
The Evolution and Future of Targeted Cancer Therapy: From Nanoparticles, Oncolytic Viruses, and Oncolytic Bacteria to the Treatment of Solid Tumors


 ,
,
Abstract
1. Introduction
The Unique and Challenging Context of Solid Tumors
2. Nanoparticles
2.1. Liposomes
2.2. Polymersomes
2.3. Exosomes
2.4. Advantages, Disadvantages, and the Future of Nanoparticle-Mediated Oncotherapy
3. Oncolytic Viruses
3.1. Mechanisms of Oncotherapy
3.2. Combinatorial Oncolytic Viral Oncotherapies
3.3. Oncolytic Virus-Assisted Tumor-Imaging
3.4. Advantages, Disadvantages, and the Future of Oncolytic Virus Therapy
4. Oncolytic Bacteria
4.1. Oncolytic Bacteria: Attenuation and Mechanisms
4.2. Targeting Safety, Delivery and Efficacy of Oncolytic Bacteria
4.3. The Optimization of Bacteria-Mediated Oncotherapeutic Payloads
4.4. Advantage, Disadvantages, and the Future of Oncolytic Bacteria
5. Comparing Nanoparticle, Oncolytic Virus and Oncolytic Bacteria: Development as Novel Oncotherapeutics
5.1. Generating Novel Therapeutics: Accomplishing Selective Targeting
5.1.1. Cell Surface Molecules
5.1.2. Intracellular Molecules
5.1.3. Endogenous Environment
5.1.4. Exogenous Stimuli
5.1.5. Carrier Cell-Mediated Selective Delivery
5.2. Modification and Characterization of Novel Therapeutics
5.3. Establishing Biodistribution and Efficacy of Novel Therapeutic
5.3.1. Small Animal Model Selection
5.3.2. Immune Clearance and Biological Barriers
5.3.3. Route of Administration
5.4. Large Animal Models and Clinical Trial Initiation
6. Overview of Clinical Trials
6.1. Nanoparticle Oncotherapeutic Trials
6.2. Oncolytic Virus Clinical Trials
6.3. Clinical Trials of Oncolytic Bacteria
6.4. The State of Nanoparticle, Oncolytic Virus, and Oncolytic Bacteria Clinical Progression
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Collado, M.; Serrano, M. Senescence in Tumours: Evidence from Mice and Humans. Nat. Rev. Cancer 2010, 10, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, D.L.; Sage, J. Cellular Mechanisms of Tumour Suppression by the Retinoblastoma Gene. Nat. Rev. Cancer 2008, 8, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Arneth, B. Tumor Microenvironment. Medicina 2019, 56, 15. [Google Scholar] [CrossRef]
- Mac Gabhann, F.; Popel, A.S. Systems Biology of Vascular Endothelial Growth Factors. Microcirculation 2008, 15, 715–738. [Google Scholar] [CrossRef]
- Ferrara, N. Vascular Endothelial Growth Factor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 789–791. [Google Scholar] [CrossRef]
- Carmeliet, P. VEGF as a Key Mediator of Angiogenesis in Cancer. Oncology 2005, 69, 4–10. [Google Scholar] [CrossRef]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the Angiogenic Switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef]
- Carreau, A.; El Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C. Why Is the Partial Oxygen Pressure of Human Tissues a Crucial Parameter? Small Molecules and Hypoxia. J. Cell. Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef]
- McKeown, S.R. Defining Normoxia, Physoxia and Hypoxia in Tumours-Implications for Treatment Response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef]
- Gujar, S.A.; Lee, P.W.K. Oncolytic Virus-Mediated Reversal of Impaired Tumor Antigen Presentation. Front. Oncol. 2014, 4, 77. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-Related Inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Awasthi, R.; Roseblade, A.; Hansbro, P.M.; Rathbone, M.J.; Dua, K.; Bebawy, M. Nanoparticles in Cancer Treatment: Opportunities and Obstacles. Curr. Drug Targets 2018, 19, 1696–1709. [Google Scholar] [CrossRef]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR Effect: Unique Features of Tumor Blood Vessels for Drug Delivery, Factors Involved, and Limitations and Augmentation of the Effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef]
- Gu, G.; Chen, C.; Zhang, S.; Yin, B.; Wang, J. Self-Assembly Dual-Responsive NO Donor Nanoparticles for Effective Cancer Therapy. ACS Appl. Mater. Interfaces 2021. [Google Scholar] [CrossRef]
- Freitas, L.F.; Ferreira, A.H.; Thipe, V.C.; Varca, G.H.C.; Lima, C.S.A.; Batista, J.G.S.; Riello, F.N.; Nogueira, K.; Cruz, C.P.C.; Mendes, G.O.A.; et al. The State of the Art of Theranostic Nanomaterials for Lung, Breast, and Prostate Cancers. Nanomaterials 2021, 11, 2579. [Google Scholar] [CrossRef]
- Krishnan, N.; Fang, R.H.; Zhang, L. Engineering of Stimuli-Responsive Self-Assembled Biomimetic Nanoparticles. Adv. Drug Deliv. Rev. 2021, 179, 114006. [Google Scholar] [CrossRef]
- Anajafi, T.; Mallik, S. Polymersome-Based Drug-Delivery Strategies for Cancer Therapeutics. Ther. Deliv. 2015, 6, 521–534. [Google Scholar] [CrossRef]
- Gao, W.; Hu, C.-M.J.; Fang, R.H.; Zhang, L. Liposome-like Nanostructures for Drug Delivery. J. Mater. Chem. B 2013, 1, 6569–6585. [Google Scholar] [CrossRef]
- Pullan, J.E.; Confeld, M.I.; Osborn, J.K.; Kim, J.; Sarkar, K.; Mallik, S. Exosomes as Drug Carriers for Cancer Therapy. Mol. Pharm. 2019, 16, 1789–1798. [Google Scholar] [CrossRef]
- Tripodi, L.; Vitale, M.; Cerullo, V.; Pastore, L. Oncolytic Adenoviruses for Cancer Therapy. Int. J. Mol. Sci. 2021, 22, 2517. [Google Scholar] [CrossRef]
- Aldrak, N.; Alsaab, S.; Algethami, A.; Bhere, D.; Wakimoto, H.; Shah, K.; Alomary, M.N.; Zaidan, N. Oncolytic Herpes Simplex Virus-Based Therapies for Cancer. Cells 2021, 10, 1541. [Google Scholar] [CrossRef]
- Guo, Z.S.; Lu, B.; Guo, Z.; Giehl, E.; Feist, M.; Dai, E.; Liu, W.; Storkus, W.J.; He, Y.; Liu, Z.; et al. Vaccinia Virus-Mediated Cancer Immunotherapy: Cancer Vaccines and Oncolytics. J. Immunother. Cancer 2019, 7, 6. [Google Scholar] [CrossRef]
- Cann, S.A.H.; van Netten, J.P.; Netten, C. Dr William Coley and Tumour Regression: A Place in History or in the Future. Postgrad. Med. J. 2003, 79, 672–680. [Google Scholar]
- Staedtke, V.; Roberts, N.J.; Bai, R.-Y.; Zhou, S. Clostridium Novyi-NT in Cancer Therapy. Genes Dis. 2016, 3, 144–152. [Google Scholar] [CrossRef]
- Yeh, M.-K.; Hsin-I, C.; Ming-Yen, C. Clinical Development of Liposome Based Drugs: Formulation, Characterization, and Therapeutic Efficacy. Int. J. Nanomed. 2011, 7, 49. [Google Scholar] [CrossRef]
- Bozzuto, G.; Molinari, A. Liposomes as Nanomedical Devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef] [PubMed]
- Schwendener, R.A. Liposomes as Vaccine Delivery Systems: A Review of the Recent Advances. Ther. Adv. Vaccines 2014, 2, 159–182. [Google Scholar] [CrossRef] [PubMed]
- Surapaneni, M.S.; Das, S.K.; Das, N.G. Designing Paclitaxel Drug Delivery Systems Aimed at Improved Patient Outcomes: Current Status and Challenges. ISRN Pharmacol. 2012, 2012, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, L.; Luo, S.; Hu, J.; Huang, X.; Li, P.-W.; Zhang, Y.; Wu, C.; Tian, B.-L. Enhancement of Antitumor Efficacy of Paclitaxel-Loaded PEGylated Liposomes by N,N-Dimethyl Tertiary Amino Moiety in Pancreatic Cancer. Drug Des. Dev. Ther. 2020, 14, 2945–2957. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Yang, Y.; Chen, J.; Tang, H.; Sun, Y.; Zhang, Z.; Wang, Z.; Li, Y.; Li, Y.; Luan, X.; et al. Preparation, Characterization, and Pharmacokinetic Study of a Novel Long-Acting Targeted Paclitaxel Liposome with Antitumor Activity. Int. J. Nanomed. 2020, 15, 553–571. [Google Scholar] [CrossRef]
