2 of Escherichia coli that inhibit DnaA in DNA replication. Nucleic Acids Res. 2011, 39, 4180–4191. [Google Scholar] [CrossRef] [PubMed]
- Chodavarapu, S.; Gomez, R.; Vicente, M.; Kaguni, J.M. Escherichia coli Dps interacts with DnaA protein to impede initiation: A model of adaptive mutation. Mol. Microbiol. 2008, 67, 1331–1346. [Google Scholar] [CrossRef] [PubMed]
- Almiron, M.; Link, A.J.; Furlong, D.; Kolter, R. A novel DNA-binding protein with regulatory and protective roles in starved Escherichia coli. Genes Dev. 1992, 6, 2646–2654. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L. The physiological role of ferritin-like compounds in bacteria. Crit. Rev. Microbiol. 2004, 30, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Schaper, S.; Messer, W. Prediction of the structure of the replication initiator protein DnaA. Proteins 1997, 28, 1–9. [Google Scholar] [CrossRef]
- Nozaki, S.; Ogawa, T. Determination of the minimum domain II size of Escherichia coli DnaA protein essential for cell viability. Microbiology 2008, 154, 3379–3384. [Google Scholar] [CrossRef] [PubMed]
- Molt, K.L.; Sutera, V.A., Jr.; Moore, K.K.; Lovett, S.T. A role for nonessential domain II of initiator protein, DnaA, in replication control. Genetics 2009, 183, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Charbon, G.; Riber, L.; Cohen, M.; Skovgaard, O.; Fujimitsu, K.; Katayama, T.; Lobner-Olesen, A. Suppressors of DnaA(ATP) imposed overinitiation in Escherichia coli. Mol. Microbiol. 2011, 79, 914–928. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. A common set of conserved motifs in a vast variety of putative nucleic acid-dependent ATPases including MCM proteins involved in the initiation of eukaryotic DNA replication. Nucleic Acids Res. 1993, 21, 2541–2547. [Google Scholar] [CrossRef] [PubMed]
- Neuwald, A.F.; Aravind, L.; Spouge, J.L.; Koonin, E.V. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999, 9, 27–43. [Google Scholar] [PubMed]
- Erzberger, J.P.; Berger, J.M. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 93–114. [Google Scholar] [CrossRef] [PubMed]
- Duderstadt, K.E.; Berger, J.M. AAA+ ATPases in the initiation of DNA replication. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 163–187. [Google Scholar] [CrossRef] [PubMed]
- Sekimizu, K.; Bramhill, D.; Kornberg, A. ATP activates dnaA protein in initiating replication of plasmids bearing the origin of the E. coli chromosome. Cell 1987, 50, 259–265. [Google Scholar] [CrossRef]
- Kawakami, H.; Ozaki, S.; Suzuki, S.; Nakamura, K.; Senriuchi, T.; Su’etsugu, M.; Fujimitsu, K.; Katayama, T. The exceptionally tight affinity of DnaA for ATP/ADP requires a unique aspartic acid residue in the AAA+ sensor 1 motif. Mol. Microbiol. 2006, 62, 1310–1324. [Google Scholar] [CrossRef] [PubMed]
- Nishida, S.; Fujimitsu, K.; Sekimizu, K.; Ohmura, T.; Ueda, T.; Katayama, T. A nucleotide switch in the Escherichia coli DnaA protein initiates chromosomal replication: Evidnece from a mutant DnaA protein defective in regulatory ATP hydrolysis in vitro and in vivo. J. Biol. Chem. 2002, 277, 14986–14995. [Google Scholar] [CrossRef] [PubMed]
- Felczak, M.M.; Kaguni, J.M. The box VII motif of Escherichia coli DnaA protein is required for DnaA oligomerization at the E. coli replication origin. J. Biol. Chem. 2004, 279, 51156–51162. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, H.; Keyamura, K.; Katayama, T. Formation of an ATP-DnaA-specific initiation complex requires DnaA arginine 285, a conserved motif in the AAA+ protein family. J. Biol. Chem. 2005, 280, 27420–27430. [Google Scholar] [CrossRef] [PubMed]
- Katayama, T. Roles for the AAA+ motifs of DnaA in the initiation of DNA replication. Biochem. Soc. Trans. 2008, 36, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Houston, D.R. (Institute of Structural and Molecular Biology. University of Edinburgh, Edinburgh, UK). Personal communication, 2017.
