Evaluation of In-Flow Magnetoresistive Chip Cell—Counter as a Diagnostic Tool

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
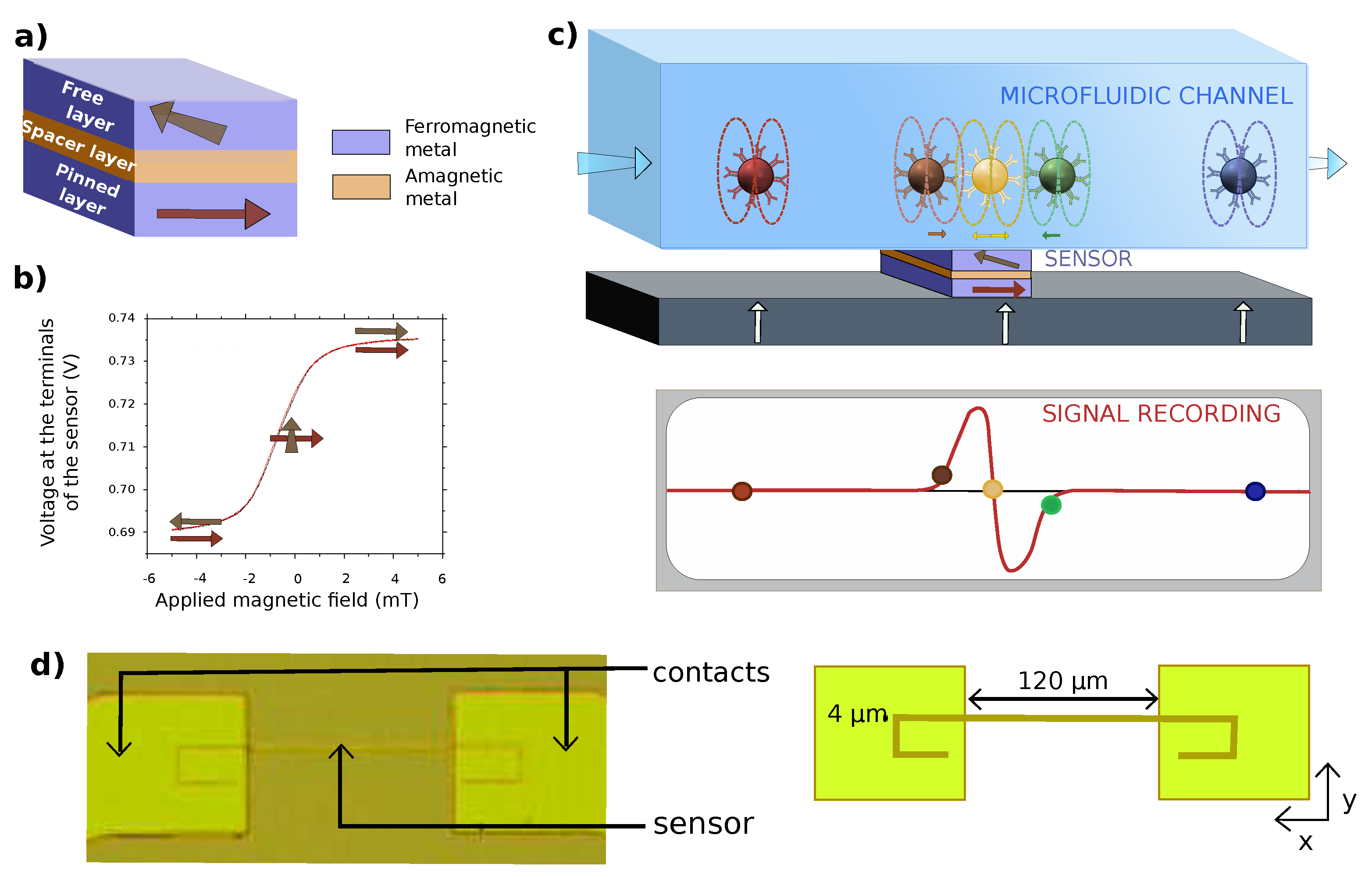
2.1. Sensor Fabrication
2.2. Microfluidic Channel Fabrication
2.3. Cell Culture
2.4. Production of IpaD-315 Antibodies
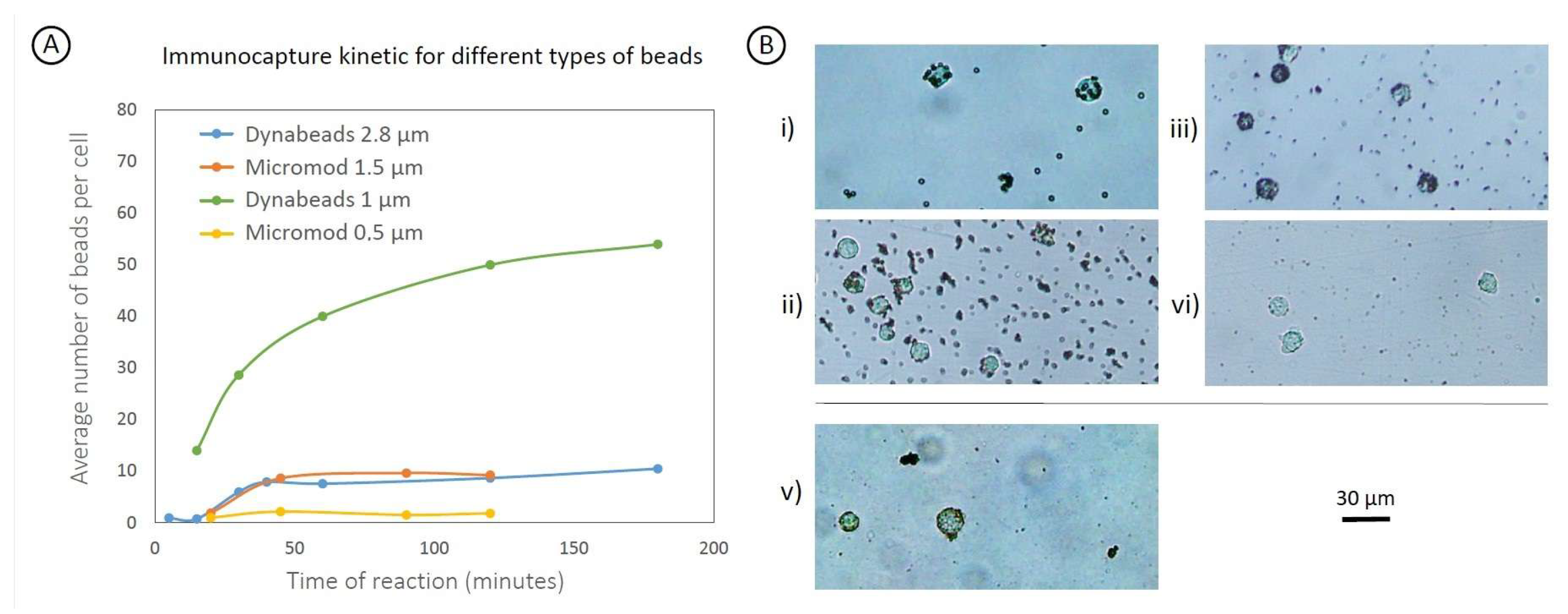
2.5. Particle Functionalization
2.6. MP Cell Labeling
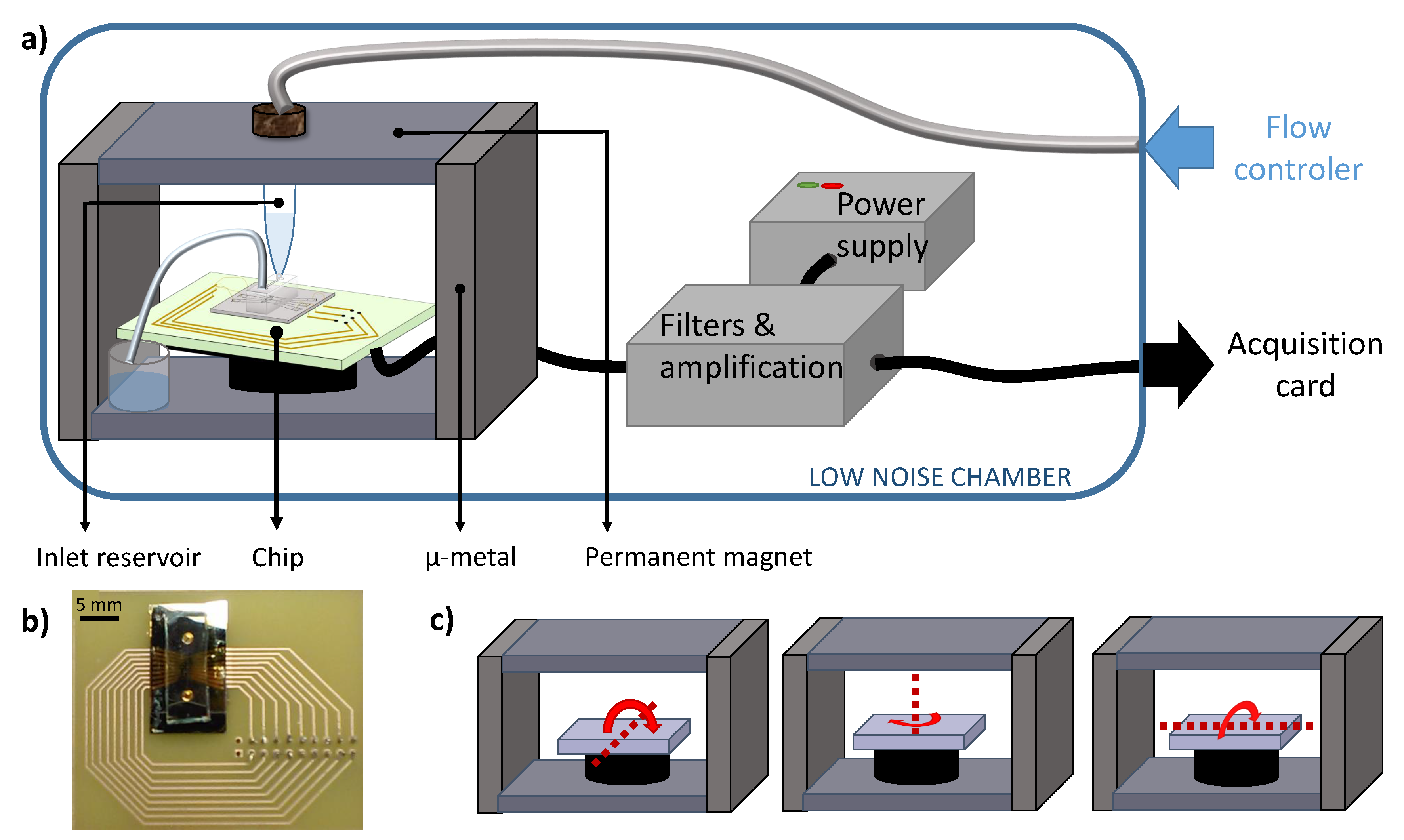
2.7. Experimental Set-Up
2.8. Electronics
2.9. Comparative ELISA Tests
2.10. Comparative Flow Cytometry Tests
3. Results and Discussion
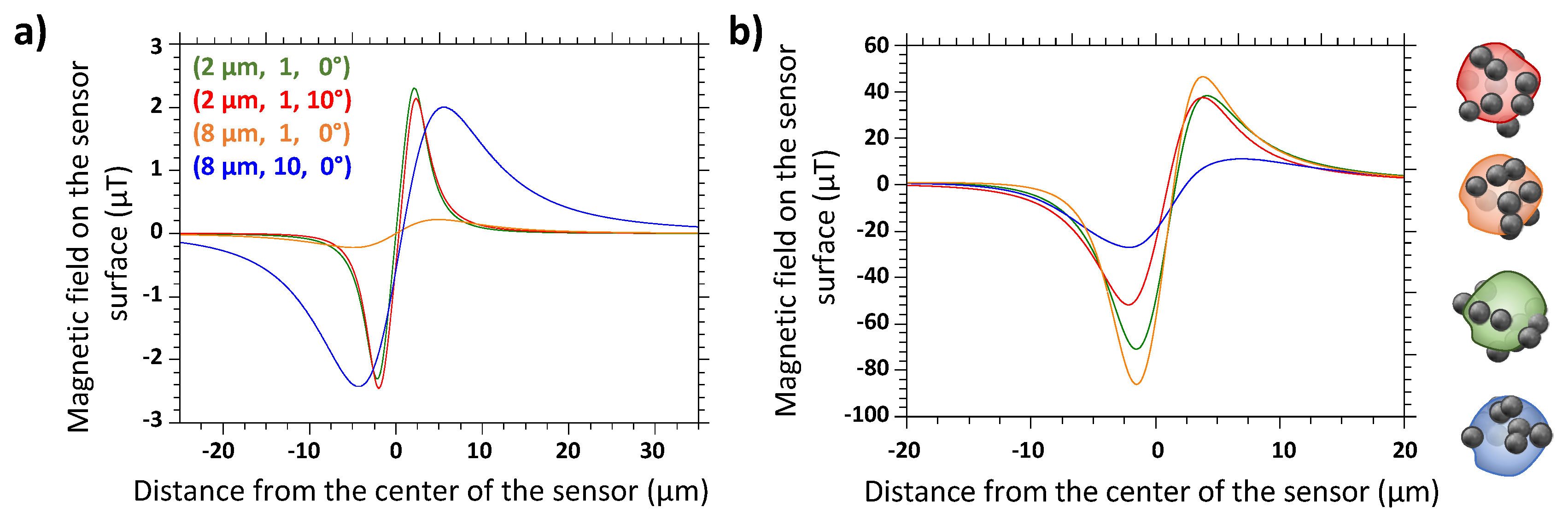
3.1. Simulations of Single Magnetic Beads and MP-Labeled Cells
3.2. Deduction of Best Experimental Conditions
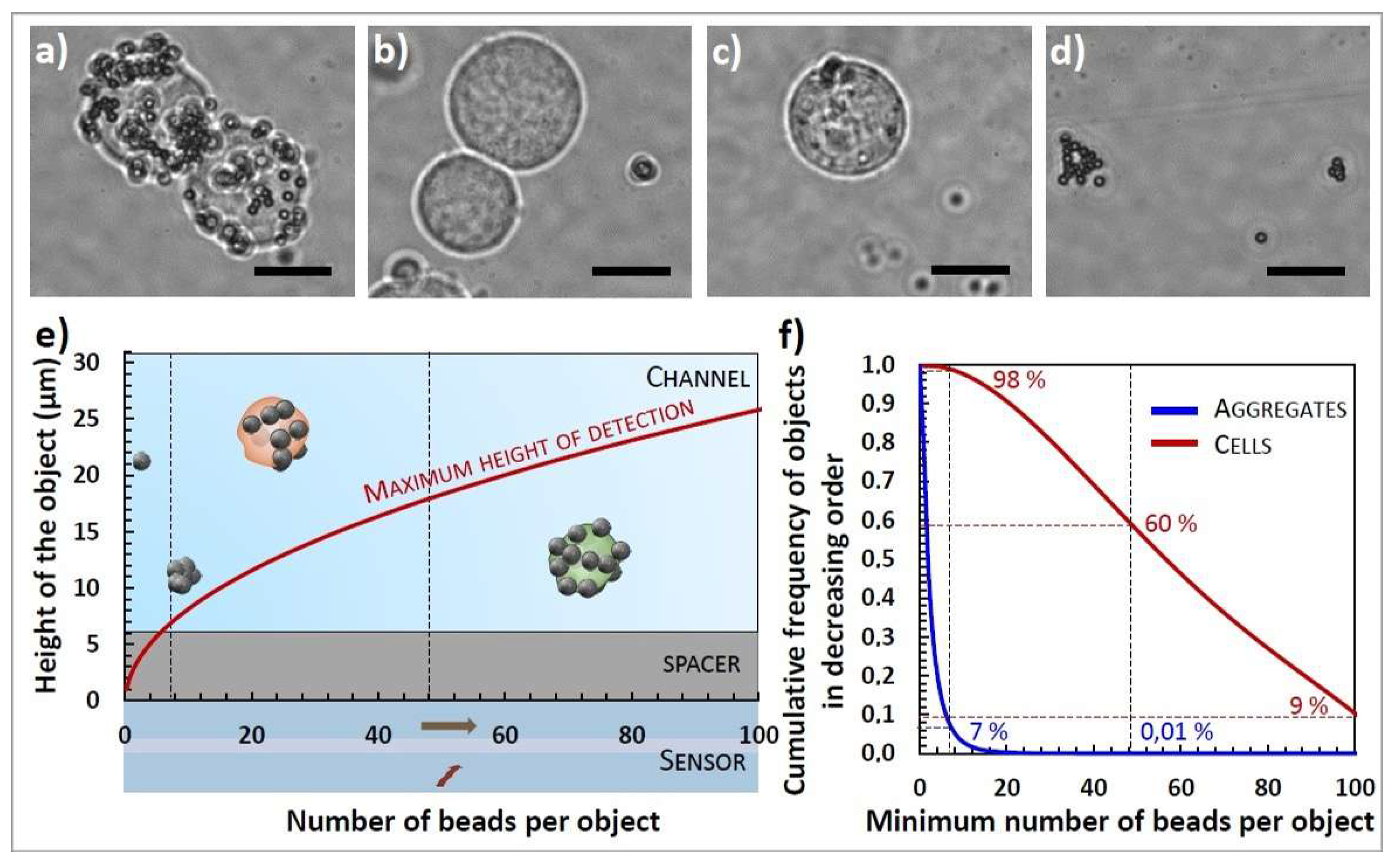
3.2.1. Sample Characterization
3.2.2. Chip Design
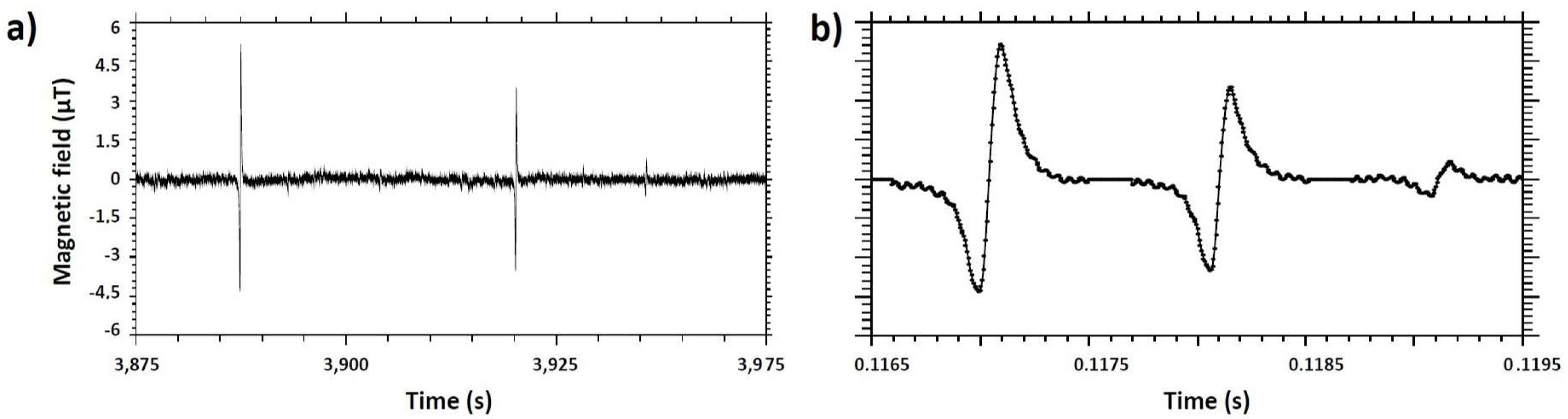
3.3. Performance of the GMR Chip Test
3.4. ELISA Test Sensitivity
3.5. Flow Cytometry Results
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BSA | Bovine Serum Albumine |
| CHO | Chineese Hamster Ovary cells |
| ELISA | Enzyme-Linked ImmunoSorbent Assay |
| GMR | Giant Magneto-Resistance |
| mAb | Monoclonal antibody |
| MP | Magnetic Particle |
| PBS | Phosphate buffer saline |
| PDMS | Polydimethylsiloxane |
Appendix A. Magnetic Particles Aggregation Study

References
- Bialy, L.; Mlynarczuk-Bialy, I. Advances in Biomedical Research—Selected Topics; Wydawnictwo Naukowe TYGIEL sp. z o.o.: Lublin, Poland, 2018. [Google Scholar]
- Cohen, S.J.; Punt, C.J.; Iannotti, N.; Saidman, B.H.; Sabbath, K.D.; Gabrail, N.Y.; Picus, J.; Morse, M.; Mitchell, E.; Miller, M.C.; et al. Relationship of Circulating Tumor Cells to Tumor Response, Progression-Free Survival, and Overall Survival in Patients With Metastatic Colorectal Cancer. J. Clin. Oncol. 2008, 26, 3213–3221. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, M.; Giordano, A.; Jackson, S.; Hess, K.R.; De Giorgi, U.; Mego, M.; Handy, B.C.; Ueno, N.T.; Alvarez, R.H.; De Laurentiis, M.; et al. Circulating tumor cells as prognostic and predictive markers in metastatic breast cancer patients receiving first-line systemic treatment. Breast Cancer Res. 2011, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sinha, M.; Jupe, J.; Mack, H.; Coleman, T.P.; Lawrence, S.M.; Fraley, S.I. Emerging Technologies for Molecular Diagnosis of Sepsis. Clin. Microbiol. Rev. 2018, 31, e00089-17. [Google Scholar] [CrossRef] [PubMed]
- Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; Dandona, L.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years LivedWith Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015 A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2017, 3, 524–548. [Google Scholar]
- Fulwyler, M.J. Electronic Separation of Biological Cells by Volume. Science 1965, 150, 910–911. [Google Scholar] [CrossRef]
- Aebisher, D.; Bartusik, D.; Tabarkiewicz, J. Laser flow cytometry as a tool for the advancement of clinical medicine. Biomed. Pharmacother. 2017, 85, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; Wittwer, C. Flow Cytometry: Principles and Clinical Applications in Hematology. Clin. Chem. 2000, 46, 1221–1229. [Google Scholar]
- Bakke, A.C. Clinical Applications of Flow Cytometry. Lab. Med. 2000, 31, 97–104. [Google Scholar] [CrossRef][Green Version]
- Wood, B.L. Principles of Minimal Residual Disease Detection for Hematopoietic Neoplasms by Flow Cytometry. Cytom. Part B 2016, 90, 47–53. [Google Scholar] [CrossRef]
- Chernecky, C.C.; Berger, B.J. Laboratory Tests and Diagnostic Procedures; Elsevier Health Sciences: Saint Louis, MO, USA, 2012. [Google Scholar]
- Féraudet-Tarisse, C.; Vaisanen-Tunkelrott, M.L.; Moreau, K.; Lamourette, P.; Créminon, C.; Volland, H. Pathogen-free screening of bacteria-specific hybridomas for selecting high-quality monoclonal antibodies against pathogen bacteria as illustrated for Legionella pneumophila. J. Immunol. Methods 2013, 391, 81–94. [Google Scholar] [CrossRef]
- Gan, S.D.; Patel, K.R. Enzyme Immunoassay and Enzyme-Linked Immunosorbent Assay. J. Investig. Dermatol. 2013, 133, e12. [Google Scholar] [CrossRef]
- Piyasena, M.E.; Graves, S.W. The intersection of flow cytometry with microfluidics and microfabrication. Lab Chip 2014, 14, 1044–1059. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Fan, N.; Yang, X.; Peng, B.; Jiang, H. New advances in microfluidic flow cytometry. Electrophoresis 2018, 40, 1212–1229. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Gonzalez, D.; Lopez-Cabarcos, E.; Rubio-Retama, J.; Laurenti, M. Sensors and bioassays powered by upconverting materials. Adv. Colloid Interface Sci. 2017, 249, 66–87. [Google Scholar] [CrossRef] [PubMed]