- Cabanes, A.; Briggs, K.E.; Gokhale, P.C.; Treat, J.A.; Rahman, A. Comparative in Vivo Studies with Paclitaxel and Liposome-Encapsulated Paclitaxel. Int. J. Oncol. 1998, 12, 1035–1075. [Google Scholar] [CrossRef]
- Irvine, D.J.; Hanson, M.C.; Rakhra, K.; Tokatlian, T. Synthetic Nanoparticles for Vaccines and Immunotherapy. Chem. Rev. 2015, 115, 11109–11146. [Google Scholar] [CrossRef]
- Franco, M.S.; Gomes, E.R.; Roque, M.C.; Oliveira, M.C. Triggered Drug Release from Liposomes: Exploiting the Outer and Inner Tumor Environment. Front. Oncol. 2021, 11, 623760. [Google Scholar] [CrossRef]
- Karanth, H.; Murthy, R.S.R. PH-Sensitive Liposomes-Principle and Application in Cancer Therapy. J. Pharm. Pharmacol. 2007, 59, 469–483. [Google Scholar] [CrossRef]
- Barbosa, M.V.; Monteiro, L.O.F.; Carneiro, G.; Malagutti, A.R.; Vilela, J.M.C.; Andrade, M.S.; Oliveira, M.C.; Carvalho-Junior, A.D.; Leite, E.A. Experimental Design of a Liposomal Lipid System: A Potential Strategy for Paclitaxel-Based Breast Cancer Treatment. Colloids Surf. B Biointerfaces 2015, 136, 553–561. [Google Scholar] [CrossRef]
- Akbarian, A.; Ebtekar, M.; Pakravan, N.; Hassan, Z.M. Folate Receptor Alpha Targeted Delivery of Artemether to Breast Cancer Cells with Folate-Decorated Human Serum Albumin Nanoparticles. Int. J. Biol. Macromol. 2020, 152, 90–101. [Google Scholar] [CrossRef]
- Gao, Y.; Yang, S.-C.; Zhu, M.-H.; Zhu, X.-D.; Luan, X.; Liu, X.-L.; Lai, X.; Yuan, Y.; Lu, Q.; Sun, P.; et al. Metal Phenolic Network-Integrated Multistage Nanosystem for Enhanced Drug Delivery to Solid Tumors. Small 2021, 17, 2100789. [Google Scholar] [CrossRef]
- Gill, P.S.; Wernz, J.; Scadden, D.T.; Cohen, P.; Mukwaya, G.M.; von Roenn, J.H.; Jacobs, M.; Kempin, S.; Silverberg, I.; Gonzales, G.; et al. Randomized Phase III Trial of Liposomal Daunorubicin versus Doxorubicin, Bleomycin, and Vincristine in AIDS-Related Kaposi’s Sarcoma. J. Clin. Oncol. 1996, 14, 2353–2364. [Google Scholar] [CrossRef]
- Shreffler, J.W.; Pullan, J.E.; Dailey, K.M.; Mallik, S.; Brooks, A.E. Overcoming Hurdles in Nanoparticle Clinical Translation: The Influence of Experimental Design and Surface Modification. Int. J. Mol. Sci. 2019, 20, 6056. [Google Scholar] [CrossRef]
- Montaner, J.; Cano-Sarabia, M.; Simats, A.; Hernández-Guillamon, M.; Rosell, A.; Maspoch, D.; Campos-Martorell, M. Charge Effect of a Liposomal Delivery System Encapsulating Simvastatin to Treat Experimental Ischemic Stroke in Rats. Int. J. Nanomed. 2016, 11, 3035–3048. [Google Scholar] [CrossRef]
- Lee, J.S. Biodegradable Polymersomes for Drug Delivery: Circulation Kinetics and Biodistribution, Modulated Drug Delivery and Cellular Uptake. Ph.D. Thesis, University of Twente, Enschede, The Netherlands, 2011. [Google Scholar]
- Mamnoon, B.; Loganathan, J.; Confeld, M.I.; De Fonseka, N.; Feng, L.; Froberg, J.; Choi, Y.; Tuvin, D.M.; Sathish, V.; Mallik, S. Targeted Polymeric Nanoparticles for Drug Delivery to Hypoxic, Triple-Negative Breast Tumors. ACS Appl. Bio Mater. 2021, 4, 1450–1460. [Google Scholar] [CrossRef]
- Confeld, M.I.; Mamnoon, B.; Feng, L.; Jensen-Smith, H.; Ray, P.; Froberg, J.; Kim, J.; Hollingsworth, M.A.; Quadir, M.; Choi, Y.; et al. Targeting the Tumor Core: Hypoxia-Responsive Nanoparticles for the Delivery of Chemotherapy to Pancreatic Tumors. Mol. Pharm. 2020, 17, 2849–2863. [Google Scholar] [CrossRef]
- Thambi, T.; Park, J.H.; Lee, D.S. Stimuli-Responsive Polymersomes for Cancer Therapy. Biomater. Sci. 2015, 4, 55–69. [Google Scholar] [CrossRef]
- Wang, C.; Su, L.; Wu, C.; Wu, J.; Zhu, C.; Yuan, G. RGD Peptide Targeted Lipid-Coated Nanoparticles for Combinatorial Delivery of Sorafenib and Quercetin against Hepatocellular Carcinoma. Drug Dev. Ind. Pharm. 2016, 42, 1938–1944. [Google Scholar] [CrossRef]
- Ouyang, J.; Jiang, Y.; Deng, C.; Zhong, Z.; Lan, Q. Doxorubicin Delivered via ApoE-Directed Reduction-Sensitive Polymersomes Potently Inhibit Orthotopic Human Glioblastoma Xenografts in Nude Mice. Int. J. Nanomed. 2021, 16, 4105–4115. [Google Scholar] [CrossRef]
- Qin, H.; Jiang, Y.; Zhang, J.; Deng, C.; Zhong, Z. Oncoprotein Inhibitor Rigosertib Loaded in ApoE-Targeted Smart Polymersomes Reveals High Safety and Potency against Human Glioblastoma in Mice. Mol. Pharm. 2019, 16, 3711–3719. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Zhang, Y.; Cheng, R.; Yuan, J.; Zhong, Z. Systemic Delivery of NAC-1 SiRNA by Neuropilin-Targeted Polymersomes Sensitizes Antiangiogenic Therapy of Metastatic Triple-Negative Breast Cancer. Biomacromolecules 2020, 21, 5119–5127. [Google Scholar] [CrossRef]
- Diaz Bessone, M.I.; Simón-Gracia, L.; Scodeller, P.; de los Ramirez, M.A.; Lago Huvelle, M.A.; Soler-Illia, G.J.A.A.; Simian, M. IRGD-Guided Tamoxifen Polymersomes Inhibit Estrogen Receptor Transcriptional Activity and Decrease the Number of Breast Cancer Cells with Self-Renewing Capacity. J. Nanobiotechnol. 2019, 17, 120. [Google Scholar] [CrossRef]
- Zou, Y.; Wei, Y.; Sun, Y.; Bao, J.; Yao, F.; Li, Z.; Meng, F.; Hu, C.; Storm, G.; Zhong, Z. Cyclic RGD-Functionalized and Disulfide-Crosslinked Iodine-Rich Polymersomes as a Robust and Smart Theranostic Agent for Targeted CT Imaging and Chemotherapy of Tumor. Theranostics 2019, 9, 8061–8072. [Google Scholar] [CrossRef]
- Zou, Y.; Wei, J.; Xia, Y.; Meng, F.; Yuan, J.; Zhong, Z. Targeted Chemotherapy for Subcutaneous and Orthotopic Non-Small Cell Lung Tumors with Cyclic RGD-Functionalized and Disulfide-Crosslinked Polymersomal Doxorubicin. Signal Transduct. Target. Ther. 2018, 3, 1–8. [Google Scholar] [CrossRef]
- Wei, Y.; Gu, X.; Sun, Y.; Meng, F.; Storm, G.; Zhong, Z. Transferrin-Binding Peptide Functionalized Polymersomes Mediate Targeted Doxorubicin Delivery to Colorectal Cancer in Vivo. J. Control. Release 2020, 319, 407–415. [Google Scholar] [CrossRef]
- Yu, Z.; Gao, L.; Chen, K.; Zhang, W.; Zhang, Q.; Li, Q.; Hu, K. Nanoparticles: A New Approach to Upgrade Cancer Diagnosis and Treatment. Nanoscale Res. Lett. 2021, 16, 88. [Google Scholar] [CrossRef]
- Hou, X.; Shou, C.; He, M.; Xu, J.; Cheng, Y.; Yuan, Z.; Lan, M.; Zhao, Y.; Yang, Y.; Chen, X.; et al. A Combination of LightOn Gene Expression System and Tumor Microenvironment-Responsive Nanoparticle Delivery System for Targeted Breast Cancer Therapy. Acta Pharm. Sin. B 2020, 10, 1741–1753. [Google Scholar] [CrossRef]
- Luo, Z.; Dai, Y.; Gao, H. Development and Application of Hyaluronic Acid in Tumor Targeting Drug Delivery. Acta Pharm. Sin. B 2019, 9, 1099–1112. [Google Scholar] [CrossRef]
- Tammam, S.N.; Azzazy, H.ME.; Breitinger, H.G.; Lamprecht, A. Chitosan Nanoparticles for Nuclear Targeting: The Effect of Nanoparticle Size and Nuclear Localization Sequence Density. Mol. Pharm. 2015, 12, 4277–4289. [Google Scholar] [CrossRef]
- Zelmer, C.; Zweifel, L.P.; Kapinos, L.E.; Craciun, I.; Güven, Z.P.; Palivan, C.G.; Lim, R.Y.H. Organelle-Specific Targeting of Polymersomes into the Cell Nucleus. Proc. Natl. Acad. Sci. USA 2020, 117, 2770–2778. [Google Scholar] [CrossRef]
- Johnsen, K.B.; Gudbergsson, J.M.; Skov, M.N.; Pilgaard, L.; Moos, T.; Duroux, M. A Comprehensive Overview of Exosomes as Drug Delivery Vehicles—Endogenous Nanocarriers for Targeted Cancer Therapy. Biochim. Biophys. Acta Rev. Cancer 2014, 1846, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Munagala, R.; Aqil, F.; Jeyabalan, J.; Gupta, R.C. Bovine Milk-Derived Exosomes for Drug Delivery. Cancer Lett. 2016, 371, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.; Wang, M. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [PubMed]
