
Click Chemistry Functionalization of Harmonic Nanoparticles with Lanthanide Complexes Towards Tunable Platforms for Multimodal Imaging

 ,
,  , ,
, ,  , and
, and 
Abstract
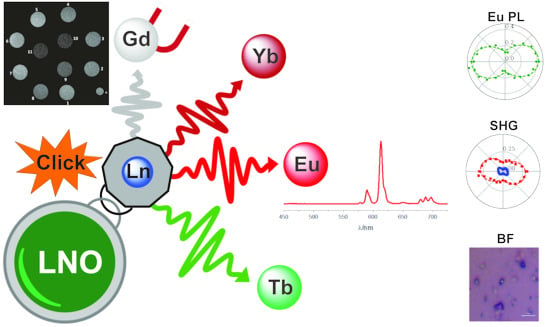
1. Introduction
2. Materials and Methods
2.1. Synthesis Procedures and Characterizations
2.1.1. Compound 1
2.1.2. Compound 2
2.1.3. Compound 3
2.1.4. Compound 4
2.1.5. Compound 5
2.1.6. Compound 6
2.1.7. H3LD Ligand
2.1.8. [LnLA] Complexes
2.1.9. [LnLD] Complexes
2.2. HNP Surface Modifications
2.2.1. Bare LNO HNP Synthesis
2.2.2. Coated LNO Intermediate Synthesis
2.2.3. LNO@[LnLA]—CuAAC Conjugation
2.2.4. LNO@[LnLD]—SPAAC Conjugation
2.2.5. LNO@[GdLD] Recycling into LNO@[TbLD]
2.3. MRI Phantom Imaging
2.4. Photophysical Property Investigation
3. Results and Discussion
3.1. Synthesis of the H3LD Ligand
3.2. H3LA and H3LD Complexes Synthesis
3.3. LNO Functionalization with Ln Complexes
3.4. Validation of MR Nonlinear Optical Dual Imaging Using LNO@[GdLD]
3.5. Investigation of Photophysical Properties
3.6. Proof-of-Concept of SHG-Induced Eu Luminescence
3.7. Recyclability of the LNO@[LnLX] System
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AcOH | Acetic acid |
| Alys | Azidolysine |
| CA | Contrast agent |
| CT | Computed tomography |
| CuAAC | Copper-catalyzed azide–alkyne [3+2]-cycloaddition |
| DCC | N,N′-Dicyclohexylcarbodiimide |
| DCE | Dichloroethane |
| DCM | Dichloromethane |
| DDQ | 2,3-dichloro-5,6-dicyano-1,4-benzoquinone |
| DH | Hydrodynamic diameter |
| DIBO | Dibenzocyclooctyne |
| DIBO-NH2 | Amino-modified dibenzocyclooctyne |
| DIPEA | Diisopropylethylamine |
| DLS | Dynamic light scattering |
| DMAc | N,N-Dimethylacetamide |
| DMF | N,N-Dimethylformamide |
| DMSO | Dimethylsulfoxide |
| EDX | Energy-dispersive X-ray spectroscopy |
| FCC | Flash column chromatography |
| FHG | Fourth harmonic generation |
| FTIR | Fourier transform infrared |
| IR | Infrared |
| Hex | Hexane |
| HNP | Harmonic nanoparticle |
| Ln | Lanthanide |
| LNO | Lithium niobate |
| MR | Magnetic resonance |
| MRI | Magnetic resonance imaging |
| MWCO | Molecular weight cut-off |
| NHS | N-Hydroxy succinimide |
| NIR | Near-infrared |
| NP | Nanoparticle |
| NPCF | Nitrophenyl chloroformate |
| PBS | Phosphate buffer saline |
| PDI | Polydispersity index |
| PEG | Poly(ethylene glycol) |
| PET | Positron emission tomography |
| PL | Photoluminescence |
| PMBBr | p-methoxybenzylbromide |
| Py | Pyridine |
| rt | Room temperature |
| SHG | Second-harmonic generation |
| SPAAC | Strain-promoted azide–alkyne [3+2]-cycloaddition |
| SPECT | Single-photon emission computed tomography |
| STEM | Scanning transmission electron microscopy |
| TACNB | 1,4-bipicolinate-1,4,7-triazacyclononane |
| Talys | Tri-azidolysine peptide |
| TEA | Triethylamine |
| THF | Tetrahydrofuran |
| THG | Third-harmonic generation |
| TLC | Thin-layer chromatography |
| Tol | Toluene |
| UV | Ultraviolet |
| Vis | Visible |
| ZP | Zeta potential |
References
- Hussain, S.; Mubeen, I.; Ullah, N.; Shah, S.S.U.D.; Khan, B.A.; Zahoor, M.; Ullah, R.; Khan, F.A.; Sultan, M.A. Modern Diagnostic Imaging Technique Applications and Risk Factors in the Medical Field: A Review. BioMed Res. Int. 2022, 2022, 5164970. [Google Scholar] [CrossRef]
- Leitgeb, R.A.; Baumann, B. Multimodal Optical Medical Imaging Concepts Based on Optical Coherence Tomography. Front. Phys. 2018, 6, 114. [Google Scholar] [CrossRef]