- MagArray. Available online: http://www.magarray.com/ (accessed on 5 February 2019).
- Zepto. Available online: http://www.zeptolife.com/ (accessed on 5 February 2019).
- Magnomics. Available online: http://www.magnomics.pt/ (accessed on 5 February 2019).
- Krishna, V.D.; Wu, K.; Perez, A.M.; Wang, J.P. Giant Magnetoresistance-based Biosensor for Detection of Influenza A Virus. Front. Microbiol. 2016, 7, 400. [Google Scholar] [CrossRef]
- Ferguson, B.S.; Buchsbaum, S.F.; Wu, T.T.; Hsieh, K.; Xiao, Y.; Sun, R.; Soh, R.T. Genetic analysis of H1N1 influenza virus from throat swab samples in a microfluidic system for point-of-care diagnostics. J. Am. Chem. Soc. 2011, 133, 9129–9135. [Google Scholar] [CrossRef]
- Pihíková, D.; Kasák, P.; Tkac, J. Glycoprofiling of cancer biomarkers: Label-free electrochemical lectin-based biosensors. Open Chem. 2015, 13, 636–655. [Google Scholar] [CrossRef]
- Serrate, D.; De Teresa, J.M.; Marquina, C.; Marzo, J.; Saurel, D.; Cardoso, F.A.; Cardoso, S.; Freitas, P.P.; Ibarra, M.R. Quantitative biomolecular sensing station based on magnetoresistive patterned arrays. Biosens. Bioelectron. 2012, 35, 206–212. [Google Scholar] [CrossRef]
- Fermon, C.; Van de Voorde, M. Nanomagnetism, Applications and Perspectives; Wiley: Weinheim, Germany, 2017. [Google Scholar]
- Lin, G.; Makarov, D.; Schmidt, O.G. Magnetic sensing platform technologies for biomedical applications. Lab Chip 2017. [Google Scholar] [CrossRef]
- Wang, T.; Yang, Z.; Lei, C.; Lei, J.; Zhou, Y. An integratedgiantmagnetoimpedancebiosensorfordetection of biomarker. Biosens. Bioelectron. 2014, 58, 338–344. [Google Scholar] [CrossRef]