- Roth, A.; Messer, W. The DNA binding domain of the initiator protein DnaA. EMBO J. 1995, 14, 2106–2111. [Google Scholar] [PubMed]
- Sutton, M.D.; Kaguni, J.M. Threonine 435 of Escherichia coli DnaA protein confers sequence- specific DNA binding activity. J. Biol. Chem. 1997, 272, 23017–23024. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Blaesing, F.; Weigel, C.; Welzeck, M.; Messer, W. Analysis of the DNA-binding domain of Escherichia coli DnaA protein. Mol. Microbiol. 2000, 36, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, N.; Kurumizaka, H.; Nureki, O.; Terada, T.; Shirouzu, M.; Katayama, T.; Yokoyama, S. Structural basis of replication origin recognition by the DnaA protein. Nucleic Acids Res. 2003, 31, 2077–2086. [Google Scholar] [CrossRef] [PubMed]
- Singleton, M.R.; Dillingham, M.S.; Wigley, D.B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007, 76, 23–50. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, N.D.; Berger, J.M. Running in reverse: The structural basis for translocation polarity in hexameric helicases. Cell 2009, 139, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Trakselis, M.A. Structural mechanisms of hexameric helicase loading, assembly, and unwinding. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Ilyina, T.V.; Gorbalenya, A.E.; Koonin, E.V. Organization and evolution of bacterial and bacteriophage primase-helicase systems. J. Mol. Evol. 1992, 34, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, S.M.; Richardson, C.C. Motors, switches, and contacts in a replisome. Annu. Rev. Biochem. 2009, 78, 205–243. [Google Scholar] [CrossRef] [PubMed]
- Jezewska, M.J.; Rajendran, S.; Bujalowska, D.; Bujalowski, W. Does single-stranded DNA pass through the inner channel of the protein hexamer in the complex with the Escherichia coli DnaB helicase?. Fluorescence energy transfer studies. J. Biol. Chem. 1998, 273, 10515–10529. [Google Scholar] [CrossRef] [PubMed]
- Itsathitphaisarn, O.; Wing, R.A.; Eliason, W.K.; Wang, J.; Steitz, T.A. The hexameric helicase DnaB adopts a nonplanar conformation during translocation. Cell 2012, 151, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Carney, S.M.; Gomathinayagam, S.; Leuba, S.H.; Trakselis, M.A. Bacterial DnaB helicase interacts with the excluded strand to regulate unwinding. J. Biol. Chem. 2017, 292, 19001–19012. [Google Scholar] [CrossRef] [PubMed]
- Donate, L.E.; Llorca, O.; Barcena, M.; Brown, S.E.; Dixon, N.E.; Carazo, J.M. pH-controlled quaternary states of hexameric DnaB helicase. J. Mol. Biol. 2000, 303, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Yu, X.; VanLoock, M.S.; Jezewska, M.J.; Bujalowski, W.; Egelman, E.H. Flexibility of the rings: Structural asymmetry in the DnaB hexameric helicase. J. Mol. Biol. 2002, 321, 839–849. [Google Scholar] [CrossRef]
- Lo, Y.H.; Tsai, K.L.; Sun, Y.J.; Chen, W.T.; Huang, C.Y.; Hsiao, C.D. The crystal structure of a replicative hexameric helicase DnaC and its complex with single-stranded DNA. Nucleic Acids Res. 2009, 37, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Eliason, W.K.; Steitz, T.A. Structure of a helicase-helicase loader complex reveals insights into the mechanism of bacterial primosome assembly. Nat. Commun. 2013, 4, 2495. [Google Scholar] [CrossRef] [PubMed]
- Arias-Palomo, E.; O’Shea, V.L.; Hood, I.V.; Berger, J.M. The bacterial DnaC helicase loader is a DnaB ring breaker. Cell 2013, 153, 438–448. [Google Scholar] [CrossRef] [PubMed]
- San Martin, C.; Radermacher, M.; Wolpensinger, B.; Engel, A.; Miles, C.S.; Dixon, N.E.; Carazo, J.M. Three-dimensional reconstructions from cryoelectron microscopy images reveal an intimate complex between helicase DnaB and its loading partner DnaC. Structure 1998, 6, 501–509. [Google Scholar] [CrossRef]
- Leipe, D.D.; Aravind, L.; Grishin, N.V.; Koonin, E.V. The bacterial replicative helicase DnaB evolved from a RecA duplication. Genome Res. 2000, 10, 5–16. [Google Scholar] [PubMed]
- Wickner, S.; Wright, M.; Hurwitz, J. Association of DNA-dependent and -independent ribonucleoside triphosphatase activities with dnaB gene product of Escherichia coli. Proc. Natl. Acad. Sci. USA 1974, 71, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Reha-Krantz, L.J.; Hurwitz, J. The dnaB gene product of Escherichia coli. II. Single stranded DNA- dependent ribonucleoside triphosphatase activity. J. Biol. Chem. 1978, 253, 4051–4057. [Google Scholar] [PubMed]
- LeBowitz, J.H.; McMacken, R. The Escherichia coli dnaB replication protein is a DNA helicase. J. Biol. Chem. 1986, 261, 4738–4748. [Google Scholar] [PubMed]
- Jezewska, M.J.; Rajendran, S.; Bujalowski, W. Strand specificity in the interactions of Escherichia coli primary replicative helicase DnaB protein with a replication fork. Biochemistry 1997, 36, 10320–10326. [Google Scholar] [CrossRef] [PubMed]