- Tichauer, K.M.; Wang, Y.; Pogue, B.W.; Liu, J.T.C. Quantitative in Vivo Cell-Surface Receptor Imaging in Oncology: Kinetic Modeling and Paired-Agent Principles from Nuclear Medicine and Optical Imaging. Phys. Med. Biol. 2015, 60, R239–R269. [Google Scholar] [CrossRef]
- Lee, S.Y.; Jeon, S.I.; Jung, S.; Chung, I.J.; Ahn, C.-H. Targeted Multimodal Imaging Modalities. Adv. Drug Deliv. Rev. 2014, 76, 60–78. [Google Scholar] [CrossRef]
- Weissleder, R.; Pittet, M.J. Imaging in the Era of Molecular Oncology. Nature 2008, 452, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Eder, A.-C.; Omrane, M.A.; Stadlbauer, S.; Roscher, M.; Khoder, W.Y.; Gratzke, C.; Kopka, K.; Eder, M.; Meyer, P.T.; Jilg, C.A.; et al. The PSMA-11-Derived Hybrid Molecule PSMA-914 Specifically Identifies Prostate Cancer by Preoperative PET/CT and Intraoperative Fluorescence Imaging. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2057–2058. [Google Scholar] [CrossRef] [PubMed]
- Baranski, A.-C.; Schäfer, M.; Bauder-Wüst, U.; Roscher, M.; Schmidt, J.; Stenau, E.; Simpfendörfer, T.; Teber, D.; Maier-Hein, L.; Hadaschik, B.; et al. PSMA-11–Derived Dual-Labeled PSMA Inhibitors for Preoperative PET Imaging and Precise Fluorescence-Guided Surgery of Prostate Cancer. J. Nucl. Med. 2018, 59, 639–645. [Google Scholar] [CrossRef]
- Xu, S.; Guo, C.; Pan, K.; Wang, L. Combined Fluorescence and MRI in Bioimaging. In Imaging Tools for Chemical Biology; Royal Society of Chemistry: London, UK, 2024. [Google Scholar] [CrossRef]
- Yang, J.; Yan, M.; Wang, Z.; Zhang, C.; Guan, M.; Sun, Z. Optical and MRI Multimodal Tracing of Stem Cells In Vivo. Mol. Imaging 2023, 2023, 4223485. [Google Scholar] [CrossRef]
- Hao, J.; Cai, H.; Gu, L.; Ma, Y.; Li, Y.; Liu, B.; Zhu, H.; Zeng, F.; Wu, M. A Transferrin Receptor Targeting Dual-Modal MR/NIR Fluorescent Imaging Probe for Glioblastoma Diagnosis. Regen. Biomater. 2024, 11, rbae015. [Google Scholar] [CrossRef]
- Gibson, A.P. Medical Imaging Applied to Heritage. Br. J. Radiol. 2023, 96, 20230611. [Google Scholar] [CrossRef]
- Belianinov, A.; Ievlev, A.V.; Lorenz, M.; Borodinov, N.; Doughty, B.; Kalinin, S.V.; Fernández, F.M.; Ovchinnikova, O.S. Correlated Materials Characterization via Multimodal Chemical and Functional Imaging. ACS Nano 2018, 12, 11798–11818. [Google Scholar] [CrossRef] [PubMed]
- Armetta, F.; Bianco, A.L.; Boiko, V.; Hreniak, D.; Saladino, M.L. Multimodal Anti-Counterfeiting Inks: Modern Use of an Ancient Pigment in Synergy with a Persistent Phosphor. J. Mater. Chem. C 2025, 13, 1188–1197. [Google Scholar] [CrossRef]
- Andres, J.; Hersch, R.D.; Moser, J.-E.; Chauvin, A.-S. A New Anti-Counterfeiting Feature Relying on Invisible Luminescent Full Color Images Printed with Lanthanide-Based Inks. Adv. Funct. Mater. 2014, 24, 5029–5036. [Google Scholar] [CrossRef]
- Calle, D.; Ballesteros, P.; Cerdán, S. Advanced Contrast Agents for Multimodal Biomedical Imaging Based on Nanotechnology. In Preclinical MRI: Methods and Protocols; García Martín, M.L., López Larrubia, P., Eds.; Springer: New York, NY, USA, 2018; pp. 441–457. ISBN 978-1-4939-7531-0. [Google Scholar]
- Bischof, J.; Fletcher, G.; Verkade, P.; Kuntner, C.; Fernandez-Rodriguez, J.; Chaabane, L.; Rose, L.A.; Walter, A.; Vandenbosch, M.; van Zandvoort, M.A.M.J.; et al. Multimodal Bioimaging across Disciplines and Scales: Challenges, Opportunities and Breaking down Barriers. npj Imaging 2024, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Staedler, D.; Magouroux, T.; Hadji, R.; Joulaud, C.; Extermann, J.; Schwung, S.; Passemard, S.; Kasparian, C.; Clarke, G.; Gerrmann, M.; et al. Harmonic Nanocrystals for Biolabeling: A Survey of Optical Properties and Biocompatibility. ACS Nano 2012, 6, 2542–2549. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, P.; Maloney, J.; Wu, D.; Fraser, S.E. Second Harmonic Generating (SHG) Nanoprobes for in Vivo Imaging. Proc. Natl. Acad. Sci. USA 2010, 107, 14535–14540. [Google Scholar] [CrossRef]