- Nam, G.-H.; Choi, Y.; Kim, G.B.; Kim, S.; Kim, S.A.; Kim, I.-S. Emerging Prospects of Exosomes for Cancer Treatment: From Conventional Therapy to Immunotherapy. Adv. Mater. 2020, 32, 2002440. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A Doxorubicin Delivery Platform Using Engineered Natural Membrane Vesicle Exosomes for Targeted Tumor Therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Zhou, Y.; Lu, M.; Si, H.; Li, L.; Tang, B. Responsive Dual-Targeting Exosome as a Drug Carrier for Combination Cancer Immunotherapy. Research 2021, 2021, 9862876. [Google Scholar] [CrossRef]
- Ong, H.T.; Timm, M.M.; Greipp, P.R.; Witzig, T.E.; Dispenzieri, A.; Russell, S.J.; Peng, K.-W. Oncolytic Measles Virus Targets High CD46 Expression on Multiple Myeloma Cells. Exp. Hematol. 2006, 34, 713–720. [Google Scholar] [CrossRef]
- Li, L.; Liu, S.; Han, D.; Tang, B.; Ma, J. Delivery and Biosafety of Oncolytic Virotherapy. Front. Oncol. 2020, 10, 475. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, F. Advances and Potential Pitfalls of Oncolytic Viruses Expressing Immunomodulatory Transgene Therapy for Malignant Gliomas. Cell Death Dis. 2020, 11, 485. [Google Scholar] [CrossRef]
- Zhou, S.; Gravekamp, C.; Bermudes, D.; Liu, K. Tumour-Targeting Bacteria Engineered to Fight Cancer. Nat. Rev. Cancer 2018, 18, 727–743. [Google Scholar] [CrossRef]
- Feng, X.; He, P.; Zeng, C.; Li, Y.-H.; Das, S.K.; Li, B.; Yang, H.-F.; Du, Y. Novel Insights into the Role of Clostridium Novyi-NT Related Combination Bacteriolytic Therapy in Solid Tumors. Oncol. Lett. 2021, 21, 1. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Andtbacka, R.H.I.; Collichio, F.A.; Wolf, M.; Zhao, Z.; Shilkrut, M.; Puzanov, I.; Ross, M. Durable Response Rate as an Endpoint in Cancer Immunotherapy: Insights from Oncolytic Virus Clinical Trials. J. Immunother. Cancer 2017, 5, 72. [Google Scholar] [CrossRef]
- Kelly, E.; Russell, S.J. History of Oncolytic Viruses: Genesis to Genetic Engineering. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 651–659. [Google Scholar] [CrossRef]
- Jun, K.-H.; Gholami, S.; Song, T.-J.; Au, J.; Haddad, D.; Carson, J.; Chen, C.-H.; Mojica, K.; Zanzonico, P.; Chen, N.G.; et al. A Novel Oncolytic Viral Therapy and Imaging Technique for Gastric Cancer Using a Genetically Engineered Vaccinia Virus Carrying the Human Sodium Iodide Symporter. J. Exp. Clin. Cancer Res. 2014, 33, 2. [Google Scholar] [CrossRef]
- Weller, T.H.; Robbins, F.C.; Enders, J.F. Cultivation of Poliomyelitis Virus in Cultures of Human Foreskin and Embryonic Tissues. Proc. Soc. Exp. Biol. Med. Soc. Exp. Biol. Med. 1949, 72, 153–155. [Google Scholar] [CrossRef]
- Dobosz, P.; Dzieciątkowski, T. The Intriguing History of Cancer Immunotherapy. Front. Immunol. 2019, 10, 2965. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic Viruses: A New Class of Immunotherapy Drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Wang, G.; Kang, X.; Chen, K.S.; Jehng, T.; Jones, L.; Chen, J.; Huang, X.F.; Chen, S.-Y. An Engineered Oncolytic Virus Expressing PD-L1 Inhibitors Activates Tumor Neoantigen-Specific T Cell Responses. Nat. Commun. 2020, 11, 1395. [Google Scholar] [CrossRef]
- Andtbacka, R.H.I.; Collichio, F.; Harrington, K.J.; Middleton, M.R.; Downey, G.; Öhrling, K.; Kaufman, H.L. Final Analyses of OPTiM: A Randomized Phase III Trial of Talimogene Laherparepvec versus Granulocyte-Macrophage Colony-Stimulating Factor in Unresectable Stage III-IV Melanoma. J. Immunother. Cancer 2019, 7, 145. [Google Scholar] [CrossRef]
- Russell, S.J.; Peng, K.-W.; Bell, J.C. Oncolytic Virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef]
- Kim, E.; Kim, J.-H.; Shin, H.-Y.; Lee, H.; Yang, J.M.; Kim, J.; Sohn, J.-H.; Kim, H.; Yun, C.-O. Ad-MTERT-Delta19, a Conditional Replication-Competent Adenovirus Driven by the Human Telomerase Promoter, Selectively Replicates in and Elicits Cytopathic Effect in a Cancer Cell-Specific Manner. Hum. Gene Ther. 2003, 14, 1415–1428. [Google Scholar] [CrossRef]
- Wirth, T.; Zender, L.; Schulte, B.; Mundt, B.; Plentz, R.; Rudolph, K.L.; Manns, M.; Kubicka, S.; Kühnel, F. A Telomerase-Dependent Conditionally Replicating Adenovirus for Selective Treatment of Cancer. Cancer Res. 2003, 63, 3181–3188. [Google Scholar]
- Parato, K.A.; Breitbach, C.J.; Le Boeuf, F.; Wang, J.; Storbeck, C.; Ilkow, C.; Diallo, J.-S.; Falls, T.; Burns, J.; Garcia, V.; et al. The Oncolytic Poxvirus JX-594 Selectively Replicates in and Destroys Cancer Cells Driven by Genetic Pathways Commonly Activated in Cancers. Mol. Ther. 2012, 20, 749–758. [Google Scholar] [CrossRef]
- Martin, N.T.; Wrede, C.; Niemann, J.; Brooks, J.; Schwarzer, D.; Kühnel, F.; Gerardy-Schahn, R. Targeting Polysialic Acid-Abundant Cancers Using Oncolytic Adenoviruses with Fibers Fused to Active Bacteriophage Borne Endosialidase. Biomaterials 2018, 158, 86–94. [Google Scholar] [CrossRef]
- Petrovic, B.; Leoni, V.; Gatta, V.; Zaghini, A.; Vannini, A.; Campadelli-Fiume, G. Dual Ligand Insertion in GB and GD of Oncolytic Herpes Simplex Viruses for Retargeting to a Producer Vero Cell Line and to Cancer Cells. J. Virol. 2018, 92, e02122-17. [Google Scholar] [CrossRef]
- Bhatia, S.; O’Bryan, S.M.; Rivera, A.A.; Curiel, D.T.; Mathis, J.M. CXCL12 Retargeting of an Adenovirus Vector to Cancer Cells Using a Bispecific Adapter. Oncolytic Virother. 2016, 5, 99–113. [Google Scholar] [CrossRef]
- Puhlmann, M.; Gnant, M.; Brown, C.K.; Alexander, H.R.; Bartlett, D.L. Thymidine Kinase-Deleted Vaccinia Virus Expressing Purine Nucleoside Phosphorylase as a Vector for Tumor-Directed Gene Therapy. Hum. Gene Ther. 1999, 10, 649–657. [Google Scholar] [CrossRef]
- Toth, K.; Dhar, D.; Wold, W.S.M. Oncolytic (Replication-Competent) Adenoviruses as Anticancer Agents. Expert Opin. Biol. Ther. 2010, 10, 353–368. [Google Scholar] [CrossRef]
- Stojdl, D.F.; Lichty, B.D.; tenOever, B.R.; Paterson, J.M.; Power, A.T.; Knowles, S.; Marius, R.; Reynard, J.; Poliquin, L.; Atkins, H.; et al. VSV Strains with Defects in Their Ability to Shutdown Innate Immunity Are Potent Systemic Anti-Cancer Agents. Cancer Cell 2003, 4, 263–275. [Google Scholar] [CrossRef]
- Miest, T.S.; Cattaneo, R. New Viruses for Cancer Therapy: Meeting Clinical Needs. Nat. Rev. Microbiol. 2014, 12, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Samson, A.; Scott, K.J.; Taggart, D.; West, E.J.; Wilson, E.; Nuovo, G.J.; Thomson, S.; Corns, R.; Mathew, R.K.; Fuller, M.J.; et al. Intravenous Delivery of Oncolytic Reovirus to Brain Tumor Patients Immunologically Primes for Subsequent Checkpoint Blockade. Sci. Transl. Med. 2018, 10, eaam7577. [Google Scholar] [CrossRef] [PubMed]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going Viral with Cancer Immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Maroun, J.; Muñoz-Alía, M.; Ammayappan, A.; Schulze, A.; Peng, K.-W.; Russell, S. Designing and Building Oncolytic Viruses. Future Virol. 2017, 12, 193–213. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, O.; Dos Santos, J.M.; Hemminki, A. Oncolytic Viruses for Cancer Immunotherapy. J. Hematol. Oncol. 2020, 13, 84. [Google Scholar] [CrossRef]
