Thermally Self-Healing Graphene-Nanoplate/Polyurethane Nanocomposites via Diels–Alder Reaction through a One-Shot Process



Abstract
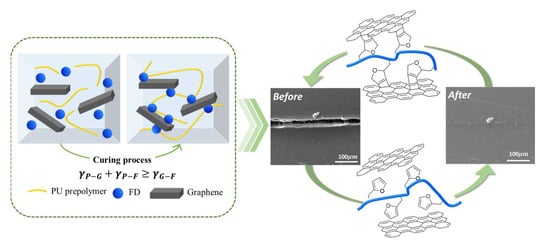
1. Introduction
2. Experimental
2.1. Materials
2.2. Preparation of GNP Masterbatch
2.3. Preparation of GNP/PU Nanocomposites
2.4. Characterizations
3. Results and Discussion
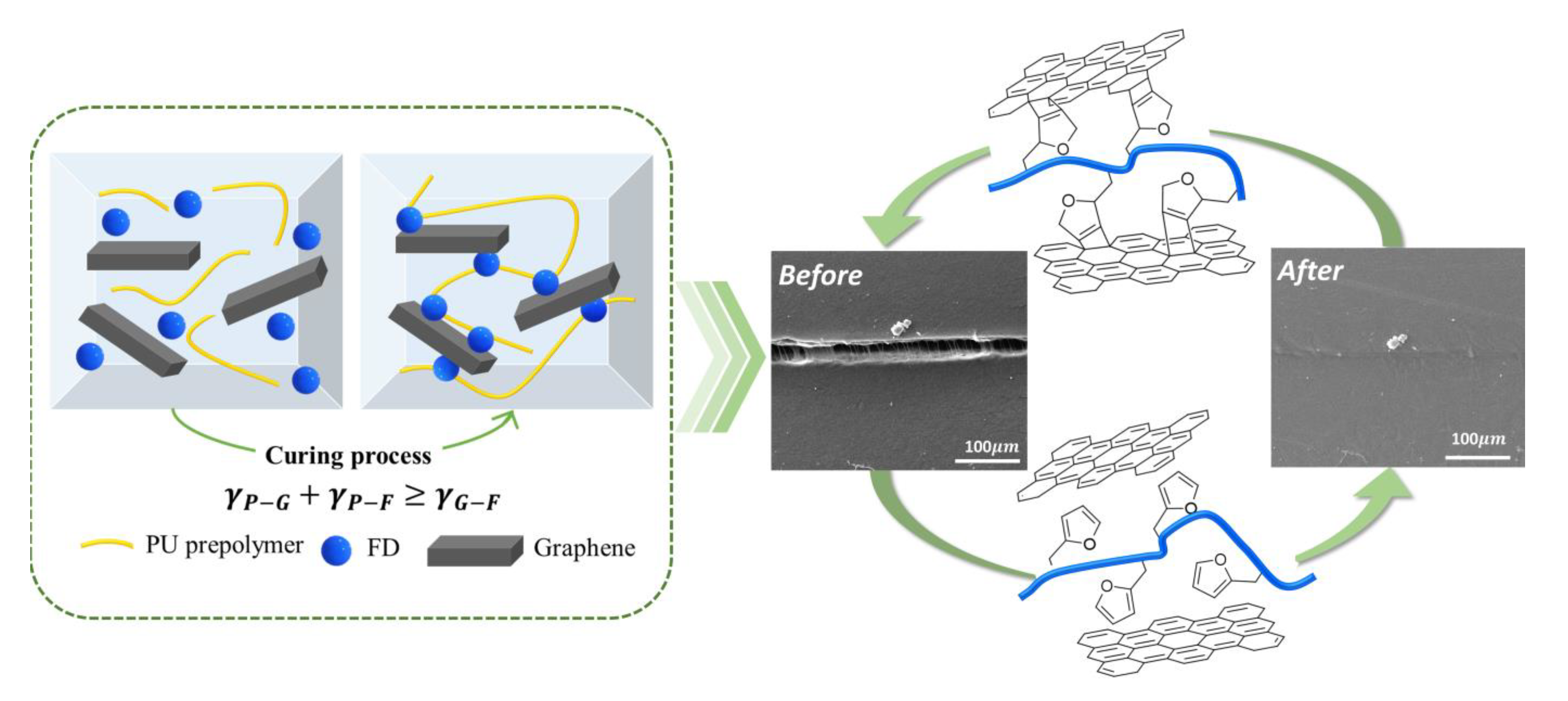
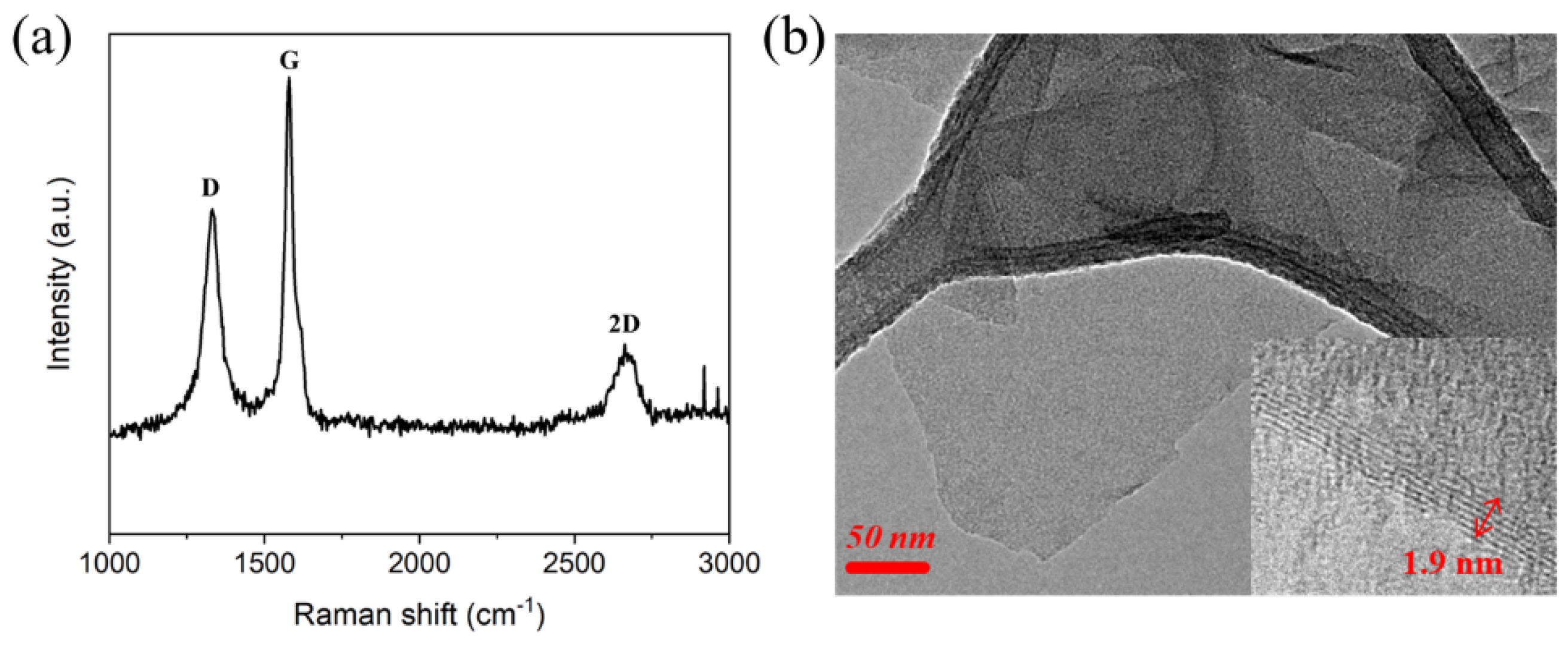
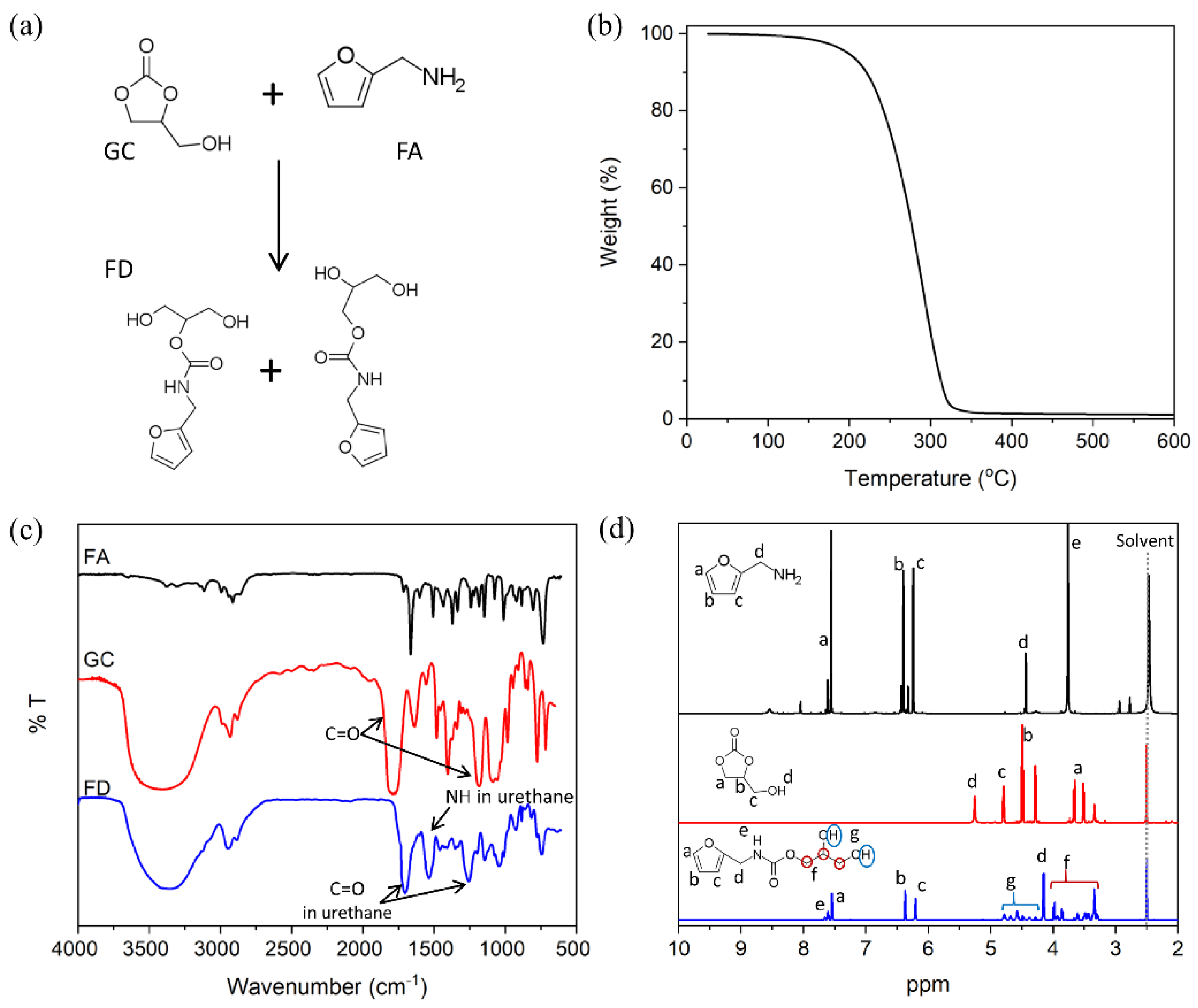
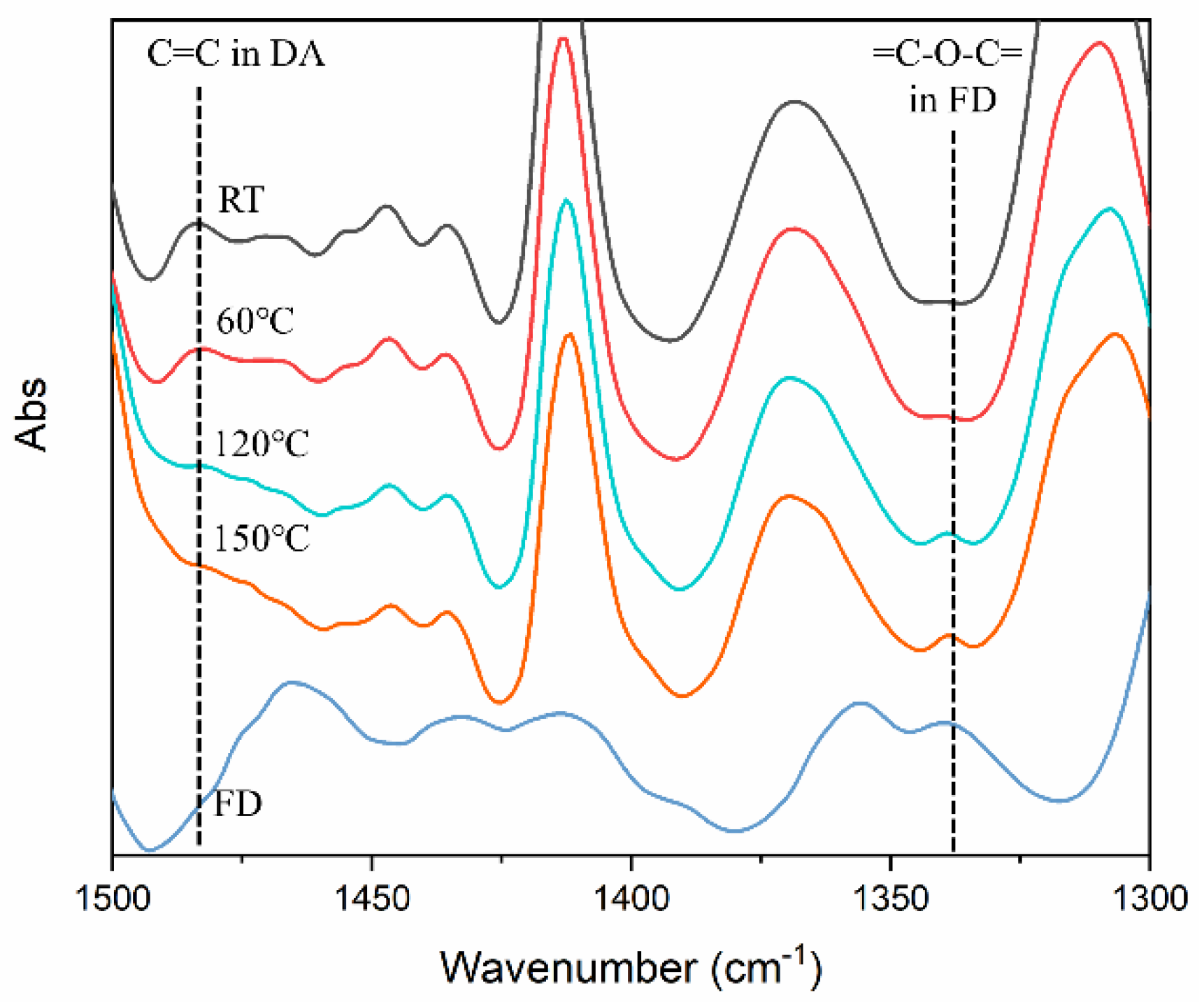
3.1. Characterization of GNP and FD
3.2. Diels–Alder Reaction in the Preparation of GNP/PU Nanocomposites
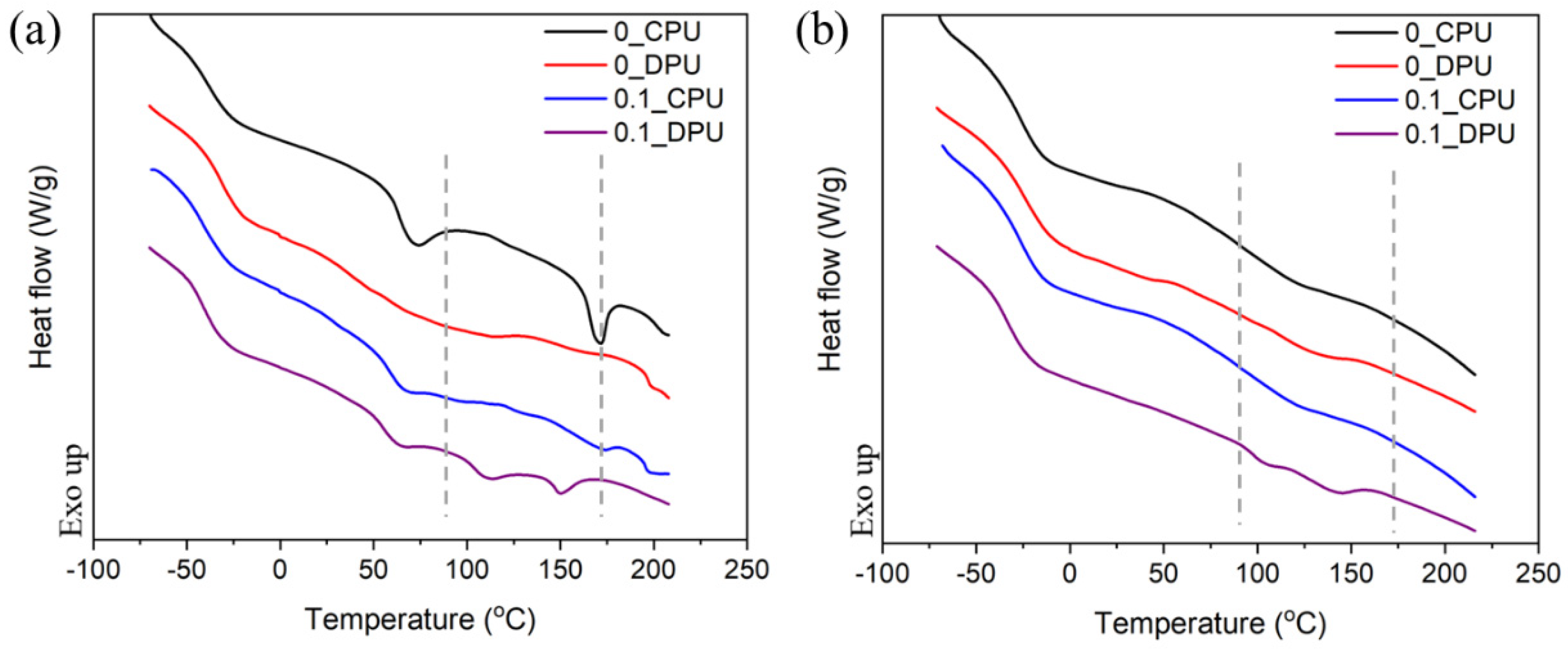
3.3. Thermally Self-Healing Properties of GNP/PU Nanocomposites
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Liu, Y.L.; Chuo, T.W. Self-healing polymers based on thermally reversible Diels-Alder chemistry. Polym. Chem. 2013, 4, 2194–2205. [Google Scholar] [CrossRef]
- Canadell, J.; Goossens, H.; Klumperman, B. Self-Healing Materials Based on Disulfide Links. Macromolecules 2011, 44, 2536–2541. [Google Scholar] [CrossRef]