- Campargue, G.; La Volpe, L.; Giardina, G.; Gaulier, G.; Lucarini, F.; Gautschi, I.; Le Dantec, R.; Staedler, D.; Diviani, D.; Mugnier, Y.; et al. Multiorder Nonlinear Mixing in Metal Oxide Nanoparticles. Nano Lett. 2020, 20, 8725–8732. [Google Scholar] [CrossRef]
- Passemard, S.; Staedler, D.; Sonego, G.; Magouroux, T.; Schneiter, G.S.; Juillerat-Jeanneret, L.; Bonacina, L.; Gerber-Lemaire, S. Functionalized Bismuth Ferrite Harmonic Nanoparticles for Cancer Cells Labeling and Imaging. J. Nanopart. Res. 2015, 17, 414. [Google Scholar] [CrossRef]
- Dubreil, L.; Leroux, I.; Ledevin, M.; Schleder, C.; Lagalice, L.; Lovo, C.; Fleurisson, R.; Passemard, S.; Kilin, V.; Gerber-Lemaire, S.; et al. Multi-Harmonic Imaging in the Second Near-Infrared Window of Nanoparticle-Labeled Stem Cells as a Monitoring Tool in Tissue Depth. ACS Nano 2017, 11, 6672–6681. [Google Scholar] [CrossRef]
- Sugiyama, N.; Sonay, A.Y.; Tussiwand, R.; Cohen, B.E.; Pantazis, P. Effective Labeling of Primary Somatic Stem Cells with BaTiO3 Nanocrystals for Second Harmonic Generation Imaging. Small 2018, 14, 1703386. [Google Scholar] [CrossRef]
- Karpf, S.; Glöckner Burmeister, N.; Dubreil, L.; Ghosh, S.; Hollandi, R.; Pichon, J.; Leroux, I.; Henkel, A.; Lutz, V.; Jurkevičius, J.; et al. Harmonic Imaging of Stem Cells in Whole Blood at GHz Pixel Rate. Small 2024, 20, 2401472. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C.; Guo, Z.; Glover, P.B.; Faulkner, S.; Pikramenou, Z. Luminescent Lanthanides in Biorelated Applications: From Molecules to Nanoparticles and Diagnostic Probes to Therapeutics. Chem. Rev. 2025, 125, 2269–2370. [Google Scholar] [CrossRef]
- Wartenberg, N.; Fries, P.; Raccurt, O.; Guillermo, A.; Imbert, D.; Mazzanti, M. A Gadolinium Complex Confined in Silica Nanoparticles as a Highly Efficient T1/T2 MRI Contrast Agent. Chem. Eur. J. 2013, 19, 6980–6983. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Choi, S.H.; Hyeon, T. Nano-Sized CT Contrast Agents. Adv. Mater. 2013, 25, 2641–2660. [Google Scholar] [CrossRef]
- Amoroso, A.J.; Fallis, I.A.; Pope, S.J.A. Chelating Agents for Radiolanthanides: Applications to Imaging and Therapy. Coord. Chem. Rev. 2017, 340, 198–219. [Google Scholar] [CrossRef]
- Huang, Y.; Li, L.; Zhang, D.; Gan, L.; Zhao, P.; Zhang, Y.; Zhang, Q.; Hua, M.; Jia, C. Gadolinium-Doped Carbon Quantum Dots Loaded Magnetite Nanoparticles as a Bimodal Nanoprobe for Both Fluorescence and Magnetic Resonance Imaging. Magn. Reson. Imaging 2020, 68, 113–120. [Google Scholar] [CrossRef]
- Eliseeva, S.V.; Song, B.; Vandevyver, C.D.B.; Chauvin, A.-S.; Wacker, J.B.; Bünzli, J.-C.G. Increasing the Efficiency of Lanthanide Luminescent Bioprobes: Bioconjugated Silica Nanoparticles as Markers for Cancerous Cells. New J. Chem. 2010, 34, 2915–2921. [Google Scholar] [CrossRef]
- Wang, W.; Song, S.; Liu, W.; Xia, T.; Du, G.; Zhai, X.; Jin, B. Two-Photon Excited Luminescence of Structural Light Enhancement in Subwavelength SiO2 Coating Europium Ion-Doped Paramagnetic Gadolinium Oxide Nanoparticle and Application for Magnetic Resonance Imaging. Discov. Nano 2023, 18, 85. [Google Scholar] [CrossRef]