- Boyle, D.S.; Hawkins, K.R.; Steele, M.S.; Singhal, M.; Cheng, X. Emerging technologies for point-of-care CD4 T-lymphocyte counting. Trends Biotechnol. 2012, 30, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Barroso, T.G.; Martins, R.C.; Fernandes, E.; Cardoso, S.; Rivas, J.; Freitas, P.P. Detection of BCG bacteria using a magnetoresistive biosensor: A step towards a fully electronic platform for tuberculosis point-of-care detection. Biosens. Bioelectron. 2018, 100, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Lange, J.; Kötitz, R.; Haller, A.; Trahms, L.; Semmler, W.; Weitschies, W. Magnetorelaxometry—A new binding specific detection method based on magnetic nanoparticles. J. Magn. Magn. Mater. 2002, 252, 381–383. [Google Scholar] [CrossRef]
- Yang, T.Q.; Abe, M.; Horiguchi, K.; Enpuku, K. Detection of magnetic nanoparticles with ac susceptibility measurement. Phys. C 2004, 412–414, 1496–1500. [Google Scholar] [CrossRef]
- Ludwig, F.; Heima, E.; Mä useleina, S.; Eberbeckb, D.; Schilling, M. Magnetorelaxometry of magnetic nanoparticles with fluxgate magnetometers for the analysis of biological targets. J. Magn. Magn. Mater. 2005, 293, 690–695. [Google Scholar] [CrossRef]
- Denmark, D.J.; Bustos-Perez, X.; Swain, A.; Phan, M.H.; Mohapatra, S.; Mohapatra, S.S. Readiness of Magnetic Nanobiosensors for Point-of-Care Commercialization. J. Electron. Mater. 2019, 48, 4749–4761. [Google Scholar] [CrossRef]
- Guo, L.; Yang, Z.; Zhi, S.; Feng, Z.; Lei, C.; Zhou, Y. A sensitive and innovative detection method for rapid C-reactive proteins analysis based on a micro-fluxgate sensor system. PLoS ONE 2018, 13, e0194631. [Google Scholar] [CrossRef]
- Pekas, N.; Porter, M.D. Giant magnetoresistance monitoring of magnetic picodroplets in an integrated microfluidic system. Appl. Phys. Lett. 2004, 85, 4783–4785. [Google Scholar] [CrossRef]
- Loureiro, J.; Ferreira, R.; Cardoso, S.; Freitas, P.P.; Germano, J.; Fermon, C.; Arrias, G.; Pannetier-Lecoeur, M.; Rivadulla, F.; Rivas, J. Toward a magnetoresistive chip cytometer: Integrated detection of magnetic beads flowing at cm/s velocities in microfluidic channels. Appl. Phys. Lett. 2009, 95, 034104. [Google Scholar] [CrossRef]
- Loureiro, J.; Andrade, P.Z.; Cardoso, S.; da Silva, C.L.; Cabral, J.M.; Freitas, P.P. Magnetoresistive chip cytometer. Lab Chip 2011, 11, 2255–2261. [Google Scholar] [CrossRef]
- Walt, D.R. Optical methods for single molecule detection and analysis. Anal. Chem. 2013, 83, 1258–1263. [Google Scholar] [CrossRef] [PubMed]