- Chao, A.; Negulescu, I.; Zhang, D. Dynamic Covalent Polymer Networks Based on Degenerative Imine Bond Exchange: Tuning the Malleability and Self-Healing Properties by Solvent. Macromolecules 2016, 49, 6277–6284. [Google Scholar] [CrossRef]
- Kuang, X.; Liu, G.; Dong, X.; Wang, D. Triple-Shape Memory Epoxy Based on Diels-Alder Adduct Molecular Switch. Polymer 2016, 84, 1–9. [Google Scholar] [CrossRef]
- Klein, R.; Übel, F.; Frey, H. Maleimide Glycidyl Ether: A Bifunctional Monomer for Orthogonal Cationic and Radical Polymerizations. Macromol. Rapid Commun. 2015, 36, 1822–1828. [Google Scholar] [CrossRef] [PubMed]
- Turkenburg, D.H.; Fischer, H.R. Diels-Alder Based, Thermo-Reversible Cross-Linked Epoxies for Use in Self-Healing Composites. Polymer 2015, 79, 187–194. [Google Scholar] [CrossRef]
- Coleman, J.N.; Lotya, M.; O’Neill, A.; Bergin, S.D.; King, P.J.; Khan, U.; Young, K.; Gaucher, A.; De, S.; Smith, R.J.; et al. Two-Dimensional Nanosheets Produced by Liquid Exfoliation of Layered Materials. Science 2011, 331, 568–571. [Google Scholar] [CrossRef]
- Paton, K.R.; Varrla, E.; Backes, C.; Smith, R.J.; Khan, U.; O’Neill, A.; Boland, C.; Lotya, M.; Istrate, O.M.; King, P.; et al. Scalable Production of Large Quantities of Defect-Free Few-Layer Graphene by Shear Exfoliation in Liquids. Nat. Mater. 2014, 13, 624–630. [Google Scholar] [CrossRef]