- Kostiv, U.; Natile, M.M.; Jirák, D.; Půlpánová, D.; Jiráková, K.; Vosmanská, M.; Horák, D. PEG-Neridronate-Modified NaYF4:Gd3+,Yb3+,Tm3+/NaGdF4 Core–Shell Upconverting Nanoparticles for Bimodal Magnetic Resonance/Optical Luminescence Imaging. ACS Omega 2021, 6, 14420–14429. [Google Scholar] [CrossRef]
- Liu, N.; Homann, C.; Morfin, S.; Kesanakurti, M.S.; Calvert, N.D.; Shuhendler, A.J.; Al, T.; Hemmer, E. Core–Multi-Shell Design: Unlocking Multimodal Capabilities in Lanthanide-Based Nanoparticles as Upconverting, T2-Weighted MRI and CT Probes. Nanoscale 2023, 15, 19546–19556. [Google Scholar] [CrossRef]
- Chen, C.; Ao, L.; Wu, Y.-T.; Cifliku, V.; Cardoso Dos Santos, M.; Bourrier, E.; Delbianco, M.; Parker, D.; Zwier, J.M.; Huang, L.; et al. Single-Nanoparticle Cell Barcoding by Tunable FRET from Lanthanides to Quantum Dots. Angew. Chem. Int. Ed. 2018, 57, 13686–13690. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Suffren, Y.; Daiguebonne, C.; Freslon, S.; Bernot, K.; Calvez, G.; Pollès, L.L.; Roiland, C.; Guillou, O. Multi-Emissive Lanthanide-Based Coordination Polymers for Potential Application as Luminescent Bar-Codes. Inorg. Chem. 2019, 58, 2659–2668. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Li, B.; Wu, Y.; He, H.; Zhu, X.; Zhang, H.; Dou, C.; Feng, L.; Fan, Y.; Zhang, F. A Tumor-Microenvironment-Responsive Lanthanide–Cyanine FRET Sensor for NIR-II Luminescence-Lifetime In Situ Imaging of Hepatocellular Carcinoma. Adv. Mater. 2020, 32, 2001172. [Google Scholar] [CrossRef]
- Smith, D.G.; McMahon, B.K.; Pal, R.; Parker, D. Live Cell Imaging of Lysosomal pH Changes with pH Responsive Ratiometric Lanthanide Probes. Chem. Commun. 2012, 48, 8520–8522. [Google Scholar] [CrossRef]
- Ezerskyte, E.; Morkvenas, A.; Venius, J.; Sakirzanovas, S.; Karabanovas, V.; Katelnikovas, A.; Klimkevicius, V. Biocompatible Upconverting Nanoprobes for Dual-Modal Imaging and Temperature Sensing. ACS Appl. Nano Mater. 2024, 7, 6185–6195. [Google Scholar] [CrossRef]
- Cheung, T.L.; Ju, Z.; Zhang, W.; Parker, D.; Deng, R. Mechanistic Investigation of Sensitized Europium Luminescence: Excited State Dynamics and Luminescence Lifetime Thermometry. ACS Appl. Mater. Interfaces 2024, 16, 43933–43941. [Google Scholar] [CrossRef]
- Su, Y.; Hao, L.-N.; Liu, K.; Zhang, J.; Dong, L.; Xu, Y.; Lu, Y.; Qian, H.-S. Epitaxial Growth of Ultrathin Layers on the Surface of Sub-10 Nm Nanoparticles: The Case of β-NaGdF4:Yb/Er@NaDyF4 Nanoparticles. RSC Adv. 2018, 8, 12944–12950. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, S.; Arancibia, D.; Rojas, M.; Carmona, F.; Ortega, A.; Valenzuela, J.; Hernández-Álvarez, C.; Martín, I.R. Simultaneous Second Harmonic Generation and Multiphoton Excited Photoluminescence in Samarium-Doped BaTiO3 Nanoparticles Functionalized with Poly(Ethylene Glycol). ACS Omega 2024, 9, 28061–28071. [Google Scholar] [CrossRef]
- Ivanov, S.A.; Sarkar, T.; Fortalnova, E.A.; Politova, E.D.; Stefanovich, S.Y.; Safronenko, M.G.; Nordblad, P.; Mathieu, R. Composition Dependence of the Multifunctional Properties of Nd-Doped Bi4Ti3O12 Ceramics. J. Mater. Sci. Mater. Electron. 2017, 28, 7692–7707. [Google Scholar] [CrossRef]
- Taiariol, L.; Chaix, C.; Farre, C.; Moreau, E. Click and Bioorthogonal Chemistry: The Future of Active Targeting of Nanoparticles for Nanomedicines? Chem. Rev. 2022, 122, 340–384. [Google Scholar] [CrossRef]
- Yang, Y.; Hu, X.; Yang, Z.; Huang, W. Insights into Molecular Lanthanide Complexes: Construction, Properties and Bioimaging and Biosensing Applications. Adv. Funct. Mater. 2025, 35, 2412970. [Google Scholar] [CrossRef]