- Guo, Z.S.; Liu, Z.; Kowalsky, S.; Feist, M.; Kalinski, P.; Lu, B.; Storkus, W.J.; Bartlett, D.L. Oncolytic Immunotherapy: Conceptual Evolution, Current Strategies, and Future Perspectives. Front. Immunol. 2017, 8, 555. [Google Scholar] [CrossRef]
- Diaz, R.M.; Galivo, F.; Kottke, T.; Wongthida, P.; Qiao, J.; Thompson, J.; Valdes, M.; Barber, G.; Vile, R.G. Oncolytic Immunovirotherapy for Melanoma Using Vesicular Stomatitis Virus. Cancer Res. 2007, 67, 2840–2848. [Google Scholar] [CrossRef]
- Moehler, M.H.; Zeidler, M.; Wilsberg, V.; Cornelis, J.J.; Woelfel, T.; Rommelaere, J.; Galle, P.R.; Heike, M. Parvovirus H-1-Induced Tumor Cell Death Enhances Human Immune Response in Vitro via Increased Phagocytosis, Maturation, and Cross-Presentation by Dendritic Cells. Hum. Gene Ther. 2005, 16, 996–1005. [Google Scholar] [CrossRef]
- Vigil, A.; Martinez, O.; Chua, M.A.; García-Sastre, A. Recombinant Newcastle Disease Virus as a Vaccine Vector for Cancer Therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 1883–1890. [Google Scholar] [CrossRef]
- Bridle, B.W.; Stephenson, K.B.; Boudreau, J.E.; Koshy, S.; Kazdhan, N.; Pullenayegum, E.; Brunellière, J.; Bramson, J.L.; Lichty, B.D.; Wan, Y. Potentiating Cancer Immunotherapy Using an Oncolytic Virus. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 1430–1439. [Google Scholar] [CrossRef]
- Harrop, R.; John, J.; Carroll, M.W. Recombinant Viral Vectors: Cancer Vaccines. Adv. Drug Deliv. Rev. 2006, 58, 931–947. [Google Scholar] [CrossRef]
- Zeh, H.J.; Downs-Canner, S.; McCart, J.A.; Guo, Z.S.; Rao, U.N.M.; Ramalingam, L.; Thorne, S.H.; Jones, H.L.; Kalinski, P.; Wieckowski, E.; et al. First-in-Man Study of Western Reserve Strain Oncolytic Vaccinia Virus: Safety, Systemic Spread, and Antitumor Activity. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 202–214. [Google Scholar] [CrossRef]
- Santos, J.M.; Havunen, R.; Hemminki, A. Modulation of the Tumor Microenvironment with an Oncolytic Adenovirus for Effective T-Cell Therapy and Checkpoint Inhibition. Methods Enzymol. 2020, 635, 205–230. [Google Scholar] [CrossRef]
- Dias, J.D.; Hemminki, O.; Diaconu, I.; Hirvinen, M.; Bonetti, A.; Guse, K.; Escutenaire, S.; Kanerva, A.; Pesonen, S.; Löskog, A.; et al. Targeted Cancer Immunotherapy with Oncolytic Adenovirus Coding for a Fully Human Monoclonal Antibody Specific for CTLA-4. Gene Ther. 2012, 19, 988–998. [Google Scholar] [CrossRef]
- Zafar, S.; Sorsa, S.; Siurala, M.; Hemminki, O.; Havunen, R.; Cervera-Carrascon, V.; Santos, J.M.; Wang, H.; Lieber, A.; De Gruijl, T.; et al. CD40L Coding Oncolytic Adenovirus Allows Long-Term Survival of Humanized Mice Receiving Dendritic Cell Therapy. Oncoimmunology 2018, 7, e1490856. [Google Scholar] [CrossRef]
- Galivo, F.; Diaz, R.M.; Wongthida, P.; Thompson, J.; Kottke, T.; Barber, G.; Melcher, A.; Vile, R. Single-Cycle Viral Gene Expression, Rather Than Progressive Replication and Oncolysis, Is Required for VSV Therapy of B16 Melanoma. Gene Ther. 2010, 17, 158–170. [Google Scholar] [CrossRef]
- Roy, D.G.; Geoffroy, K.; Marguerie, M.; Khan, S.T.; Martin, N.T.; Kmiecik, J.; Bobbala, D.; Aitken, A.S.; de Souza, C.T.; Stephenson, K.B.; et al. Adjuvant Oncolytic Virotherapy for Personalized Anti-Cancer Vaccination. Nat. Commun. 2021, 12, 2626. [Google Scholar] [CrossRef]
- Watanabe, K.; Luo, Y.; Da, T.; Guedan, S.; Ruella, M.; Scholler, J.; Keith, B.; Young, R.M.; Engels, B.; Sorsa, S.; et al. Pancreatic Cancer Therapy with Combined Mesothelin-Redirected Chimeric Antigen Receptor T Cells and Cytokine-Armed Oncolytic Adenoviruses. JCI Insight 2015, 3, e99573. [Google Scholar] [CrossRef]
- Nishio, N.; Diaconu, I.; Liu, H.; Cerullo, V.; Caruana, I.; Hoyos, V.; Bouchier-Hayes, L.; Savoldo, B.; Dotti, G. Armed Oncolytic Virus Enhances Immune Functions of Chimeric Antigen Receptor-Modified T Cells in Solid Tumors. Cancer Res. 2014, 74, 5195–5205. [Google Scholar] [CrossRef]
- Kanerva, A.; Nokisalmi, P.; Diaconu, I.; Koski, A.; Cerullo, V.; Liikanen, I.; Tähtinen, S.; Oksanen, M.; Heiskanen, R.; Pesonen, S.; et al. Antiviral and Antitumor T-Cell Immunity in Patients Treated with GM-CSF-Coding Oncolytic Adenovirus. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 2734–2744. [Google Scholar] [CrossRef]
- Bhattacharya, P.; Thiruppathi, M.; Elshabrawy, H.A.; Alharshawi, K.; Kumar, P.; Prabhakar, B.S. GM-CSF: An Immune Modulatory Cytokine That Can Suppress Autoimmunity. Cytokine 2015, 75, 261–271. [Google Scholar] [CrossRef]
- Burke, S.; Shergold, A.; Elder, M.J.; Whitworth, J.; Cheng, X.; Jin, H.; Wilkinson, R.W.; Harper, J.; Carroll, D.K. Oncolytic Newcastle Disease Virus Activation of the Innate Immune Response and Priming of Antitumor Adaptive Responses in Vitro. Cancer Immunol. Immunother. 2020, 69, 1015–1027. [Google Scholar] [CrossRef]
- Mastrangelo, M.J.; Maguire, H.C.; Lattime, E.C. Intralesional Vaccinia/GM-CSF Recombinant Virus in the Treatment of Metastatic Melanoma. Adv. Exp. Med. Biol. 2000, 465, 391–400. [Google Scholar] [CrossRef]
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Kelly, C.M.; Antonescu, C.R.; Bowler, T.; Munhoz, R.; Chi, P.; Dickson, M.A.; Gounder, M.M.; Keohan, M.L.; Movva, S.; Dholakia, R.; et al. Objective Response Rate Among Patients with Locally Advanced or Metastatic Sarcoma Treated With Talimogene Laherparepvec in Combination With Pembrolizumab: A Phase 2 Clinical Trial. JAMA Oncol. 2020, 6, 402–408. [Google Scholar] [CrossRef]
- Harrington, K.J.; Kong, A.; Mach, N.; Chesney, J.A.; Fernandez, B.C.; Rischin, D.; Cohen, E.E.W.; Radcliffe, H.-S.; Gumuscu, B.; Cheng, J.; et al. Talimogene Laherparepvec and Pembrolizumab in Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck (MASTERKEY-232): A Multicenter, Phase 1b Study. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 5153–5161. [Google Scholar] [CrossRef]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e10. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Harrington, K.J.; Hingorani, M.; Tanay, M.A.; Hickey, J.; Bhide, S.A.; Clarke, P.M.; Renouf, L.C.; Thway, K.; Sibtain, A.; McNeish, I.A.; et al. Phase I/II Study of Oncolytic HSVGM-CSF in Combination with Radiotherapy and Cisplatin in Untreated Stage III/IV Squamous Cell Cancer of the Head and Neck. Clin. Cancer Res. 2010, 16, 4005–4015. [Google Scholar] [CrossRef]
- Harrington, K.J.; Karapanagiotou, E.M.; Roulstone, V.; Twigger, K.R.; White, C.L.; Vidal, L.; Beirne, D.; Prestwich, R.; Newbold, K.; Ahmed, M.; et al. Two-Stage Phase I Dose-Escalation Study of Intratumoral Reovirus Type 3 Dearing and Palliative Radiotherapy in Patients with Advanced Cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 3067–3077. [Google Scholar] [CrossRef]
- Heo, J.; Breitbach, C.J.; Moon, A.; Kim, C.W.; Patt, R.; Kim, M.K.; Lee, Y.K.; Oh, S.Y.; Woo, H.Y.; Parato, K.; et al. Sequential Therapy with JX-594, a Targeted Oncolytic Poxvirus, Followed by Sorafenib in Hepatocellular Carcinoma: Preclinical and Clinical Demonstration of Combination Efficacy. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1170–1179. [Google Scholar] [CrossRef]