- Jezewska, M.J.; Rajendran, S.; Bujalowski, W. Complex of Escherichia coli primary replicative helicase DnaB protein with a replication fork: Recognition and structure. Biochemistry 1998, 37, 3116–3136. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.L. The 3′-tail of a forked-duplex sterically determines whether one or two DNA strands pass through the central channel of a replication-fork helicase. J. Mol. Biol. 2000, 301, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Galletto, R.; Jezewska, M.J.; Bujalowski, W. Unzipping mechanism of the double-stranded DNA unwinding by a hexameric helicase: The effect of the 3′ arm and the stability of the dsDNA on the unwinding activity of the Escherichia coli DnaB helicase. J. Mol. Biol. 2004, 343, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Enemark, E.J.; Joshua-Tor, L. Mechanism of DNA translocation in a replicative hexameric helicase. Nature 2006, 442, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Enemark, E.J.; Joshua-Tor, L. On helicases and other motor proteins. Curr. Opin. Struct. Biol. 2008, 18, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Lyubimov, A.Y.; Strycharska, M.; Berger, J.M. The nuts and bolts of ring-translocase structure and mechanism. Curr. Opin. Struct. Biol. 2011, 21, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Wickner, S.; Hurwitz, J. Interaction of Escherichia coli dnaB and dnaC(D) gene products in vitro. Proc. Natl. Acad. Sci. USA 1975, 72, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Wahle, E.; Lasken, R.S.; Kornberg, A. The dnaB-dnaC replication protein complex of Escherichia coli. II. Role of the complex in mobilizing dnaB functions. J. Biol. Chem. 1989, 264, 2469–2475. [Google Scholar] [PubMed]
- Wahle, E.; Lasken, R.S.; Kornberg, A. The dnaB-dnaC replication protein complex of Escherichia coli. I. Formation and properties. J. Biol. Chem. 1989, 264, 2463–2468. [Google Scholar] [PubMed]
- Wu, C.A.; Zechner, E.L.; Reems, J.A.; McHenry, C.S.; Marians, K.J. Coordinated leading- and lagging-strand synthesis at the Escherichia coli DNA replication fork. V. Primase action regulates the cycle of Okazaki fragment synthesis. J. Biol. Chem. 1992, 267, 4074–4083. [Google Scholar] [PubMed]
- Tougu, K.; Marians, K.J. The interaction between helicase and primase sets the replication fork clock. J. Biol. Chem. 1996, 271, 21398–21405. [Google Scholar] [CrossRef] [PubMed]
- Corn, J.E.; Berger, J.M. Regulation of bacterial priming and daughter strand synthesis through helicase-primase interactions. Nucleic Acids Res. 2006, 34, 4082–4088. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Dallmann, H.G.; McHenry, C.S.; Marians, K.J. Coupling of a replicative polymerase and helicase: A tau-DnaB interaction mediates rapid replication fork movement. Cell 1996, 84, 643–650. [Google Scholar] [CrossRef]
- Graham, J.E.; Marians, K.J.; Kowalczykowski, S.C. Independent and stochastic action of DNA polymerases in the replisome. Cell 2017, 169, 1201–1213. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.H.; Huang, C.Y. Characterization of flavonol inhibition of DnaB helicase: Real-time monitoring, structural modeling, and proposed mechanism. J. Biomed. Biotechnol. 2012, 2012, 735368. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Gorbalenya, A.E. The superfamily of UvrA-related ATPases includes three more subunits of putative ATP-dependent nucleases. Protein Seq. Data Anal. 1992, 5, 43–45. [Google Scholar] [PubMed]
- Iyer, L.M.; Leipe, D.D.; Koonin, E.V.; Aravind, L. Evolutionary history and higher order classification of AAA+ ATPases. J. Struct. Biol. 2004, 146, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Davey, M.J.; Fang, L.; McInerney, P.; Georgescu, R.E.; O’Donnell, M. The DnaC helicase loader is a dual ATP/ADP switch protein. EMBO J. 2002, 21, 3148–3159. [Google Scholar] [CrossRef] [PubMed]
- Mott, M.L.; Erzberger, J.P.; Coons, M.M.; Berger, J.M. Structural synergy and molecular crosstalk between bacterial helicase loaders and replication initiators. Cell 2008, 135, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Dueber, E.L.; Corn, J.E.; Bell, S.D.; Berger, J.M. Replication origin recognition and deformation by a heterodimeric archaeal ORC1 complex. Science 2007, 317, 1210–1213. [Google Scholar] [CrossRef] [PubMed]
- Gaudier, M.; Schuwirth, B.S.; Westcott, S.L.; Wigley, D.B. Structural basis of DNA replication origin recognition by an ORC protein. Science 2007, 317, 1213–1216. [Google Scholar] [CrossRef] [PubMed]
- Bleichert, F.; Botchan, M.R.; Berger, J.M. Crystal structure of the eukaryotic origin recognition complex. Nature 2015, 519, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Ludlam, A.V.; McNatt, M.W.; Carr, K.M.; Kaguni, J.M. Essential amino acids of Escherichia coli DnaC protein in an N-terminal domain interact with DnaB helicase. J. Biol. Chem. 2001, 276, 27345–27353. [Google Scholar] [CrossRef] [PubMed]
- Hupert-Kocurek, K.; Sage, J.M.; Makowska-Grzyska, M.; Kaguni, J.M. Genetic method to analyze essential genes of Escherichia coli. Appl. Environ. Microbiol. 2007, 73, 7075–7082. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Galletto, R.; Jezewska, M.J.; Bujalowski, W. Interactions of the Escherichia coli DnaB helicase hexamer with the replication factor the DnaC protein. Effect of nucleotide cofactors and the ssDNA on protein-protein interactions and the topology of the complex. J. Mol. Biol. 2003, 329, 441–465. [Google Scholar] [CrossRef]