- Parker, D.; Fradgley, J.D.; Wong, K.-L. The Design of Responsive Luminescent Lanthanide Probes and Sensors. Chem. Soc. Rev. 2021, 50, 8193–8213. [Google Scholar] [CrossRef]
- Caravan, P.; Ellison, J.J.; McMurry, T.J.; Lauffer, R.B. Gadolinium(III) Chelates as MRI Contrast Agents: Structure, Dynamics, and Applications. Chem. Rev. 1999, 99, 2293–2352. [Google Scholar] [CrossRef]
- Wang, B.; Hai, J.; Wang, Q.; Li, T.; Yang, Z. Coupling of Luminescent Terbium Complexes to Fe3O4 Nanoparticles for Imaging Applications. Angew. Chem. Int. Ed. 2011, 50, 3063–3066. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, B.; Wang, B.; Yang, Z.; Wang, Q.; Li, T.; Qin, D.; Li, Y.; Wang, M.; Yan, M. Magnetic Nanoparticles Modified with DTPA-AMC-Rare Earth for Fluorescent and Magnetic Resonance Dual Mode Imaging. Dalton Trans. 2012, 41, 8723–8728. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, C.S.; Massue, J.; Quinn, S.J.; Gunnlaugsson, T. Lanthanide Luminescent Gold Nanoparticles: pH-Driven Self-Assembly Formation between Eu(III)-Cyclen Conjugated AuNPs and Sensitising β-Diketonate Antenna in Water. Org. Biomol. Chem. 2009, 7, 3074–3078. [Google Scholar] [CrossRef]
- Xu, J.; Sun, Z.; Jia, L.; Li, B.; Zhao, L.; Liu, X.; Ma, Y.; Tian, H.; Wang, Q.; Liu, W.; et al. Visible Light Sensitized Attapulgite-Based Lanthanide Composites: Microstructure, Photophysical Behaviour and Biological Application. Dalton Trans. 2011, 40, 12909–12916. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, D.; Hu, Y.; Wang, Y.; Yang, W.J.; Wang, L. Synthesis of Water-Soluble Europium-Containing Nanoprobes via Polymerization-Induced Self-Assembly and Their Cellular Imaging Applications. Talanta 2021, 232, 122182. [Google Scholar] [CrossRef]
- Nonat, A.; Gateau, C.; Fries, P.H.; Mazzanti, M. Lanthanide Complexes of a Picolinate Ligand Derived from 1,4,7-Triazacyclononane with Potential Application in Magnetic Resonance Imaging and Time-Resolved Luminescence Imaging. Chem. Eur. J. 2006, 12, 7133–7150. [Google Scholar] [CrossRef]
- Walton, J.W.; Carr, R.; Evans, N.H.; Funk, A.M.; Kenwright, A.M.; Parker, D.; Yufit, D.S.; Botta, M.; De Pinto, S.; Wong, K.-L. Isostructural Series of Nine-Coordinate Chiral Lanthanide Complexes Based on Triazacyclononane. Inorg. Chem. 2012, 51, 8042–8056. [Google Scholar] [CrossRef] [PubMed]
- Neil, E.R.; Funk, A.M.; Yufit, D.S.; Parker, D. Synthesis, Stereocontrol and Structural Studies of Highly Luminescent Chiral Tris-Amidepyridyl-Triazacyclononane Lanthanide Complexes. Dalton Trans. 2014, 43, 5490–5504. [Google Scholar] [CrossRef] [PubMed]
- Soulié, M.; Latzko, F.; Bourrier, E.; Placide, V.; Butler, S.J.; Pal, R.; Walton, J.W.; Baldeck, P.L.; Le Guennic, B.; Andraud, C.; et al. Comparative Analysis of Conjugated Alkynyl Chromophore–Triazacyclononane Ligands for Sensitized Emission of Europium and Terbium. Chem. Eur. J. 2014, 20, 8636–8646. [Google Scholar] [CrossRef]
- Nonat, A.; Giraud, M.; Gateau, C.; Fries, P.H.; Helm, L.; Mazzanti, M. Gadolinium(III) Complexes of 1,4,7-Triazacyclononane Based Picolinate Ligands: Simultaneous Optimization of Water Exchange Kinetics and Electronic Relaxation. Dalton Trans. 2009, 8033–8046. [Google Scholar] [CrossRef]
- De Matos, R.; Gheata, A.; Campargue, G.; Vuilleumier, J.; Nicolle, L.; Pierzchala, K.; Jelescu, I.; Lucarini, F.; Gautschi, I.; Riporto, F.; et al. Gd3+-Functionalized Lithium Niobate Nanoparticles for Dual Multiphoton and Magnetic Resonance Bioimaging. ACS Appl. Nano Mater. 2022, 5, 2912–2922. [Google Scholar] [CrossRef]