- Cerullo, V.; Diaconu, I.; Kangasniemi, L.; Rajecki, M.; Escutenaire, S.; Koski, A.; Romano, V.; Rouvinen, N.; Tuuminen, T.; Laasonen, L.; et al. Immunological Effects of Low-Dose Cyclophosphamide in Cancer Patients Treated with Oncolytic Adenovirus. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Histed, S.N.; Lindenberg, M.L.; Mena, E.; Turkbey, B.; Choyke, P.L.; Kurdziel, K.A. Review of Functional/ Anatomic Imaging in Oncology. Nucl. Med. Commun. 2012, 33, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Mariani, G.; Bruselli, L.; Kuwert, T.; Kim, E.E.; Flotats, A.; Israel, O.; Dondi, M.; Watanabe, N. A Review on the Clinical Uses of SPECT/CT. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1959–1985. [Google Scholar] [CrossRef]
- Weber, W.A.; Avril, N.; Schwaiger, M. Relevance of Positron Emission Tomography (PET) in Oncology. Strahlenther. Onkol. Organ. Dtsch. Rontgenges. 1999, 175, 356–373. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.W. Positron Emission Tomography/Computed Tomography. Semin. Nucl. Med. 2008, 38, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Bockisch, A.; Freudenberg, L.S.; Schmidt, D.; Kuwert, T. Hybrid Imaging by SPECT/CT and PET/CT: Proven Outcomes in Cancer Imaging. Semin. Nucl. Med. 2009, 39, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.W. Combined Positron Emission Tomography-Computed Tomography: The Historical Perspective. Semin. Ultrasound 2008, 29, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Maurer, A.H. Combined Imaging Modalities: PET/CT and SPECT/CT. Health Phys. 2008, 95, 571–576. [Google Scholar] [CrossRef]
- Muñoz-Álvarez, K.A.; Altomonte, J.; Laitinen, I.; Ziegler, S.; Steiger, K.; Esposito, I.; Schmid, R.M.; Ebert, O. PET Imaging of Oncolytic VSV Expressing the Mutant HSV-1 Thymidine Kinase Transgene in a Preclinical HCC Rat Model. Mol. Ther. 2015, 23, 728–736. [Google Scholar] [CrossRef]
- Rogers, B.E.; Parry, J.J.; Andrews, R.; Cordopatis, P.; Nock, B.A.; Maina, T. MicroPET Imaging of Gene Transfer with a Somatostatin Receptor-Based Reporter Gene and (94m)Tc-Demotate 1. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2005, 46, 1889–1897. [Google Scholar]
- McCart, J.A.; Mehta, N.; Scollard, D.; Reilly, R.M.; Carrasquillo, J.A.; Tang, N.; Deng, H.; Miller, M.; Xu, H.; Libutti, S.K.; et al. Oncolytic Vaccinia Virus Expressing the Human Somatostatin Receptor SSTR2: Molecular Imaging after Systemic Delivery Using 111In-Pentetreotide. Mol. Ther. J. Am. Soc. Gene Ther. 2004, 10, 553–561. [Google Scholar] [CrossRef]
- Warner, S.G.; Kim, S.-I.; Chaurasiya, S.; O’Leary, M.P.; Lu, J.; Sivanandam, V.; Woo, Y.; Chen, N.G.; Fong, Y. A Novel Chimeric Poxvirus Encoding HNIS Is Tumor-Tropic, Imageable, and Synergistic with Radioiodine to Sustain Colon Cancer Regression. Mol. Ther. Oncolytics 2019, 13, 82–92. [Google Scholar] [CrossRef]
- Concilio, S.C.; Russell, S.J.; Peng, K.-W. A Brief Review of Reporter Gene Imaging in Oncolytic Virotherapy and Gene Therapy. Mol. Ther. Oncolytics 2021, 21, 98–109. [Google Scholar] [CrossRef]
- Van Sande, J.; Massart, C.; Beauwens, R.; Schoutens, A.; Costagliola, S.; Dumont, J.E.; Wolff, J. Anion Selectivity by the Sodium Iodide Symporter. Endocrinology 2003, 144, 247–252. [Google Scholar] [CrossRef]
- Wolff, J.; Maurey, J.R. Thyroidal Iodide Transport. IV. The Role of Ion Size. Biochim. Biophys. Acta 1963, 69, 58–67. [Google Scholar] [CrossRef]
- Msaouel, P.; Opyrchal, M.; Dispenzieri, A.; Peng, K.W.; Federspiel, M.J.; Russell, S.J.; Galanis, E. Clinical Trials with Oncolytic Measles Virus: Current Status and Future Prospects. Curr. Cancer Drug Targets 2018, 18, 177–187. [Google Scholar] [CrossRef]
- Cho, J.Y.; Xing, S.; Liu, X.; Buckwalter, T.L.; Hwa, L.; Sferra, T.J.; Chiu, I.M.; Jhiang, S.M. Expression and Activity of Human Na+/I− Symporter in Human Glioma Cells by Adenovirus-Mediated Gene Delivery. Gene Ther. 2000, 7, 740–749. [Google Scholar] [CrossRef][Green Version]
- Li, H.; Nakashima, H.; Decklever, T.D.; Nace, R.A.; Russell, S.J. HSV-NIS, an Oncolytic Herpes Simplex Virus Type 1 Encoding Human Sodium Iodide Symporter for Preclinical Prostate Cancer Radiovirotherapy. Cancer Gene Ther. 2013, 20, 478–485. [Google Scholar] [CrossRef]
- Dingli, D.; Peng, K.-W.; Harvey, M.E.; Greipp, P.R.; O’Connor, M.K.; Cattaneo, R.; Morris, J.C.; Russell, S.J. Image-Guided Radiovirotherapy for Multiple Myeloma Using a Recombinant Measles Virus Expressing the Thyroidal Sodium Iodide Symporter. Blood 2004, 103, 1641–1646. [Google Scholar] [CrossRef]
- Haddad, D.; Chen, C.-H.; Carlin, S.; Silberhumer, G.; Chen, N.G.; Zhang, Q.; Longo, V.; Carpenter, S.G.; Mittra, A.; Carson, J.; et al. Imaging Characteristics, Tissue Distribution, and Spread of a Novel Oncolytic Vaccinia Virus Carrying the Human Sodium Iodide Symporter. PLoS ONE 2012, 7, e41647. [Google Scholar] [CrossRef]
- Bishnoi, S.; Tiwari, R.; Gupta, S.; Byrareddy, S.N.; Nayak, D. Oncotargeting by Vesicular Stomatitis Virus (VSV): Advances in Cancer Therapy. Viruses 2018, 10, 90. [Google Scholar] [CrossRef]
- Goel, A.; Carlson, S.K.; Classic, K.L.; Greiner, S.; Naik, S.; Power, A.T.; Bell, J.C.; Russell, S.J. Radioiodide Imaging and Radiovirotherapy of Multiple Myeloma Using VSV(Δ51)-NIS, an Attenuated Vesicular Stomatitis Virus Encoding the Sodium Iodide Symporter Gene. Blood 2007, 110, 2342–2350. [Google Scholar] [CrossRef]
- Miller, A.; Russell, S.J. The Use of the NIS Reporter Gene for Optimizing Oncolytic Virotherapy. Expert Opin. Biol. Ther. 2016, 16, 15–32. [Google Scholar] [CrossRef]
- Warner, S.G.; O’Leary, M.P.; Fong, Y. Therapeutic Oncolytic Viruses: Clinical Advances and Future Directions. Curr. Opin. Oncol. 2017, 29, 359–365. [Google Scholar] [CrossRef]
- Gong, J.; Sachdev, E.; Mita, A.C.; Mita, M.M. Clinical Development of Reovirus for Cancer Therapy: An Oncolytic Virus with Immune-Mediated Antitumor Activity. World J. Methodol. 2016, 6, 25–42. [Google Scholar] [CrossRef]
- Jin, K.-T.; Du, W.-L.; Liu, Y.-Y.; Lan, H.-R.; Si, J.-X.; Mou, X.-Z. Oncolytic Virotherapy in Solid Tumors: The Challenges and Achievements. Cancers 2021, 13, 588. [Google Scholar] [CrossRef]
- He, X.; Yang, Y.; Li, L.; Zhang, P.; Guo, H.; Liu, N.; Yang, X.; Xu, F. Engineering Extracellular Matrix to Improve Drug Delivery for Cancer Therapy. Drug Discov. Today 2020, 25, 1727–1734. [Google Scholar] [CrossRef] [PubMed]
- Goradel, N.H.; Negahdari, B.; Ghorghanlu, S.; Jahangiri, S.; Arashkia, A. Strategies for Enhancing Intratumoral Spread of Oncolytic Adenoviruses. Pharmacol. Ther. 2020, 213, 107586. [Google Scholar] [CrossRef] [PubMed]
- Berkey, S.E.; Thorne, S.H.; Bartlett, D.L. Oncolytic Virotherapy and the Tumor Microenvironment. Adv. Exp. Med. Biol. 2017, 1036, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.R. Exploring the Genomes of Cancer Cells: Progress and Promise. Science 2011, 331, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Coley, W.B. The Treatment of Malignant Tumors by Repeated Inoculations of Erysipelas. With a Report of Ten Original Cases. 1893. Clin. Orthop. 1991, 262, 3–11. [Google Scholar]
- Coley, W.B. The Treatment of Inoperable Sarcoma by Bacterial Toxins (the Mixed Toxins of the Streptococcus Erysipelas and the Bacillus Prodigiosus). Proc. R. Soc. Med. 1910, 3, 1–48. [Google Scholar] [CrossRef]