- Chodavarapu, S.; Jones, A.D.; Feig, M.; Kaguni, J.M. DnaC traps DnaB as an open ring and remodels the domain that binds primase. Nucleic Acids Res. 2016, 44, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Felczak, M.M.; Chodavarapu, S.; Kaguni, J.M. DnaC, the indispensable companion of DnaB helicase, controls the accessibility of DnaB helicase by primase. J. Biol. Chem. 2017, 292, 20871–20882. [Google Scholar] [CrossRef] [PubMed]
- Barcena, M.; Ruiz, T.; Donate, L.E.; Brown, S.E.; Dixon, N.E.; Radermacher, M.; Carazo, J.M. The DnaB.DnaC complex: A structure based on dimers assembled around an occluded channel. EMBO J. 2001, 20, 1462–1468. [Google Scholar] [CrossRef] [PubMed]
- Learn, B.A.; Um, S.J.; Huang, L.; McMacken, R. Cryptic single-stranded-DNA binding activities of the phage lambda P and Escherichia coli DnaC replication initiation proteins facilitate the transfer of E. coli DnaB helicase onto DNA. Proc. Natl. Acad. Sci. USA 1997, 94, 1154–1159. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.B.; Flowers, S.; Biswas-Fiss, E.E. Quantitative analysis of nucleotide modulation of DNA binding by the DnaC protein of Escherichia coli. Biochem. J. 2004, 379, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Makowska-Grzyska, M.; Kaguni, J.M. Primase directs the release of DnaC from DnaB. Mol. Cell 2010, 37, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Felczak, M.M.; Sage, J.M.; Hupert-Kocurek, K.; Aykul, S.; Kaguni, J.M. Substitutions of conserved residues in the C-terminal region of DnaC cause thermolability in helicase loading. J. Biol. Chem. 2016, 291, 4803–4812. [Google Scholar] [CrossRef] [PubMed]
- Larson, M.A.; Griep, M.A.; Bressani, R.; Chintakayala, K.; Soultanas, P.; Hinrichs, S.H. Class-specific restrictions define primase interactions with DNA template and replicative helicase. Nucleic Acids Res. 2010, 38, 7167–7178. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yoda, K.; Okazaki, T. Specificity of recognition sequence for Escherichia coli primase. Mol. Gen. Genet. 1991, 227, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Swart, J.R.; Griep, M.A. Primase from Escherichia coli primes single-stranded templates in the absence of single-stranded DNA-binding protein or other auxiliary proteins. Template sequence requirements based on the bacteriophage G4 complementary strand origin and Okazaki fragment initiation sites. J. Biol. Chem. 1993, 268, 12970–12976. [Google Scholar] [PubMed]
- Bird, L.E.; Pan, H.; Soultanas, P.; Wigley, D.B. Mapping protein-protein interactions within a stable complex of DNA primase and DnaB helicase from Bacillus stearothermophilus. Biochemistry 2000, 39, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Wigley, D.B. Structure of the zinc-binding domain of Bacillus stearothermophilus DNA primase. Structure 2000, 8, 231–239. [Google Scholar] [CrossRef]
- Zhou, Y.; Luo, H.; Liu, Z.; Yang, M.; Pang, X.; Sun, F.; Wang, G. Structural insight into the specific DNA template binding to DnaG primase in bacteria. Sci. Rep. 2017, 7, 659. [Google Scholar] [CrossRef] [PubMed]
- Syson, K.; Thirlway, J.; Hounslow, A.M.; Soultanas, P.; Waltho, J.P. Solution structure of the helicase-interaction domain of the primase DnaG: A model for helicase activation. Structure 2005, 13, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Chintakayala, K.; Larson, M.A.; Grainger, W.H.; Scott, D.J.; Griep, M.A.; Hinrichs, S.H.; Soultanas, P. Domain swapping reveals that the C- and N-terminal domains of DnaG and DnaB, respectively, are functional homologues. Mol. Microbiol. 2007, 63, 1629–1639. [Google Scholar] [CrossRef] [PubMed]
- Strycharska, M.S.; Arias-Palomo, E.; Lyubimov, A.Y.; Erzberger, J.P.; O’Shea, V.L.; Bustamante, C.J.; Berger, J.M. Nucleotide and partner-protein control of bacterial replicative helicase structure and function. Mol. Cell 2013, 52, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Mitkova, A.V.; Khopde, S.M.; Biswas, S.B. Mechanism and stoichiometry of interaction of DnaG primase with DnaB helicase of Escherichia coli in RNA primer synthesis. J. Biol. Chem. 2003, 278, 52253–52261. [Google Scholar] [CrossRef] [PubMed]
- Yuzhakov, A.; Turner, J.; O’Donnell, M. Replisome assembly reveals the basis for asymmetric function in leading and lagging strand replication. Cell 1996, 86, 877–886. [Google Scholar] [CrossRef]
- Chintakayala, K.; Larson, M.A.; Griep, M.A.; Hinrichs, S.H.; Soultanas, P. Conserved residues of the C-terminal p16 domain of primase are involved in modulating the activity of the bacterial primosome. Mol. Microbiol. 2008, 68, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lee, S.J.; Zhu, B.; Tran, N.Q.; Tabor, S.; Richardson, C.C. Helicase-DNA polymerase interaction is critical to initiate leading-strand DNA synthesis. Proc. Natl. Acad. Sci. USA 2011, 108, 9372–9377. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Richardson, C.C. Choreography of bacteriophage T7 DNA replication. Curr. Opin. Chem. Biol. 2011, 15, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Allen, W.J.; Li, Y.; Waksman, G. Bacterial DNA polymerase I. In eLS; John Wiley & Sons Ltd.: Chichester, UK, 2010. [Google Scholar]
- McHenry, C.S. Bacterial replicases and related polymerases. Curr. Opin. Chem. Biol. 2011, 15, 587–594. [Google Scholar] [CrossRef] [PubMed]
- McHenry, C.S. DNA replicases from a bacterial perspective. Annu. Rev. Biochem. 2011, 80, 403–436. [Google Scholar] [CrossRef] [PubMed]