- Sinha Ray, S.; Okamoto, M. Polymer/Layered Silicate Nanocomposites: A Review from Preparation to Processing. Prog. Polym. Sci. 2003, 28, 1539–1641. [Google Scholar] [CrossRef]
- Wu, Q.; Xu, Y.; Yao, Z.; Liu, A.; Shi, G. Supercapacitors Based on Flexible Graphene/Polyaniline Nanofiber Composite Films. ACS Nano 2010, 4, 1963–1970. [Google Scholar] [CrossRef]
- Sarkar, S.; Bekyarova, E.; Niyogi, S.; Haddon, R.C. Diels-Alder Chemistry of Graphite and Graphene: Graphene as Diene and Dienophile. J. Am. Chem. Soc. 2011, 133, 3324–3327. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.-M.; Jeon, I.-Y.; Baek, J.-B. Mechanochemically Driven Solid-State Diels-Alder Reaction of Grpahite into Graphene Nanoplatelets. Chem. Sci. 2013, 4, 4273–4277. [Google Scholar] [CrossRef]
- Ji, Z.; Chen, J.; Huang, L.; Shi, G. High-Yield Production of Highly Conductive Graphene via Reversible Covalent Chemistry. Chem. Commun. 2015, 51, 2806–2809. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.M.; Liu, Y.L. Functionalization of Multi-Walled Carbon Nanotubes with Furan and Maleimide Compounds through Diels-Alder Cycloaddition. Carbon N. Y. 2009, 47, 3041–3049. [Google Scholar] [CrossRef]