- Helou, M.; Reisbeck, M.; Tedde, S.F.; Richter, L.; Bär, L.; Bosch, J.J.; Stauber, R.H.; Quandt, E.; Hayden, O. Time-of-flight magnetic flow cytometry in whole blood with integrated sample preparation. Lab Chip 2013, 13, 1035–1038. [Google Scholar] [CrossRef] [PubMed]
- Muluneh, M.; Issadore, D. Microchip-based detection of magnetically labeled cancer biomarkers. Adv. Drug Deliv. Rev. 2014, 66, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Murali, P.; Niknejad, A.M.; Boser, B.E. CMOS Microflow Cytometer for Magnetic Label Detection and Classification. IEEE J. Solid-State Circuits 2017, 52, 543–555. [Google Scholar] [CrossRef]
- Giouroudi, I.; Kokkinis, G. Recent Advances in Magnetic Microfluidic Biosensors. Nanomaterials 2017, 7, 171. [Google Scholar] [CrossRef] [PubMed]
- Fodil, K.; Denoual, M.; Dolabdjian, C.; Treizebre, A.; Senez, V. In-flow detection of ultra-small magnetic particles by an integrated giant magnetic impedance sensor. Appl. Phys. Lett. 2016, 108, 173701. [Google Scholar] [CrossRef]
- García-Arribas, A.; Martínez, F.; Fernández, E.; Ozaeta, I.; Kurlyandskaya, G.V.; Svalov, A.V.; Berganzo, J.; Barandiaran, J.M. GMI detection of magnetic-particle concentration in continuous flow. Sens. Actuators A 2011, 172, 103–108. [Google Scholar] [CrossRef]
- Blanc-Béguin, F.; Nabily, S.; Gieraltowski, J.; Turzo, A.; Querellou, S.; Salaun, P. Cytotoxicity and GMI bio-sensor detection of maghemite nanoparticles internalized into cells. J. Magn. Magn. Mater. 2009, 321, 192–197. [Google Scholar] [CrossRef]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Delahaut, P. Immunisation—Choice of host, adjuvants and boosting schedules with emphasis on polyclonal antibody production. Methods 2017, 116, 4–11. [Google Scholar] [CrossRef]
- Giouroudi, I.; Hristoforou, E. Perspective: Magnetoresistive sensors for biomedicine. J. Appl. Phys. 2018, 124, 030902. [Google Scholar] [CrossRef]
- Lenz, J.E. A Review of Magnetic Sensors. Proc. IEEE 1990, 78, 973–989. [Google Scholar] [CrossRef]
- Nabaei, V.; Chandrawati, R.; Heidari, H. Magnetic biosensors: Modelling and simulation. Biosens. Bioelectron. 2018, 103, 69–86. [Google Scholar] [CrossRef]