- Dohrmann, P.R.; Correa, R.; Frisch, R.L.; Rosenberg, S.M.; McHenry, C.S. The DNA polymerase III holoenzyme contains gamma and is not a trimeric polymerase. Nucleic Acids Res. 2016, 44, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.; Wing, R.A.; Steitz, T.A. The structure of T. aquaticus DNA polymerase III is distinct from eukaryotic replicative DNA polymerases. Cell 2006, 126, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Wing, R.A.; Bailey, S.; Steitz, T.A. Insights into the replisome from the structure of a ternary complex of the DNA polymerase III alpha-subunit. J. Mol. Biol. 2008, 382, 859–869. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, D.R.; McHenry, C.S. Identification of the beta-binding domain of the alpha subunit of Escherichia coli polymerase III holoenzyme. J. Biol. Chem. 1996, 271, 20699–20704. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.R.; McHenry, C.S. Biotin tagging deletion analysis of domain limits involved in protein-macromolecular interactions. Mapping the tau binding domain of the DNA polymerase III alpha subunit. J. Biol. Chem. 1996, 271, 20690–20698. [Google Scholar] [CrossRef] [PubMed]
- Reems, J.A.; Wood, S.; McHenry, C.S. Escherichia coli DNA polymerase III holoenzyme subunits alpha, beta, and gamma directly contact the primer-template. J. Biol. Chem. 1995, 270, 5606–5613. [Google Scholar] [CrossRef] [PubMed]
- Lopez de Saro, F.J.; Georgescu, R.E.; Goodman, M.F.; O’Donnell, M. Competitive processivity-clamp usage by DNA polymerases during DNA replication and repair. EMBO J. 2003, 22, 6408–6418. [Google Scholar] [CrossRef] [PubMed]
- Dervyn, E.; Suski, C.; Daniel, R.; Bruand, C.; Chapuis, J.; Errington, J.; Janniere, L.; Ehrlich, S.D. Two essential DNA polymerases at the bacterial replication fork. Science 2001, 294, 1716–1719. [Google Scholar] [CrossRef] [PubMed]
- Sanders, G.M.; Dallmann, H.G.; McHenry, C.S. Reconstitution of the B. subtilis replisome with 13 proteins including two distinct replicases. Mol. Cell 2010, 37, 273–281. [Google Scholar] [CrossRef] [PubMed]
- McHenry, C.S. Breaking the rules: Bacteria that use several DNA polymerase IIIs. EMBO Rep. 2011, 12, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Johansson, E.; Dixon, N. Replicative DNA polymerases. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Li, Y.; Schroeder, J.W.; Simmons, L.A.; Biteen, J.S. Single-molecule DNA polymerase dynamics at a bacterial replisome in live cells. Biophys. J. 2016, 111, 2562–2569. [Google Scholar] [CrossRef] [PubMed]
- Jameson, K.H.; Wilkinson, A.J. Control of initiation of DNA replication in Bacillus subtilis and Escherichia coli. Genes 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Taft-Benz, S.A.; Schaaper, R.M. The theta subunit of Escherichia coli DNA polymerase III: A role in stabilizing the epsilon proofreading subunit. J. Bacteriol. 2004, 186, 2774–2780. [Google Scholar] [CrossRef] [PubMed]
- DeRose, E.F.; Darden, T.; Harvey, S.; Gabel, S.; Perrino, F.W.; Schaaper, R.M.; London, R.E. Elucidation of the epsilon-theta subunit interface of Escherichia coli DNA polymerase III by NMR spectroscopy. Biochemistry 2003, 42, 3635–3644. [Google Scholar] [CrossRef] [PubMed]
- Keniry, M.A.; Park, A.Y.; Owen, E.A.; Hamdan, S.M.; Pintacuda, G.; Otting, G.; Dixon, N.E. Structure of the theta subunit of Escherichia coli DNA polymerase III in complex with the epsilon subunit. J. Bacteriol. 2006, 188, 4464–4473. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M. Replisome architecture and dynamics in Escherichia coli. J. Biol. Chem. 2006, 281, 10653–10656. [Google Scholar] [CrossRef] [PubMed]
- Scheuermann, R.; Tam, S.; Burgers, P.M.; Lu, C.; Echols, H. Identification of the epsilon-subunit of Escherichia coli DNA polymerase III holoenzyme as the dnaQ gene product: A fidelity subunit for DNA replication. Proc. Natl. Acad. Sci. USA 1983, 80, 7085–7089. [Google Scholar] [CrossRef] [PubMed]
- Fijalkowska, I.J.; Schaaper, R.M. Mutants in the Exo I motif of Escherichia coli dnaQ: Defective proofreading and inviability due to error catastrophe. Proc. Natl. Acad. Sci. USA 1996, 93, 2856–2861. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.P.; Onrust, R.; O’Donnell, M.; Kuriyan, J. Three-dimensional structure of the beta subunit of E. coli DNA polymerase III holoenzyme: A sliding DNA clamp. Cell 1992, 69, 425–437. [Google Scholar] [CrossRef]
- Fay, P.J.; Johanson, K.O.; McHenry, C.S.; Bambara, R.A. Size classes of products synthesized processively by DNA polymerase III and DNA polymerase III holoenzyme of Escherichia coli. J. Biol. Chem. 1981, 256, 976–983. [Google Scholar] [PubMed]
- Maki, S.; Kornberg, A. DNA polymerase III holoenzyme of Escherichia coli. III. Distinctive processive polymerases reconstituted from purified subunits. J. Biol. Chem. 1988, 263, 6561–6569. [Google Scholar] [PubMed]
- Stukenberg, P.T.; Studwell, V.P.; O’Donnell, M. Mechanism of the sliding beta-clamp of DNA polymerase III holoenzyme. J. Biol. Chem. 1991, 266, 11328–11334. [Google Scholar] [PubMed]
- Bloom, L.B.; Goodman, M.F. Polymerase processivity: Measurement and mechanisms. In eLS; John Wiley & Sons Ltd.: Chichester, UK, 2001. [Google Scholar]