- Le, C.M.Q.; Cao, X.T.; Lim, K.T. Ultrasound-Promoted Direct Functionalization of Multi-Walled Carbon Nanotubes in Water via Diels-Alder “Click Chemistry”. Ultrason. Sonochem. 2017, 39, 321–329. [Google Scholar] [CrossRef]
- Bai, J.; He, Q.; Shi, Z.; Tian, M.; Xu, H.; Ma, X.; Yin, J. Self-Assembled Elastomer Nanocomposites Utilizing C60 and Poly(Styrene-b-Butadiene-b-Styrene) via Thermally Reversible Diels-Alder Reaction with Self-Healing and Remolding Abilities. Polymer 2017, 116, 268–277. [Google Scholar] [CrossRef]
- Kötteritzsch, J.; Geitner, R.; Ahner, J.; Abend, M.; Zechel, S.; Vitz, J.; Hoeppener, S.; Dietzek, B.; Schmitt, M.; Popp, J.; et al. Remendable Polymers via Reversible Diels–Alder Cycloaddition of Anthracene-Containing Copolymers with Fullerenes. J. Appl. Polym. Sci. 2018, 135, 1–14. [Google Scholar] [CrossRef]
- Pokharel, P.; Lee, D.S. High Performance Polyurethane Nanocomposite Films Prepared from a Masterbatch of Graphene Oxide in Polyether Polyol. Chem. Eng. J. 2014, 253, 356–365. [Google Scholar] [CrossRef]