- Coley, W.B. Late Results of the Treatment of Inoperable Sarcoma by the Mixed Toxins of Erysipelas and Bacillus Prodigiosus. Am. J. Med. Sci. 1906, 131, 375. [Google Scholar]
- Ozdemir, T.; Fedorec, A.J.H.; Danino, T.; Barnes, C.P. Synthetic Biology and Engineered Live Biotherapeutics: Toward Increasing System Complexity. Cell Syst. 2018, 7, 5–16. [Google Scholar] [CrossRef]
- Felgner, S.; Kocijancic, D.; Frahm, M.; Heise, U.; Rohde, M.; Zimmermann, K.; Falk, C.; Erhardt, M.; Weiss, S. Engineered Salmonella Enterica Serovar Typhimurium Overcomes Limitations of Anti-Bacterial Immunity in Bacteria-Mediated Tumor Therapy. Oncoimmunology 2018, 7, e1382791. [Google Scholar] [CrossRef]
- Dailey, K.M.; Jacobson, R.I.; Johnson, P.R.; Woolery, T.J.; Kim, J.; Jansen, R.J.; Mallik, S.; Brooks, A.E. Methods and Techniques to Facilitate the Development of Clostridium Novyi NT as an Effective, Therapeutic Oncolytic Bacteria. Front. Microbiol. 2021, 12, 624618. [Google Scholar] [CrossRef]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 Signal Transduction Pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Eklund, M.W.; Poysky, F.T.; Meyers, J.A.; Pelroy, G.A. Interspecies Conversion of Clostridium Botulinum Type C to Clostridium Novyi Type A by Bacteriophage. Science 1974, 186, 456–458. [Google Scholar] [CrossRef]
- Eklund, M.W.; Poysky, F.T.; Peterson, M.E.; Meyers, J.A. Relationship of Bacteriophages to Alpha Toxin Production in Clostridium Novyi Types A and B. Infect. Immun. 1976, 14, 793–803. [Google Scholar] [CrossRef]
- Dang, L.H.; Bettegowda, C.; Huso, D.L.; Kinzler, K.W.; Vogelstein, B. Combination Bacteriolytic Therapy for the Treatment of Experimental Tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 15155–15160. [Google Scholar] [CrossRef]
- Andino, A.; Hanning, I. Salmonella Enterica: Survival, Colonization, and Virulence Differences among Serovars. Sci. World J. 2015, 2015, 520179. [Google Scholar] [CrossRef]
- Na, H.S.; Kim, H.J.; Lee, H.-C.; Hong, Y.; Rhee, J.H.; Choy, H.E. Immune Response Induced by Salmonella Typhimurium Defective in PpGpp Synthesis. Vaccine 2006, 24, 2027–2034. [Google Scholar] [CrossRef]
- Jeong, J.-H.; Song, M.; Park, S.-I.; Cho, K.-O.; Rhee, J.H.; Choy, H.E. Salmonella Enterica Serovar Gallinarum Requires PpGpp for Internalization and Survival in Animal Cells. J. Bacteriol. 2008, 190, 6340–6350. [Google Scholar] [CrossRef]
- Toso, J.F.; Gill, V.J.; Hwu, P.; Marincola, F.M.; Restifo, N.P.; Schwartzentruber, D.J.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Stock, F.; et al. Phase I Study of the Intravenous Administration of Attenuated Salmonella Typhimurium to Patients with Metastatic Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2002, 20, 142–152. [Google Scholar] [CrossRef]
- Morales, A. BCG: A Throwback from the Stone Age of Vaccines Opened the Path for Bladder Cancer Immunotherapy. Can. J. Urol. 2017, 24, 8788–8793. [Google Scholar]
- Shintani, Y.; Sawada, Y.; Inagaki, T.; Kohjimoto, Y.; Uekado, Y.; Shinka, T. Intravesical Instillation Therapy with Bacillus Calmette-Guérin for Superficial Bladder Cancer: Study of the Mechanism of Bacillus Calmette-Guérin Immunotherapy. Int. J. Urol. 2007, 14, 140–146. [Google Scholar] [CrossRef]
- Eisenstark, A.; Kazmierczak, R.A.; Dino, A.; Khreis, R.; Newman, D.; Schatten, H. Development of Salmonella Strains as Cancer Therapy Agents and Testing in Tumor Cell Lines. Methods Mol. Biol. 2007, 394, 323–354. [Google Scholar] [CrossRef]
- Forbes, N.S. Engineering the Perfect (Bacterial) Cancer Therapy. Nat. Rev. Cancer 2010, 10, 785–794. [Google Scholar] [CrossRef]
- Lee, C.-H.; Lin, S.-T.; Liu, J.-J.; Chang, W.-W.; Hsieh, J.-L.; Wang, W.-K. Salmonella Induce Autophagy in Melanoma by the Downregulation of AKT/MTOR Pathway. Gene Ther. 2014, 21, 309–316. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, L.; Hoffman, R.M.; Zhao, M. Vessel Destruction by Tumor-Targeting Salmonella Typhimurium A1-R Is Enhanced by High Tumor Vascularity. Cell Cycle Georget. Tex 2010, 9, 4518–4524. [Google Scholar] [CrossRef] [PubMed]
- Al-Ramadi, B.K.; Fernandez-Cabezudo, M.J.; El-Hasasna, H.; Al-Salam, S.; Bashir, G.; Chouaib, S. Potent Anti-Tumor Activity of Systemically-Administered IL2-Expressing Salmonella Correlates with Decreased Angiogenesis and Enhanced Tumor Apoptosis. Clin. Immunol. 2009, 130, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Spector, M.P.; Garcia Del Portillo, F.; Bearson, S.M.D.; Mahmud, A.; Magut, M.; Finlay, B.B.; Dougan, G.; Foster, J.W.; Pallen, M.J. The RpoS-Dependent Starvation-Stress Response Locus StiA Encodes a Nitrate Reductase (NarZYWV) Required for Carbon-Starvation-Inducible Thermotolerance and Acid Tolerance in Salmonella Typhimurium. Microbiol. Read. Engl. 1999, 145 Pt 11, 3035–3045. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Barak, Y.; Schreiber, F.; Thorne, S.H.; Contag, C.H.; Debeer, D.; Matin, A. Role of Nitric Oxide in Salmonella Typhimurium-Mediated Cancer Cell Killing. BMC Cancer 2010, 10, 146. [Google Scholar] [CrossRef] [PubMed]
- Kaimala, S.; Al-Sbiei, A.; Cabral-Marques, O.; Fernandez-Cabezudo, M.J.; Al-Ramadi, B.K. Attenuated Bacteria as Immunotherapeutic Tools for Cancer Treatment. Front. Oncol. 2018, 8, 136. [Google Scholar] [CrossRef]
- Chen, J.; Qiao, Y.; Tang, B.; Chen, G.; Liu, X.; Yang, B.; Wei, J.; Zhang, X.; Cheng, X.; Du, P.; et al. Modulation of Salmonella Tumor-Colonization and Intratumoral Anti-Angiogenesis by Triptolide and Its Mechanism. Theranostics 2017, 7, 2250–2260. [Google Scholar] [CrossRef]
- Grille, S.; Moreno, M.; Bascuas, T.; Marqués, J.M.; Muñoz, N.; Lens, D.; Chabalgoity, J.A. Salmonella Enterica Serovar Typhimurium Immunotherapy for B-Cell Lymphoma Induces Broad Anti-Tumour Immunity with Therapeutic Effect. Immunology 2014, 143, 428–437. [Google Scholar] [CrossRef]
- Cai, Z.; Sanchez, A.; Shi, Z.; Zhang, T.; Liu, M.; Zhang, D. Activation of Toll-like Receptor 5 on Breast Cancer Cells by Flagellin Suppresses Cell Proliferation and Tumor Growth. Cancer Res. 2011, 71, 2466–2475. [Google Scholar] [CrossRef]
- Chang, S.-Y.; Kim, Y.-J.; Ko, H.-J. Potential Therapeutic Anti-Tumor Effect of a Salmonella-Based Vaccine. Hum. Vaccines Immunother. 2013, 9, 1654–1660. [Google Scholar] [CrossRef][Green Version]
- Quispe-Tintaya, W.; Chandra, D.; Jahangir, A.; Harris, M.; Casadevall, A.; Dadachova, E.; Gravekamp, C. Nontoxic Radioactive Listeriaat Is a Highly Effective Therapy against Metastatic Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 8668–8673. [Google Scholar] [CrossRef]
- Forbes, N.S.; Munn, L.L.; Fukumura, D.; Jain, R.K. Sparse Initial Entrapment of Systemically Injected Salmonella Typhimurium Leads to Heterogeneous Accumulation within Tumors. Cancer Res. 2003, 63, 5188–5193. [Google Scholar]
- Hoffman, R.M. Tumor-Targeting Amino Acid Auxotrophic Salmonella Typhimurium. Amino Acids 2009, 37, 509–521. [Google Scholar] [CrossRef]
- Kasinskas, R.W.; Forbes, N.S. Salmonella Typhimurium Lacking Ribose Chemoreceptors Localize in Tumor Quiescence and Induce Apoptosis. Cancer Res. 2007, 67, 3201–3209. [Google Scholar] [CrossRef]
- Zhao, M.; Yang, M.; Ma, H.; Li, X.; Tan, X.; Li, S.; Yang, Z.; Hoffman, R.M. Targeted Therapy with a Salmonella Typhimurium Leucine-Arginine Auxotroph Cures Orthotopic Human Breast Tumors in Nude Mice. Cancer Res. 2006, 66, 7647–7652. [Google Scholar] [CrossRef]