- Indiani, C.; O’Donnell, M. The replication clamp-loading machine at work in the three domains of life. Nat. Rev. Mol. Cell Biol. 2006, 7, 751–761. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.; Kuriyan, J. Clamp loaders and replication initiation. Curr. Opin. Struct. Biol. 2006, 16, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Hedglin, M.; Kumar, R.; Benkovic, S.J. Replication clamps and clamp loaders. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. A superfamily of ATPases with diverse functions containing either classical or deviant ATP-binding motif. J. Mol. Biol. 1993, 229, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Davey, M.J.; Jeruzalmi, D.; Kuriyan, J.; O’Donnell, M. Motors and switches: AAA+ machines within the replisome. Nat. Rev. Mol. Cell Biol. 2002, 3, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Flower, A.M.; McHenry, C.S. The gamma subunit of DNA polymerase III holoenzyme of Escherichia coli is produced by ribosomal frameshifting. Proc. Natl. Acad. Sci. USA 1990, 87, 3713–3717. [Google Scholar] [CrossRef] [PubMed]
- Tsuchihashi, Z.; Kornberg, A. Translational frameshifting generates the gamma subunit of DNA polymerase III holoenzyme. Proc. Natl. Acad. Sci. USA 1990, 87, 2516–2520. [Google Scholar] [CrossRef] [PubMed]
- Blinkowa, A.L.; Walker, J.R. Programmed ribosomal frameshifting generates the Escherichia coli DNA polymerase III gamma subunit from within the tau subunit reading frame. Nucleic Acids Res. 1990, 18, 1725–1729. [Google Scholar] [CrossRef] [PubMed]
- Simonetta, K.R.; Kazmirski, S.L.; Goedken, E.R.; Cantor, A.J.; Kelch, B.A.; McNally, R.; Seyedin, S.N.; Makino, D.L.; O’Donnell, M.; Kuriyan, J. The mechanism of ATP-dependent primer-template recognition by a clamp loader complex. Cell 2009, 137, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Jeruzalmi, D.; Yurieva, O.; Zhao, Y.; Young, M.; Stewart, J.; Hingorani, M.; O’Donnell, M.; Kuriyan, J. Mechanism of processivity clamp opening by the delta subunit wrench of the clamp loader complex of E. coli DNA polymerase III. Cell 2001, 106, 417–428. [Google Scholar] [CrossRef]
- Naktinis, V.; Onrust, R.; Fang, L.; O’Donnell, M. Assembly of a chromosomal replication machine: Two DNA polymerases, a clamp loader, and sliding clamps in one holoenzyme particle. II. Intermediate complex between the clamp loader and its clamp. J. Biol. Chem. 1995, 270, 13358–13365. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.A.; Paschall, C.O.; O’Donnell, M.; Bloom, L.B. A slow ATP-induced conformational change limits the rate of DNA binding but not the rate of beta clamp binding by the Escherichia coli gamma complex clamp loader. J. Biol. Chem. 2009, 284, 32147–32157. [Google Scholar] [CrossRef] [PubMed]
- Douma, L.G.; Yu, K.K.; England, J.K.; Levitus, M.; Bloom, L.B. Mechanism of opening a sliding clamp. Nucleic Acids Res. 2017, 45, 10178–10189. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Engen, J.R.; Beuning, P.J. Escherichia coli processivity clamp beta from DNA polymerase III is dynamic in solution. Biochemistry 2011, 50, 5958–5968. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; McHenry, C.S. tau binds and organizes Escherichia coli replication through distinct domains. Partial proteolysis of terminally tagged tau to determine candidate domains and to assign domain V as the alpha binding domain. J. Biol. Chem. 2001, 276, 4433–4440. [Google Scholar] [CrossRef] [PubMed]
- Mok, M.; Marians, K.J. The Escherichia coli preprimosome and DNA B helicase can form replication forks that move at the same rate. J. Biol. Chem. 1987, 262, 16644–16654. [Google Scholar] [PubMed]
- Mok, M.; Marians, K.J. Formation of rolling-circle molecules during phi X174 complementary strand DNA replication. J. Biol. Chem. 1987, 262, 2304–2309. [Google Scholar] [PubMed]
- Gao, D.; McHenry, C.S. tau binds and organizes Escherichia coli replication proteins through distinct domains. Domain IV, located within the unique C terminus of tau, binds the replication fork, helicase, DnaB. J. Biol. Chem. 2001, 276, 4441–4446. [Google Scholar] [CrossRef] [PubMed]
- Dalrymple, B.P.; Kongsuwan, K.; Wijffels, G.; Dixon, N.E.; Jennings, P.A. A universal protein-protein interaction motif in the eubacterial DNA replication and repair systems. Proc. Natl. Acad. Sci. USA 2001, 98, 11627–11632. [Google Scholar] [CrossRef] [PubMed]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.D.; Berger, J.M. Structure, molecular mechanisms, and evolutionary relationships in DNA topoisomerases. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Forterre, P.; Gribaldo, S.; Gadelle, D.; Serre, M.C. Origin and evolution of DNA topoisomerases. Biochimie 2007, 89, 427–446. [Google Scholar] [CrossRef] [PubMed]
- Vos, S.M.; Tretter, E.M.; Schmidt, B.H.; Berger, J.M. All tangled up: How cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 2011, 12, 827–841. [Google Scholar] [CrossRef] [PubMed]
- Ketron, A.C.; Osheroff, N. DNA Topology and topoisomerases. In Molecular Life Sciences: An Encyclopedic Reference; Bell, E., Ed.; Springer: New York, NY, USA, 2014; pp. 1–19. [Google Scholar]
- Drlica, K.; Hiasa, H.; Kerns, R.; Malik, M.; Mustaev, A.; Zhao, X. Quinolones: Action and resistance updated. Curr. Top. Med. Chem. 2009, 9, 981–998. [Google Scholar] [CrossRef] [PubMed]