- Pannetier-Lecoeur, M. Superconducting-Magnetoresistive Sensor: Reaching the Femtotesla at 77 K; Habilitation à Diriger des Recherches en Physique, Condensed Matter, Université Pierre et Marie Curie–Paris VI: Paris, France, 2010. [Google Scholar]
- Issadore, D.; Chung, J.; Shao, H.; Liong, M.; Ghazani, A.A.; Castro, C.M.; Weissleder, R.; Lee, H. Ultrasensitive clinical enumeration of rare cells ex vivo using a micro-Hall detector. Sci. Transl. Med. 2012, 4, 141ra92. [Google Scholar] [CrossRef] [PubMed]
- Issadore, D.; Chung, H.J.; Chung, J.; Budin, G.; Weissleder, R.; Lee, H. μHall chip for sensitive detection of bacteria. Adv. Healthc. Mater. 2013, 2, 1224–1228. [Google Scholar] [CrossRef] [PubMed]
- Chícharo, A.; Martins, M.; Barnsley, L.C.; Taouallah, A.; Fernandes, J.; Silva, B.F.B.; Cardoso, S.; Diéguez, L.; Espiña, B.; Freitas, P.P. Enhanced magnetic microcytometer with 3D flow focusing for cell enumeration. Lab Chip 2018, 18, 2593–2603. [Google Scholar] [CrossRef] [PubMed]
- Reisbeck, M.; Richter, L.; Helou, M.J.; Arlinghaus, S.; Anton, B.; van Dommelen, I.; Nitzsche, M.; Baßler, M.; Kappes, B.; Friedrich, O.; et al. Hybrid integration of scalable mechanical and magnetophoretic focusing for magnetic flow cytometry. Biosens. Bioelectron. 2018, 109, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Duffy, D.C.; McDonald, J.C.; Schueller, O.J.A.; Whitesides, G.M. Rapid Prototyping of Microfluidic Systems in Poly(dimethylsiloxane). Ana. Chem. 1998, 70, 4974–4984. [Google Scholar] [CrossRef] [PubMed]
- Sierocki, R.; Jneid, B.; Rouaix, A.; Plaisance, M.; Féraudet-Tarisse, C.; Maillère, B.; Nozach, H.; Simon, S. An antibody targeting type III secretion system induces broad protection against Salmonella and Shigella infections. Unpublished manuscript. 2019. [Google Scholar]
- Reisbeck, M.; Helou, M.J.; Richter, L.; Kappes, B.; Friedrich, O.; Hayden, O. Magnetic fingerprints of rolling cells for quantitative flow cytometry in whole blood. Sci. Rep. 2016, 6, 32838. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.P.; Lai, M.F.; Huang, H.T.; Lin, C.W.; Wei, Z.H. Wheatstone bridge giant-magnetoresistance based cell counter. Biosens. Bioelectron. 2014, 57, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Pannetier, M.; Fermon, C.; Le Goff, G.; Simola, J.; Kerr, E. Low noise magnetoresistive sensors for current measurement and compasses. J. Magn. Magn Mater. 2007, 316, 246–248. [Google Scholar] [CrossRef]