- Zhao, M.; Yang, M.; Li, X.-M.; Jiang, P.; Baranov, E.; Li, S.; Xu, M.; Penman, S.; Hoffman, R.M. Tumor-Targeting Bacterial Therapy with Amino Acid Auxotrophs of GFP-Expressing Salmonella Typhimurium. Proc. Natl. Acad. Sci. USA 2005, 102, 755–760. [Google Scholar] [CrossRef]
- Sun, C.; Wang, H.; Mao, S.; Liu, J.; Li, S.; Wang, J. Reactive Oxygen Species Involved in CT26 Immunogenic Cell Death Induced by Clostridium Difficile Toxin B. Immunol. Lett. 2015, 164, 65–71. [Google Scholar] [CrossRef]
- Ganai, S.; Arenas, R.B.; Sauer, J.P.; Bentley, B.; Forbes, N.S. In Tumors Salmonella Migrate Away from Vasculature toward the Transition Zone and Induce Apoptosis. Cancer Gene Ther. 2011, 18, 457–466. [Google Scholar] [CrossRef]
- Kim, S.H.; Castro, F.; Paterson, Y.; Gravekamp, C. High Efficacy of a Listeria-Based Vaccine against Metastatic Breast Cancer Reveals a Dual Mode of Action. Cancer Res. 2009, 69, 5860–5866. [Google Scholar] [CrossRef]
- Diaz, L.A., Jr.; Cheong, I.; Foss, C.A.; Zhang, X.; Peters, B.A.; Agrawal, N.; Bettegowda, C.; Karim, B.; Liu, G.; Khan, K.; et al. Pharmacologic and Toxicologic Evaluation of C. Novyi-NT Spores. Toxicol. Sci. 2005, 88, 562–575. [Google Scholar] [CrossRef]
- Staedtke, V.; Bai, R.-Y.; Sun, W.; Huang, J.; Kibler, K.K.; Tyler, B.M.; Gallia, G.L.; Kinzler, K.; Vogelstein, B.; Zhou, S.; et al. Clostridium Novyi-NT Can Cause Regression of Orthotopically Implanted Glioblastomas in Rats. Oncotarget 2015, 6, 5536–5546. [Google Scholar] [CrossRef]
- Sznol, M.; Lin, S.L.; Bermudes, D.; Zheng, L.M.; King, I. Use of Preferentially Replicating Bacteria for the Treatment of Cancer. J. Clin. Investig. 2000, 105, 1027–1030. [Google Scholar] [CrossRef]
- DeClue, A.E.; Axiak-Bechtel, S.M.; Zhang, Y.; Saha, S.; Zhang, L.; Tung, D.; Bryan, J.N. Immune Response to C. Novyi-NT Immunotherapy. Vet. Res. 2018, 49, 38. [Google Scholar] [CrossRef]
- Dang, L.H.; Bettegowda, C.; Agrawal, N.; Cheong, I.; Huso, D.; Frost, P.; Loganzo, F.; Greenberger, L.; Barkoczy, J.; Pettit, G.R.; et al. Targeting Vascular and Avascular Compartments of Tumors with C. Novyi-NT and Anti-Microtubule Agents. Cancer Biol. Ther. 2004, 3, 326–337. [Google Scholar] [CrossRef][Green Version]
- Agrawal, N.; Bettegowda, C.; Cheong, I.; Geschwind, J.-F.; Drake, C.G.; Hipkiss, E.L.; Tatsumi, M.; Dang, L.H.; Diaz, L.A.; Pomper, M.; et al. Bacteriolytic Therapy Can Generate a Potent Immune Response against Experimental Tumors. Proc. Natl. Acad. Sci. USA 2004, 101, 15172–15177. [Google Scholar] [CrossRef]
- Roberts, N.J.; Zhang, L.; Janku, F.; Collins, A.; Bai, R.-Y.; Staedtke, V.; Rusk, A.W.; Tung, D.; Miller, M.; Roix, J.; et al. Intratumoral Injection of Clostridium Novyi-NT Spores Induces Antitumor Responses. Sci. Transl. Med. 2014, 6, 249ra111. [Google Scholar] [CrossRef]
- Al-Saafeen, B.H.; Fernandez-Cabezudo, M.J.; al-Ramadi, B.K. Integration of Salmonella into Combination Cancer Therapy. Cancers 2021, 13, 3228. [Google Scholar] [CrossRef]
- Low, K.B.; Ittensohn, M.; Luo, X.; Zheng, L.-M.; King, I.; Pawelek, J.M.; Bermudes, D. Construction of VNP20009: A Novel, Genetically Stable Antibiotic-Sensitive Strain of Tumor-Targeting Salmonella for Parenteral Administration in Humans. Methods Mol. Med. 2004, 90, 47–60. [Google Scholar]
- Clairmont, C.; Lee, K.C.; Pike, J.; Ittensohn, M.; Low, K.B.; Pawelek, J.; Bermudes, D.; Brecher, S.M.; Margitich, D.; Turnier, J.; et al. Biodistribution and Genetic Stability of the Novel Antitumor Agent VNP20009, a Genetically Modified Strain of Salmonella Typhimurium. J. Infect. Dis. 2000, 181, 1996–2002. [Google Scholar] [CrossRef]
- Chang, C.-H.; Cheng, W.-J.; Chen, S.-Y.; Kao, M.-C.; Chiang, C.-J.; Chao, Y.-P. Engineering of Escherichia Coli for Targeted Delivery of Transgenes to HER2/Neu-Positive Tumor Cells. Biotechnol. Bioeng. 2011, 108, 1662–1672. [Google Scholar] [CrossRef] [PubMed]
- Massa, P.E.; Paniccia, A.; Monegal, A.; de Marco, A.; Rescigno, M. Salmonella Engineered to Express CD20-Targeting Antibodies and a Drug-Converting Enzyme Can Eradicate Human Lymphomas. Blood 2013, 122, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-H.; Zheng, J.H.; Nguyen, V.H.; Jiang, S.-N.; Kim, D.-Y.; Szardenings, M.; Min, J.H.; Hong, Y.; Choy, H.E.; Min, J.-J. RGD Peptide Cell-Surface Display Enhances the Targeting and Therapeutic Efficacy of Attenuated Salmonella-Mediated Cancer Therapy. Theranostics 2016, 6, 1672–1682. [Google Scholar] [CrossRef] [PubMed]
- Badie, F.; Ghandali, M.; Tabatabaei, S.A.; Safari, M.; Khorshidi, A.; Shayestehpour, M.; Mahjoubian-Tehran, M.; Morshedi, K.; Jalili, A.; Tajiknia, V.; et al. Use of Salmonella Bacteria in Cancer Therapy: Direct, Drug Delivery and Combination Approaches. Front. Oncol. 2021, 11, 624759. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, X.; Guo, Y.; Wang, J.; Zhang, D.; Mei, Y.; Shi, J.; Tan, W.; Zheng, J.H. Genetically Engineered Oncolytic Bacteria as Drug Delivery Systems for Targeted Cancer Theranostics. Acta Biomater. 2021, 124, 72–87. [Google Scholar] [CrossRef]
- Liang, K.; Liu, Q.; Li, P.; Han, Y.; Bian, X.; Tang, Y.; Kong, Q. Endostatin Gene Therapy Delivered by Attenuated Salmonella Typhimurium in Murine Tumor Models. Cancer Gene Ther. 2018, 25, 167–183. [Google Scholar] [CrossRef]
- Wall, D.M.; Srikanth, C.V.; McCormick, B.A. Targeting Tumors with Salmonella Typhimurium—Potential for Therapy. Oncotarget 2011, 1, 721–728. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Cunningham, C.; Senzer, N.; Kuhn, J.; Cramm, J.; Litz, C.; Cavagnolo, R.; Cahill, A.; Clairmont, C.; Sznol, M. Pilot Trial of Genetically Modified, Attenuated Salmonella Expressing the E. Coli Cytosine Deaminase Gene in Refractory Cancer Patients. Cancer Gene Ther. 2003, 10, 737–744. [Google Scholar] [CrossRef]
- Vacchelli, E.; Aranda, F.; Obrist, F.; Eggermont, A.; Galon, J.; Cremer, I.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Immunostimulatory Cytokines in Cancer Therapy. Oncoimmunology 2014, 3, e29030. [Google Scholar] [CrossRef]
- Camacho, E.M.; Mesa-Pereira, B.; Medina, C.; Flores, A.; Santero, E. Engineering Salmonella as Intracellular Factory for Effective Killing of Tumour Cells. Sci. Rep. 2016, 6, 30591. [Google Scholar] [CrossRef]
- Royo, J.L.; Becker, P.D.; Camacho, E.M.; Cebolla, A.; Link, C.; Santero, E.; Guzmán, C.A. In Vivo Gene Regulation in Salmonella Spp. by a Salicylate-Dependent Control Circuit. Nat. Methods 2007, 4, 937–942. [Google Scholar] [CrossRef]
- Loessner, H.; Endmann, A.; Leschner, S.; Westphal, K.; Rohde, M.; Miloud, T.; Hämmerling, G.; Neuhaus, K.; Weiss, S. Remote Control of Tumour-Targeted Salmonella Enterica Serovar Typhimurium by the Use of L-Arabinose as Inducer of Bacterial Gene Expression in Vivo. Cell. Microbiol. 2007, 9, 1529–1537. [Google Scholar] [CrossRef]
- Stritzker, J.; Weibel, S.; Hill, P.J.; Oelschlaeger, T.A.; Goebel, W.; Szalay, A.A. Tumor-Specific Colonization, Tissue Distribution, and Gene Induction by Probiotic Escherichia Coli Nissle 1917 in Live Mice. Int. J. Med. Microbiol. IJMM 2007, 297, 151–162. [Google Scholar] [CrossRef]
- Ryan, R.M.; Green, J.; Williams, P.J.; Tazzyman, S.; Hunt, S.; Harmey, J.H.; Kehoe, S.C.; Lewis, C.E. Bacterial Delivery of a Novel Cytolysin to Hypoxic Areas of Solid Tumors. Gene Ther. 2009, 16, 329–339. [Google Scholar] [CrossRef]