- Hiasa, H.; Marians, K.J. Topoisomerase III, but not topoisomerase I, can support nascent chain elongation during theta-type DNA replication. J. Biol. Chem. 1994, 269, 32655–32659. [Google Scholar] [PubMed]
- Hiasa, H.; DiGate, R.J.; Marians, K.J. Decatenating activity of Escherichia coli DNA gyrase and topoisomerases I and III during oriC and pBR322 DNA replication in vitro. J. Biol. Chem. 1994, 269, 2093–2099. [Google Scholar] [PubMed]
- Perez-Cheeks, B.A.; Lee, C.; Hayama, R.; Marians, K.J. A role for topoisomerase III in Escherichia coli chromosome segregation. Mol. Microbiol. 2012, 86, 1007–1022. [Google Scholar] [CrossRef] [PubMed]
- Terekhova, K.; Gunn, K.H.; Marko, J.F.; Mondragon, A. Bacterial topoisomerase I and topoisomerase III relax supercoiled DNA via distinct pathways. Nucleic Acids Res. 2012, 40, 10432–10440. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.F.; Wang, J.C. Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. USA 1987, 84, 7024–7027. [Google Scholar] [CrossRef] [PubMed]
- Masse, E.; Drolet, M. Relaxation of transcription-induced negative supercoiling is an essential function of Escherichia coli DNA topoisomerase I. J. Biol. Chem. 1999, 274, 16654–16658. [Google Scholar] [CrossRef] [PubMed]
- Zechiedrich, E.L.; Khodursky, A.B.; Bachellier, S.; Schneider, R.; Chen, D.; Lilley, D.M.; Cozzarelli, N.R. Roles of topoisomerases in maintaining steady-state DNA supercoiling in Escherichia coli. J. Biol. Chem. 2000, 275, 8103–8113. [Google Scholar] [CrossRef] [PubMed]
- Hiasa, H. DNA Topoisomerases as targets for antibacterial agents. Methods Mol. Biol. 2018, 1703, 47–62. [Google Scholar] [PubMed]
- Gellert, M.; O’Dea, M.H.; Itoh, T.; Tomizawa, J. Novobiocin and coumermycin inhibit DNA supercoiling catalyzed by DNA gyrase. Proc. Natl. Acad. Sci. USA 1976, 73, 4474–4478. [Google Scholar] [CrossRef] [PubMed]
- Sugino, A.; Peebles, C.L.; Kreuzer, K.N.; Cozzarelli, N.R. Mechanism of action of nalidixic acid: Purification of Escherichia coli nalA gene product and its relationship to DNA gyrase and a novel nicking-closing enzyme. Proc. Natl. Acad. Sci. USA 1977, 74, 4767–4771. [Google Scholar] [CrossRef] [PubMed]
- Anderson, V.E.; Zaniewski, R.P.; Kaczmarek, F.S.; Gootz, T.D.; Osheroff, N. Quinolones inhibit DNA religation mediated by Staphylococcus aureus topoisomerase IV. Changes in drug mechanism across evolutionary boundaries. J. Biol. Chem. 1999, 274, 35927–35932. [Google Scholar] [CrossRef] [PubMed]
- Collin, F.; Karkare, S.; Maxwell, A. Exploiting bacterial DNA gyrase as a drug target: Current state and perspectives. Appl. Microbiol. Biotechnol. 2011, 92, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Wohlkonig, A.; Chan, P.F.; Fosberry, A.P.; Homes, P.; Huang, J.; Kranz, M.; Leydon, V.R.; Miles, T.J.; Pearson, N.D.; Perera, R.L.; et al. Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat. Struct. Mol. Biol. 2010, 17, 1152–1153. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.F.; Srikannathasan, V.; Huang, J.; Cui, H.; Fosberry, A.P.; Gu, M.; Hann, M.M.; Hibbs, M.; Homes, P.; Ingraham, K.; et al. Structural basis of DNA gyrase inhibition by antibacterial QPT-1, anticancer drug etoposide and moxifloxacin. Nat. Commun. 2015, 6, 10048. [Google Scholar] [CrossRef] [PubMed]
- Black, M.T.; Stachyra, T.; Platel, D.; Girard, A.M.; Claudon, M.; Bruneau, J.M.; Miossec, C. Mechanism of action of the antibiotic NXL101, a novel nonfluoroquinolone inhibitor of bacterial type II topoisomerases. Antimicrob. Agents Chemother. 2008, 52, 3339–3349. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, T.J.; Nayar, A.; Newman, J.V.; Hopkins, S.; Stone, G.G.; Johnstone, M.; Shapiro, A.B.; Cronin, M.; Reck, F.; Ehmann, D.E. NBTI 5463 is a novel bacterial type II topoisomerase inhibitor with activity against gram-negative bacteria and in vivo efficacy. Antimicrob. Agents Chemother. 2014, 58, 2657–2664. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gomez, L.; Hack, M.D.; Wu, J.; Wiener, J.J.; Venkatesan, H.; Santillan, A., Jr.; Pippel, D.J.; Mani, N.; Morrow, B.J.; Motley, S.T.; et al. Novel pyrazole derivatives as potent inhibitors of type II topoisomerases. Part 1: Synthesis and preliminary SAR analysis. Bioorg. Med. Chem. Lett. 2007, 17, 2723–2727. [Google Scholar] [CrossRef] [PubMed]
- Foss, M.H.; Hurley, K.A.; Sorto, N.; Lackner, L.L.; Thornton, K.M.; Shaw, J.T.; Weibel, D.B. N-Benzyl-3-sulfonamidopyrrolidines are a new class of bacterial DNA gyrase inhibitors. ACS Med. Chem. Lett. 2011, 2, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; McPherson, S.A.; Turnbough, C.L., Jr.; Kerns, R.J.; Osheroff, N. Topoisomerase IV-quinolone interactions are mediated through a water-metal ion bridge: Mechanistic basis of quinolone resistance. Nucleic Acids Res. 2013, 41, 4628–4639. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, A.; Lawson, D.M. The ATP-binding site of type II topoisomerases as a target for antibacterial drugs. Curr. Top. Med. Chem. 2003, 3, 283–303. [Google Scholar] [CrossRef] [PubMed]