- Stasiuk, G.J.; Lowe, M.P. Click Chemistry with Lanthanide Complexes: A Word of Caution. Dalton Trans. 2009, 9725–9727. [Google Scholar] [CrossRef]
- Ševčík, R.; Vaněk, J.; Michalicová, R.; Lubal, P.; Hermann, P.; Santos, I.C.; Santos, I.; Campello, M.P.C. Formation and Decomplexation Kinetics of Copper(II) Complexes with Cyclen Derivatives Having Mixed Carboxylate and Phosphonate Pendant Arms. Dalton Trans. 2016, 45, 12723–12733. [Google Scholar] [CrossRef]
- Tosato, M.; Dalla Tiezza, M.; May, N.V.; Isse, A.A.; Nardella, S.; Orian, L.; Verona, M.; Vaccarin, C.; Alker, A.; Mäcke, H.; et al. Copper Coordination Chemistry of Sulfur Pendant Cyclen Derivatives: An Attempt to Hinder the Reductive-Induced Demetalation in 64/67Cu Radiopharmaceuticals. Inorg. Chem. 2021, 60, 11530–11547. [Google Scholar] [CrossRef] [PubMed]
- Presolski, S.I.; Hong, V.P.; Finn, M.G. Copper-Catalyzed Azide–Alkyne Click Chemistry for Bioconjugation. Curr. Protoc. Chem. Biol. 2011, 3, 153–162. [Google Scholar] [CrossRef]
- Kennedy, D.C.; McKay, C.S.; Legault, M.C.B.; Danielson, D.C.; Blake, J.A.; Pegoraro, A.F.; Stolow, A.; Mester, Z.; Pezacki, J.P. Cellular Consequences of Copper Complexes Used To Catalyze Bioorthogonal Click Reactions. J. Am. Chem. Soc. 2011, 133, 17993–18001. [Google Scholar] [CrossRef]
- Fawcett, C.; Watson, J.; Richards, S.; Doherty, A.E.; Seki, H.; Love, E.A.; Coles, C.H.; Coe, D.M.; Jamieson, C. Comparative Study of Click Handle Stability in Common Ligation Conditions. Bioconjug. Chem. 2025, 36, 1054–1065. [Google Scholar] [CrossRef]
- Luu, T.; Gristwood, K.; Knight, J.C.; Jörg, M. Click Chemistry: Reaction Rates and Their Suitability for Biomedical Applications. Bioconjug. Chem. 2024, 35, 715–731. [Google Scholar] [CrossRef]
- De Matos, R.; Vuilleumier, J.; Mas, C.; Constant, S.; Staedler, D.; Gerber-Lemaire, S. Inhibitor-Conjugated Harmonic Nanoparticles Targeting Fibroblast Activation Protein. RSC Adv. 2019, 9, 31659–31669. [Google Scholar] [CrossRef]
- Vuilleumier, J.; Gaulier, G.; De Matos, R.; Mugnier, Y.; Campargue, G.; Wolf, J.-P.; Bonacina, L.; Gerber-Lemaire, S. Photocontrolled Release of the Anticancer Drug Chlorambucil with Caged Harmonic Nanoparticles. Helv. Chim. Acta 2020, 103, e1900251. [Google Scholar] [CrossRef]
- Li, Y.; Liu, X. Tunable Acid-Sensitive Ester Protecting Groups in Oligosaccharide Synthesis. Chem. Commun. 2014, 50, 3155–3158. [Google Scholar] [CrossRef]
- Riporto, F.; Dhouib, A.; Gheata, A.; Beauquis, S.; Molina, E.; Guené-Girard, S.; Galez, C.; Bornet, A.; Gautier-Luneau, I.; Gerber-Lemaire, S.; et al. Nonclassical Nucleation and Crystallization of LiNbO3 Nanoparticles from the Aqueous Solvothermal Alkoxide Route. Small 2024, 20, 2306417. [Google Scholar] [CrossRef]
- Gheata, A.; Spada, A.; Wittwer, M.; Dhouib, A.; Molina, E.; Mugnier, Y.; Gerber-Lemaire, S. Modulating the Surface Properties of Lithium Niobate Nanoparticles by Multifunctional Coatings Using Water-in-Oil Microemulsions. Nanomaterials 2023, 13, 522. [Google Scholar] [CrossRef]
- Multian, V.; Teyssier, J. Beam Splitting/Mixing Module for an Optical System and an Associated Optical System. U.S. Patent 18/735,863, 12 December 2024. [Google Scholar]
- Vuilleumier, J.; Gaulier, G.; De Matos, R.; Ortiz, D.; Menin, L.; Campargue, G.; Mas, C.; Constant, S.; Le Dantec, R.; Mugnier, Y.; et al. Two-Photon-Triggered Photorelease of Caged Compounds from Multifunctional Harmonic Nanoparticles. ACS Appl. Mater. Interfaces 2019, 11, 27443–27452. [Google Scholar] [CrossRef]