- Lee, S.-H.; Oh, C.-R.; Lee, D.-S. Large Improvement in the Mechanical Properties of Polyurethane Nanocomposites Based on a Highly Concentrated Graphite Nanoplate/Polyol Masterbatch. Nanomaterials 2019, 9, 389. [Google Scholar] [CrossRef]
- Fowkes, F.M. Dispersion Force Contributions to Surface and Interfacial Tensions, Contact Angles, and Heats of Immersion. Contact Angle, Wettability Adhes. 1964, 43, 99–111. [Google Scholar]
- Owens, D.K.; Wendt, R.C. Estimation of the Surface Free Energy of Polymers. J. Appl. Polym. Sci. 1969, 13, 1741–1747. [Google Scholar] [CrossRef]
- Wu, S. Calculation of Interfacial Tension in Polymer Systems. J. Polym. Sci. Part C Polym. Symp. 2007, 34, 19–30. [Google Scholar] [CrossRef]
- Khan, U.; O’Neill, A.; Porwal, H.; May, P.; Nawaz, K.; Coleman, J.N. Size Selection of Dispersed, Exfoliated Graphene Flakes by Controlled Centrifugation. Carbon N. Y. 2012, 50, 470–475. [Google Scholar] [CrossRef]
- Ferrari, A.C. Raman Spectroscopy of Graphene and Graphite: Disorder, Electron-Phonon Coupling, Doping and Nonadiabatic Effects. Solid State Commun. 2007, 143, 47–57. [Google Scholar] [CrossRef]