- Li, G.; Wang, S.X.; Sun, S. Model and Experiment of Detecting Multiple Magnetic Nanoparticles as Biomolecular Labels by Spin Valve Sensors. IEEE Trans. Magn. 2004, 40, 3000–3002. [Google Scholar] [CrossRef]
- Huang, L.R.; Cox, E.C.; Austin, R.H.; Sturm, J.C. Continuous particle separation through deterministic lateral displacement. Science 2004, 304, 987–990. [Google Scholar] [CrossRef] [PubMed]
- El Hasnia, A.; Göbbels, K.; Thiebes, A.L.; Bräunig, P.; Mokwa, W.; Schnakenberg, U. Focusing and sorting of particles in spiral microfluidic channels. Procedia Eng. 2011, 25, 1197–1200. [Google Scholar] [CrossRef]
- Salafi, T.; Zeming, K.K.; Zhang, Y. Advancements in microfluidics for nanoparticle separation. Lab Chip 2017, 17, 11–33. [Google Scholar] [CrossRef]
- McGrath, J.; Jimenez, M.; Bridle, H. Deterministic lateral displacement for particle separation: A review. Lab Chip 2014, 14, 4139–4158. [Google Scholar] [CrossRef] [PubMed]
- Pamme, N.; Manz, A. On-Chip Free-Flow Magnetophoresis: Continuous Flow Separation of Magnetic Particles and Agglomerates. Anal. Chem. 2004, 76, 7250–7256. [Google Scholar] [CrossRef]
- Illés, E.; Tombácz, E.; Szekeres, M.; Tóth, I.Y.; Szabó, A.; Iván, B. Novel carboxylated PEG-coating on magnetite nanoparticles designed for biomedical applications. J. Magn. Magn. Mater. 2015, 380, 132–139. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Type | Cells/mL | Antibody Beads Coating |
|---|---|---|
| 1 × 105 NS1 | anti-CD138 | |
| 3 × 104 NS1 | anti-CD138 | |
| Positive | 1 × 104 NS1 | anti-CD138 |
| 3 × 103 NS1 | anti-CD138 | |
| 1 × 103 NS1 | anti-CD138 | |
| 1 × 105 NS1 | IpaD315 | |
| Negative | 1 × 105 CHO | anti-CD138 |
| No cell | anti-CD138 |
| Day 1 | Day 2 | Day 3 | Day 4 | Day 5 | Day 6 | Summary | |||
|---|---|---|---|---|---|---|---|---|---|
| Sensor | Sensor A | Sensor B | Sensor C | Sensor D | Different | ||||
| Separation layer thickness | 6.2 m | 6.4 m | 5.7 m | 5.5 m | devices, | ||||
| Channel height | 26.5 m | 27.4 m | 25.3 m | 23.1 m | samples and | ||||