- Gheata, A.; Gaulier, G.; Campargue, G.; Vuilleumier, J.; Kaiser, S.; Gautschi, I.; Riporto, F.; Beauquis, S.; Staedler, D.; Diviani, D.; et al. Photoresponsive Nanocarriers Based on Lithium Niobate Nanoparticles for Harmonic Imaging and On-Demand Release of Anticancer Chemotherapeutics. ACS Nanosci. Au 2022, 2, 355–366. [Google Scholar] [CrossRef]
- Kijatkin, C.; Eggert, J.; Bock, S.; Berben, D.; Oláh, L.; Szaller, Z.; Kis, Z.; Imlau, M. Nonlinear Diffuse Fs-Pulse Reflectometry of Harmonic Upconversion Nanoparticles. Photonics 2017, 4, 11. [Google Scholar] [CrossRef]
- Klenen, J.; Sauerwein, F.; Vittadello, L.; Kömpe, K.; Hreb, V.; Sydorchuk, V.; Yakhnevych, U.; Sugak, D.; Vasylechko, L.; Imlau, M. Gap-Free Tuning of Second and Third Harmonic Generation in Mechanochemically Synthesized Nanocrystalline LiNb1−xTaxO3 (0 ≤ x ≤ 1) Studied with Nonlinear Diffuse Femtosecond-Pulse Reflectometry. Nanomaterials 2024, 14, 317. [Google Scholar] [CrossRef]
- Zheng, X.-Y.; Zhao, K.; Tang, J.; Wang, X.-Y.; Li, L.-D.; Chen, N.-X.; Wang, Y.-J.; Shi, S.; Zhang, X.; Malaisamy, S.; et al. Gd-Dots with Strong Ligand–Water Interaction for Ultrasensitive Magnetic Resonance Renography. ACS Nano 2017, 11, 3642–3650. [Google Scholar] [CrossRef]
- Supkowski, R.M.; Horrocks, W.D., Jr. On the Determination of the Number of Water Molecules, q, Coordinated to Europium(III) Ions in Solution from Luminescence Decay Lifetimes. Inorganica Chim. Acta 2002, 340, 44–48. [Google Scholar] [CrossRef]
- Brasselet, S.; Le Floc’h, V.; Treussart, F.; Roch, J.-F.; Zyss, J.; Botzung-Appert, E.; Ibanez, A. In Situ Diagnostics of the Crystalline Nature of Single Organic Nanocrystals by Nonlinear Microscopy. Phys. Rev. Lett. 2004, 92, 207401. [Google Scholar] [CrossRef] [PubMed]
- Bonacina, L.; Mugnier, Y.; Courvoisier, F.; Le Dantec, R.; Extermann, J.; Lambert, Y.; Boutou, V.; Galez, C.; Wolf, J.-P. Polar Fe(IO3)3 Nanocrystals as Local Probes for Nonlinear Microscopy. Appl. Phys. B 2007, 87, 399–403. [Google Scholar] [CrossRef]
- Hsieh, C.-L.; Pu, Y.; Grange, R.; Psaltis, D. Second Harmonic Generation from Nanocrystals under Linearly and Circularly Polarized Excitations. Opt. Express 2010, 18, 11917–11932. [Google Scholar] [CrossRef]
- Worku, N.; Gross, H. Vectorial Field Propagation through High NA Objectives Using Polarized Gaussian Beam Decomposition. In Proceedings of the Optical Trapping and Optical Micromanipulation XIV; SPIE: Bellingham, DC, USA, 2017; Volume 10347, pp. 107–117. [Google Scholar]
- Placide, V.; Bui, A.T.; Grichine, A.; Duperray, A.; Pitrat, D.; Andraud, C.; Maury, O. Two-Photon Multiplexing Bio-Imaging Using a Combination of Eu- and Tb-Bioprobes. Dalton Trans. 2015, 44, 4918–4924. [Google Scholar] [CrossRef]
- Runowski, M.; Marcinkowski, D.; Soler-Carracedo, K.; Gorczyński, A.; Ewert, E.; Woźny, P.; Martín, I.R. Noncentrosymmetric Lanthanide-Based MOF Materials Exhibiting Strong SHG Activity and NIR Luminescence of Er3+: Application in Nonlinear Optical Thermometry. ACS Appl. Mater. Interfaces 2023, 15, 3244–3252. [Google Scholar] [CrossRef]
- Whetter, J.N.; Marlin, A.; Amason, E.K.; Aluicio-Sarduy, E.; Koller, A.J.; Tran, P.N.; Becker, K.V.; Engle, J.W.; Boros, E. Mono-Ethyl-Phosphonates: Functional Group Masking Strategy to Stabilize and Optimize the Pharmacokinetics of Rare Earth Radiochelates. ChemistryEurope 2025, 3, e202500079. [Google Scholar] [CrossRef]