- Caņado, L.G.; Takai, K.; Enoki, T.; Endo, M.; Kim, Y.A.; Mizusaki, H.; Jorio, A.; Coelho, L.N.; Magalhães-Paniago, R.; Pimenta, M.A. General Equation for the Determination of the Crystallite Size La of Nanographite by Raman Spectroscopy. Appl. Phys. Lett. 2006, 88, 2–5. [Google Scholar]
- Tung, V.C.; Allen, M.J.; Yang, Y.; Kaner, R.B. High-Throughput Solution Processing of Large-Scale Graphene. Nat. Nanotechnol. 2009, 4, 25–29. [Google Scholar] [CrossRef]
- Green, A.A.; Hersam, M.C. Solution Phase Production of Graphene with Controlled Thickness via Density Differentiation. Nano Lett. 2009, 9, 4031–4036. [Google Scholar] [CrossRef]
- Chen, Z.; Lu, H. Constructing Sacrificial Bonds and Hidden Lengths for Ductile Graphene/Polyurethane Elastomers with Improved Strength and Toughness. J. Mater. Chem. 2012, 22, 12479–12490. [Google Scholar] [CrossRef]
- Stankovich, S.; Dikin, D.A.; Piner, R.D.; Kohlhaas, K.A.; Kleinhammes, A.; Jia, Y.; Wu, Y.; Nguyen, S.T.; Ruoff, R.S. Synthesis of Graphene-Based Nanosheets via Chemical Reduction of Exfoliated Graphite Oxide. Carbon N. Y. 2007, 45, 1558–1565. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | Interfacial Tension (dyne/cm) | |
|---|---|---|
| Wu | Owen–Wendt | |
| Prepolymer/FD | 5.12 | 7.60 |
| GNP/FD | 3.31 | 4.25 |
| GNP/Prepolymer | 0.56 | 1.11 |