| Beads batch | 1 | 2 | 3 | 4 | 5 | 5 | conditions. | ||
| Threshold (T) | 1.6 | 1.8 | 2.2 | 0.47 | 0.46 | 0.48 | Same | ||
| Sample volume (L) | 400 | 200 | 300 | 300 | 300 | 300 | 300 | experimenters | |
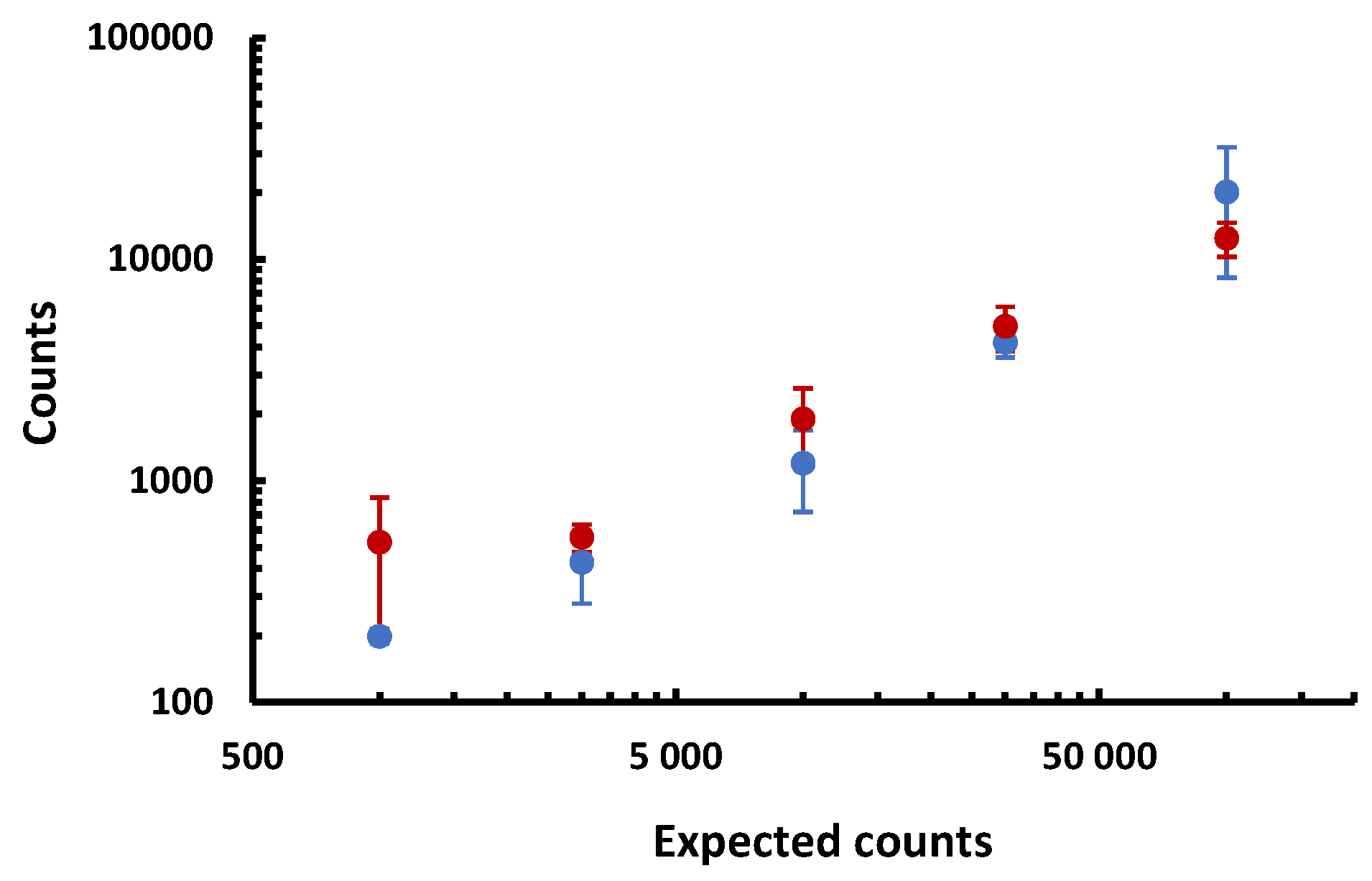
| Cells | mAbs | Counts of signals above 2.2 microteslas per milliliter | Average ± SD | ||||||
| 105 NS1 | anti-CD138 | 12,123 | 14,700 | 10,367 | 1.2 × 104 ± 1.8 103 | ||||
| 3 104 NS1 | anti-CD138 | 3463 | 5173 | 6180 | 5070 | 5.0 × 103 ± 9.7 102 | |||
| 104 NS1 | anti-CD138 | 2867 | 1223 | 1927 | 1597 | 1.9 × 103 ± 6.1 102 | |||
| 3 103 NS1 | anti-CD138 | 467 | 630 | 517 | 607 | 5.6 × 102 ± 6.6 101 | |||
| 103 NS1 | anti-CD138 | 500 | 977 | 280 | 353 | 5.3 × 102 ± 2.7 102 | |||
| 105 NS1 | IpaD315 | 895 | 60 | 660 | 637 | 723 | 313 | 520 | 5.4 × 102 ± 2.6 102 |
| 105 CHO | anti-CD138 | 690 | 380 | 367 | 4.8 × 102 ± 1.5 102 | ||||
| ∅ | anti-CD138 | 1665 | 375 | 2213 | 310 | 1.1 × 103 ± 8.2 102 | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giraud, M.; Delapierre, F.-D.; Wijkhuisen, A.; Bonville, P.; Thévenin, M.; Cannies, G.; Plaisance, M.; Paul, E.; Ezan, E.; Simon, S.; et al. Evaluation of In-Flow Magnetoresistive Chip Cell—Counter as a Diagnostic Tool. Biosensors 2019, 9, 105. https://doi.org/10.3390/bios9030105
Giraud M, Delapierre F-D, Wijkhuisen A, Bonville P, Thévenin M, Cannies G, Plaisance M, Paul E, Ezan E, Simon S, et al. Evaluation of In-Flow Magnetoresistive Chip Cell—Counter as a Diagnostic Tool. Biosensors. 2019; 9(3):105. https://doi.org/10.3390/bios9030105
Chicago/Turabian StyleGiraud, Manon, François-Damien Delapierre, Anne Wijkhuisen, Pierre Bonville, Mathieu Thévenin, Gregory Cannies, Marc Plaisance, Elodie Paul, Eric Ezan, Stéphanie Simon, and et al. 2019. "Evaluation of In-Flow Magnetoresistive Chip Cell—Counter as a Diagnostic Tool" Biosensors 9, no. 3: 105. https://doi.org/10.3390/bios9030105
APA StyleGiraud, M., Delapierre, F.-D., Wijkhuisen, A., Bonville, P., Thévenin, M., Cannies, G., Plaisance, M., Paul, E., Ezan, E., Simon, S., Fermon, C., Féraudet-Tarisse, C., & Jasmin-Lebras, G. (2019). Evaluation of In-Flow Magnetoresistive Chip Cell—Counter as a Diagnostic Tool. Biosensors, 9(3), 105. https://doi.org/10.3390/bios9030105