- Runowski, M.; Woźny, P.; Martín, I.R.; Soler-Carracedo, K.; Zheng, T.; Hemmerich, H.; Rivera-López, F.; Moszczyński, J.; Kulpiński, P.; Feldmann, S. Multimodal Optically Nonlinear Nanoparticles Exhibiting Simultaneous Higher Harmonics Generation and Upconversion Luminescence for Anticounterfeiting and 8-Bit Optical Coding. Adv. Funct. Mater. 2024, 34, 2307791. [Google Scholar] [CrossRef]
- Pointel, Y.; Daiguebonne, C.; Suffren, Y.; Natur, F.L.; Freslon, S.; Calvez, G.; Bernot, K.; Jacob, D.; Guillou, O. Colloidal Suspensions of Highly Luminescent Lanthanide-Based Coordination Polymer Molecular Alloys for Ink-Jet Printing and Tagging of Technical Liquids. Inorg. Chem. Front. 2021, 8, 2125–2135. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Sample | EtOH | PBS 0.1X | Zeta Potential (mV) | |||
|---|---|---|---|---|---|---|---|
| DH [a] (nm) | PDI | DH [a] (nm) | PDI | pH 7.4 | pH 3 | ||
| 1 | LNO@Si-Talys | 364.5 ± 54.1 | 0.16 ± 0.02 | 86.9 ± 3.1 | 0.16 ± 0.01 | −35.5 ± 0.8 | −9.9 ± 0.3 |
| 2 | LNO@[GdLA] | 191.5 ± 16.8 | 0.26 ± 0.01 | 143.6 ± 13.5 | 0.24 ± 0.01 | −35.1 ± 1.1 | 0.0 ± 0.0 |
| 3 | LNO@[EuLA] | 221.9 ± 52.8 | 0.30 ± 0.02 | 138.3 ± 4.8 | 0.17 ± 0.03 | −24.7 ± 1.9 | −0.3 ± 1.2 |
| 4 | LNO@[YbLA] | 182.3 ± 11.0 | 0.19 ± 0.03 | 144.0 ± 17.6 | 0.25 ± 0.07 | −32.3 ± 1.8 | 4.3 ± 0.4 |
| 5 | LNO@[GdLD] | 272.7 ± 2.2 | 0.22 ± 0.02 | 509.8 ± 86.0 | 0.22 ± 0.02 | −31.7 ± 1.7 | −10.1 ± 6.4 |
| 6 | LNO@[EuLD] | 296.6 ± 3.6 | 0.19 ± 0.01 | 518.0 ± 29.0 | 0.41 ± 0.06 | −23.1 ± 0.5 | 0.0 ± 0.3 |
| 7 | LNO@[YbLD] | 359.1 ± 13.2 | 0.23 ± 0.02 | 675.6 ± 82.1 | 0.50 ± 0.31 | −22.1 ± 1.2 | 3.5 ± 0.2 |
| 8 | LNO@[TbLD] | 176.3 ± 11.9 | 0.16 ± 0.03 | 864.6 ± 29.0 | 0.30 ± 0.06 | −26.9 ± 2.8 | 8.1 ± 0.2 |
| Entry | Sample | τ1/μs | τ2/μs |
|---|---|---|---|
| 1 | [EuLA] | 171.6 ± 2.2 | - |
| 2 | LNO@[EuLA] | 84.3 ± 2.3 | 14.4 ± 1.2 |
| 3 | [EuLD] | 293.5 ± 4.5 | - |
| 4 | LNO@[EuLD] | 119.9 ± 3.4 | 51.1 ± 3.3 |
| 5 | [TbLA] | 243.6 ± 1.2 | 56.2 ± 2.3 |
| 6 | LNO@[TbLA] | 81.9 ± 0.5 | - |
| 7 | [TbLD] | 503.3 ± 10.6 | 77.3 ± 6.3 |
| 8 | LNO@[TbLD] | 257.1 ± 1.4 | 56.2 ± 2.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Dumolard, S.; Multian, V.; Gheata, A.; Spada, A.; Pierzchala, K.; Lanz, B.; Dhouib, A.; Mugnier, Y.; Teyssier, J.; Bonacina, L.; et al. Click Chemistry Functionalization of Harmonic Nanoparticles with Lanthanide Complexes Towards Tunable Platforms for Multimodal Imaging. Nanomaterials 2026, 16, 591. https://doi.org/10.3390/nano16100591
Dumolard S, Multian V, Gheata A, Spada A, Pierzchala K, Lanz B, Dhouib A, Mugnier Y, Teyssier J, Bonacina L, et al. Click Chemistry Functionalization of Harmonic Nanoparticles with Lanthanide Complexes Towards Tunable Platforms for Multimodal Imaging. Nanomaterials. 2026; 16(10):591. https://doi.org/10.3390/nano16100591
Chicago/Turabian StyleDumolard, Simon, Volodymyr Multian, Adrian Gheata, Alessandra Spada, Katarzyna Pierzchala, Bernard Lanz, Ameni Dhouib, Yannick Mugnier, Jérémie Teyssier, Luigi Bonacina, and et al. 2026. "Click Chemistry Functionalization of Harmonic Nanoparticles with Lanthanide Complexes Towards Tunable Platforms for Multimodal Imaging" Nanomaterials 16, no. 10: 591. https://doi.org/10.3390/nano16100591
APA StyleDumolard, S., Multian, V., Gheata, A., Spada, A., Pierzchala, K., Lanz, B., Dhouib, A., Mugnier, Y., Teyssier, J., Bonacina, L., Chauvin, A.-S., & Gerber-Lemaire, S. (2026). Click Chemistry Functionalization of Harmonic Nanoparticles with Lanthanide Complexes Towards Tunable Platforms for Multimodal Imaging. Nanomaterials, 16(10), 591. https://doi.org/10.3390/nano16100591