| Elements | C | N | O |
|---|---|---|---|
| (wt%) | (wt%) | (wt%) | |
| GNP | 97.2 | 0 | 3.3 |
| 0.1_CPU | 90.2 | 1.5 | 8.4 |
| 0.1_DPU | 79.1 | 2.9 | 18.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, C.-R.; Lee, S.-H.; Park, J.-H.; Lee, D.-S. Thermally Self-Healing Graphene-Nanoplate/Polyurethane Nanocomposites via Diels–Alder Reaction through a One-Shot Process. Nanomaterials 2019, 9, 434. https://doi.org/10.3390/nano9030434
Oh C-R, Lee S-H, Park J-H, Lee D-S. Thermally Self-Healing Graphene-Nanoplate/Polyurethane Nanocomposites via Diels–Alder Reaction through a One-Shot Process. Nanomaterials. 2019; 9(3):434. https://doi.org/10.3390/nano9030434
Chicago/Turabian StyleOh, Cho-Rong, Sang-Hyub Lee, Jun-Hong Park, and Dai-Soo Lee. 2019. "Thermally Self-Healing Graphene-Nanoplate/Polyurethane Nanocomposites via Diels–Alder Reaction through a One-Shot Process" Nanomaterials 9, no. 3: 434. https://doi.org/10.3390/nano9030434
APA StyleOh, C.-R., Lee, S.-H., Park, J.-H., & Lee, D.-S. (2019). Thermally Self-Healing Graphene-Nanoplate/Polyurethane Nanocomposites via Diels–Alder Reaction through a One-Shot Process. Nanomaterials, 9(3), 434. https://doi.org/10.3390/nano9030434