- Flatman, R.H.; Eustaquio, A.; Li, S.M.; Heide, L.; Maxwell, A. Structure-activity relationships of aminocoumarin-type gyrase and topoisomerase IV inhibitors obtained by combinatorial biosynthesis. Antimicrob. Agents Chemother. 2006, 50, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Heide, L. New aminocoumarin antibiotics as gyrase inhibitors. Int. J. Med. Microbiol. 2014, 304, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Flatman, R.H.; Howells, A.J.; Heide, L.; Fiedler, H.P.; Maxwell, A. Simocyclinone D8, an inhibitor of DNA gyrase with a novel mode of action. Antimicrob. Agents Chemother. 2005, 49, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.J.; Flatman, R.H.; Mitchenall, L.A.; Stevenson, C.E.; Le, T.B.; Clarke, T.A.; McKay, A.R.; Fiedler, H.P.; Buttner, M.J.; Lawson, D.M.; et al. A crystal structure of the bifunctional antibiotic simocyclinone D8, bound to DNA gyrase. Science 2009, 326, 1415–1418. [Google Scholar] [CrossRef] [PubMed]
- Buttner, M.J.; Schäfer, M.; Lawson, D.M.; Maxwell, A. Structural insights into simocyclinone as an antibiotic, effector ligand and substrate. FEMS Microbiol. Rev. 2018, 42. [Google Scholar] [CrossRef] [PubMed]
- Shuman, S. DNA ligases: Progress and prospects. J. Biol. Chem. 2009, 284, 17365–17369. [Google Scholar] [CrossRef] [PubMed]
- Swift, R.V.; Amaro, R.E. Discovery and design of DNA and RNA ligase inhibitors in infectious microorganisms. Expert Opin. Drug Discov. 2009, 4, 1281–1294. [Google Scholar] [CrossRef] [PubMed]
- Pergolizzi, G.; Wagner, G.K.; Bowater, R.P. Biochemical and Structural Characterisation of DNA ligases from bacteria and archaea. Biosci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, N.; Dube, D.; Pandey, J.; Singh, B.; Kukshal, V.; Ramachandran, R.; Tripathi, R.P. NAD(+)-dependent DNA ligase: A novel target waiting for the right inhibitor. Med. Res. Rev. 2008, 28, 545–568. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.; Amin, N.; Benowitz, A.B.; Chiarparin, E.; Cui, H.; Deng, X.; Heightman, T.D.; Holmes, D.J.; Hopkins, A.; Huang, J.; et al. Fragment-based discovery of 6-azaindazoles as inhibitors of bacterial DNA ligase. ACS Med. Chem. Lett. 2013, 4, 1208–1212. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.W.; Rees, D.C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.T.; Dawes, I.W. The preferential inhibition of Bacillus subtilis spore outgrowth by chloroquine. Arch. Microbiol. 1989, 152, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Van Eijk, E.; Wittekoek, B.; Kuijper, E.J.; Smits, W.K. DNA replication proteins as potential targets for antimicrobials in drug-resistant bacterial pathogens. J. Antimicrob. Chemother. 2017, 72, 1275–1284. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| E. coli Protein | Function | Inhibitor and Reference | |
|---|---|---|---|
| SSB | ssDNA binding | CFAM (2-[2-chloro-5-(trifluoromethyl)anilino]-5-methoxybenzoic acid) [27,28] BCBP (3-(tert-butyl)-1-(6-chloro-1,3-benzothiazol-2-yl)-4,5-dihydro-1H- pyrazol-5-one) [27,28] BOTP (2-[5-(3-bromobenzylidene)-4-oxo-2-thioxo-1,3,thiazoli- din-3-yl]-3-phenyl-propanoic acid) [27,28] MPTA ([5-(2-methyl-3-phenyl-2-propen-1-ylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl] (phenyl)acetic acid) [27,28] | |
| DnaA | recognition and binding to the E. coli replication origin (oriC) | bis-indoles (derivatives of 3-acetoxy-2,2′-bi-1H-indol) [29] | |
| DnaB | replicative DNA helicase | myricetin, a flavonol [30] | |
| Primase (DnaG) | primer synthesis | bicyclic 10-membered macrolide [31] phenolic monosaccharides [32] benzo[d]pyrimido[5,4-b]furans [33] benzo[d]imidazo[2,1-b]imidazoles [33] pyrido[3′,2′:4,5]thieno[3,2-d]pyrimidines [33] | |
| DNA polymerase I | removal of primers used for DNA synthesis | ||
| DNA polymerase III holoenzyme, a DnaE-type DNA polymerase 1 | DNA replicase | Subunit and Subassembly | Inhibitor and Reference |
| DnaN or β subunit of the sliding clamp subassembly | RU7 [34], biphenyloxime [35], and nonsteroidal anti-inflamatory drugs [36] | ||
| DnaN or β subunit of the sliding clamp subassembly of S. aureus | cyclic peptides [37] | ||
| DnaN or β subunit of the sliding clamp subassembly of M. tuberculosis | griselimycins [38] | ||
| α or DnaE subunit of the core subassembly of E. coli and S. aureus DNA polymerase III | nargencin [39] | ||
| DNA ligase A | ligation of Okazaki fragments | pyridochromanones [40] pyridopyrimidines [41] N-substituted tetracyclic indole [42,43] diamino-dimethylamino-pyrimido-pyrimidine [44] arylamino compounds (quinolones, quinacrines, bisquinolines) [45] adenosine analogues [46,47] | |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaguni, J.M. The Macromolecular Machines that Duplicate the Escherichia coli Chromosome as Targets for Drug Discovery. Antibiotics 2018, 7, 23. https://doi.org/10.3390/antibiotics7010023
Kaguni JM. The Macromolecular Machines that Duplicate the Escherichia coli Chromosome as Targets for Drug Discovery. Antibiotics. 2018; 7(1):23. https://doi.org/10.3390/antibiotics7010023
Chicago/Turabian StyleKaguni, Jon M. 2018. "The Macromolecular Machines that Duplicate the Escherichia coli Chromosome as Targets for Drug Discovery" Antibiotics 7, no. 1: 23. https://doi.org/10.3390/antibiotics7010023
APA StyleKaguni, J. M. (2018). The Macromolecular Machines that Duplicate the Escherichia coli Chromosome as Targets for Drug Discovery. Antibiotics, 7(1), 23. https://doi.org/10.3390/antibiotics7010023