- Mengesha, A.; Dubois, L.; Lambin, P.; Landuyt, W.; Chiu, R.K.; Wouters, B.G.; Theys, J. Development of a Flexible and Potent Hypoxia-Inducible Promoter for Tumor-Targeted Gene Expression in Attenuated Salmonella. Cancer Biol. Ther. 2006, 5, 1120–1128. [Google Scholar] [CrossRef]
- Arrach, N.; Zhao, M.; Porwollik, S.; Hoffman, R.M.; McClelland, M. Salmonella Promoters Preferentially Activated inside Tumors. Cancer Res. 2008, 68, 4827–4832. [Google Scholar] [CrossRef]
- Din, M.O.; Danino, T.; Prindle, A.; Skalak, M.; Selimkhanov, J.; Allen, K.; Julio, E.; Atolia, E.; Tsimring, L.S.; Bhatia, S.N.; et al. Synchronized Cycles of Bacterial Lysis for in Vivo Delivery. Nature 2016, 536, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Saltzman, D.; Augustin, L.; Leonard, A.; Mertensotto, M.; Schottel, J. Low Dose Chemotherapy Combined with Attenuated Salmonella Decreases Tumor Burden and Is Less Toxic than High Dose Chemotherapy in an Autochthonous Murine Model of Breast Cancer. Surgery 2018, 163, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Wu, C.-L.; Tai, Y.-S.; Shiau, A.-L. Systemic Administration of Attenuated Salmonella Choleraesuis in Combination with Cisplatin for Cancer Therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2005, 11, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, K.; Miyake, K.; Zhao, M.; Kiyuna, T.; Igarashi, K.; Miyake, M.; Higuchi, T.; Oshiro, H.; Bouvet, M.; Unno, M.; et al. Tumor Targeting Salmonella Typhimurium A1-R in Combination with Gemcitabine (GEM) Regresses Partially GEM-Resistant Pancreatic Cancer Patient-Derived Orthotopic Xenograft (PDOX) Nude Mouse Models. Cell Cycle Georget. 2018, 17, 2019–2026. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.-J.; Wei, D.-P.; Sun, Q.-M.; Jin, G.-H.; Li, S.-F.; Huang, Y.; Hua, Z.-C. Tumor-Targeting Salmonella Typhimurium Improves Cyclophosphamide Chemotherapy at Maximum Tolerated Dose and Low-Dose Metronomic Regimens in a Murine Melanoma Model. Int. J. Cancer 2007, 121, 666–674. [Google Scholar] [CrossRef]
- Bascuas, T.; Moreno, M.; Grille, S.; Chabalgoity, J.A. Salmonella Immunotherapy Improves the Outcome of CHOP Chemotherapy in Non-Hodgkin Lymphoma-Bearing Mice. Front. Immunol. 2018, 9, 7. [Google Scholar] [CrossRef]
- Yoon, W.S.; Kim, S.; Seo, S.; Park, Y. Salmonella Typhimurium with γ-Radiation Induced H2AX Phosphorylation and Apoptosis in Melanoma. Biosci. Biotechnol. Biochem. 2014, 78, 1082–1085. [Google Scholar] [CrossRef]
- Bettegowda, C.; Dang, L.H.; Abrams, R.; Huso, D.L.; Dillehay, L.; Cheong, I.; Agrawal, N.; Borzillary, S.; McCaffery, J.M.; Watson, E.L.; et al. Overcoming the Hypoxic Barrier to Radiation Therapy with Anaerobic Bacteria. Proc. Natl. Acad. Sci. USA 2003, 100, 15083–15088. [Google Scholar] [CrossRef]
- Chen, W.; Wang, Y.; Qin, M.; Zhang, X.; Zhang, Z.; Sun, X.; Gu, Z. Bacteria-Driven Hypoxia Targeting for Combined Biotherapy and Photothermal Therapy. ACS Nano 2018, 12, 5995–6005. [Google Scholar] [CrossRef]
- Kefayat, A.; Ghahremani, F.; Motaghi, H.; Rostami, S.; Mehrgardi, M.A. Alive Attenuated Salmonella as a Cargo Shuttle for Smart Carrying of Gold Nanoparticles to Tumour Hypoxic Regions. J. Drug Target. 2019, 27, 315–324. [Google Scholar] [CrossRef]
- Chen, M.-C.; Pangilinan, C.R.; Lee, C.-H. Salmonella Breaks Tumor Immune Tolerance by Downregulating Tumor Programmed Death-Ligand 1 Expression. Cancers 2019, 12, 57. [Google Scholar] [CrossRef]
- Ebelt, N.D.; Zuniga, E.; Marzagalli, M.; Zamloot, V.; Blazar, B.R.; Salgia, R.; Manuel, E.R. Salmonella-Based Therapy Targeting Indoleamine 2,3-Dioxygenase Restructures the Immune Contexture to Improve Checkpoint Blockade Efficacy. Biomedicines 2020, 8, 617. [Google Scholar] [CrossRef]
- Zhao, T.; Wei, T.; Guo, J.; Wang, Y.; Shi, X.; Guo, S.; Jia, X.; Jia, H.; Feng, Z. PD-1-SiRNA Delivered by Attenuated Salmonella Enhances the Antimelanoma Effect of Pimozide. Cell Death Dis. 2019, 10, 164. [Google Scholar] [CrossRef]
- Zhao, T.; Feng, Y.; Guo, M.; Zhang, C.; Wu, Q.; Chen, J.; Guo, S.; Liu, S.; Zhou, Q.; Wang, Z.; et al. Combination of Attenuated Salmonella Carrying PD-1 SiRNA with Nifuroxazide for Colon Cancer Therapy. J. Cell. Biochem. 2020, 121, 1973–1985. [Google Scholar] [CrossRef]
- Mignon, C.; Sodoyer, R.; Werle, B. Antibiotic-Free Selection in Biotherapeutics: Now and Forever. Pathogens 2015, 4, 157–181. [Google Scholar] [CrossRef]
- Farrow, B.; Albo, D.; Berger, D.H. The Role of the Tumor Microenvironment in the Progression of Pancreatic Cancer. J. Surg. Res. 2008, 149, 319–328. [Google Scholar] [CrossRef]
- Tai, Y.; Chen, K.; Hsieh, J.; Shen, T. Exosomes in Cancer Development and Clinical Applications. Cancer Sci. 2018, 109, 2364–2374. [Google Scholar] [CrossRef]
- Dai, J.; Su, Y.; Zhong, S.; Cong, L.; Liu, B.; Yang, J.; Tao, Y.; He, Z.; Chen, C.; Jiang, Y. Exosomes: Key Players in Cancer and Potential Therapeutic Strategy. Signal Transduct. Target. Ther. 2020, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, M.; Le’Negrate, G.; Krajewska, M.; Reed, J.C. Inhibition of Tumor Growth Using Salmonella Expressing Fas Ligand. JNCI J. Natl. Cancer Inst. 2008, 100, 1113–1116. [Google Scholar] [CrossRef] [PubMed]
- Shafiee, F.; Aucoin, M.G.; Jahanian-Najafabadi, A. Targeted Diphtheria Toxin-Based Therapy: A Review Article. Front. Microbiol. 2019, 10, 2340. [Google Scholar] [CrossRef] [PubMed]
- Cheung, L.S.; Fu, J.; Kumar, P.; Kumar, A.; Urbanowski, M.E.; Ihms, E.A.; Parveen, S.; Bullen, C.K.; Patrick, G.J.; Harrison, R.; et al. Second-Generation IL-2 Receptor-Targeted Diphtheria Fusion Toxin Exhibits Antitumor Activity and Synergy with Anti–PD-1 in Melanoma. Proc. Natl. Acad. Sci. USA 2019, 116, 3100–3105. [Google Scholar] [CrossRef]
- Cassavaugh, J.; Lounsbury, K.M. Hypoxia-Mediated Biological Control. J. Cell. Biochem. 2011, 112, 735–744. [Google Scholar] [CrossRef]
- Cooper, J.; Giancotti, F.G. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in Cancer: Biological Implications and Therapeutic Opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef]
- Chen, K.; Chen, X. Integrin Targeted Delivery of Chemotherapeutics. Theranostics 2011, 1, 189–200. [Google Scholar] [CrossRef]
- Wu, P.-H.; Opadele, A.E.; Onodera, Y.; Nam, J.-M. Targeting Integrins in Cancer Nanomedicine: Applications in Cancer Diagnosis and Therapy. Cancers 2019, 11, 1783. [Google Scholar] [CrossRef]
- Aumailley, M.; Gurrath, M.; Müller, G.; Calvete, J.; Timpl, R.; Kessler, H. Arg-Gly-Asp Constrained within Cyclic Pentapoptides Strong and Selective Inhibitors of Cell Adhesion to Vitronectin and Laminin Fragment P1. FEBS Lett. 1991, 291, 50–54. [Google Scholar] [CrossRef]
- Haubner, R.; Gratias, R.; Diefenbach, B.; Goodman, S.L.; Jonczyk, A.; Kessler, H. Structural and Functional Aspects of RGD-Containing Cyclic Pentapeptides as Highly Potent and Selective Integrin AVβ3 Antagonists. J. Am. Chem. Soc. 1996, 118, 7461–7472. [Google Scholar] [CrossRef]
- Koivunen, E.; Wang, B.; Ruoslahti, E. Phage Libraries Displaying Cyclic Peptides with Different Ring Sizes: Ligand Specificities of the RGD-Directed Integrins. Biotechnol. Nat. 1995, 13, 265–270. [Google Scholar] [CrossRef]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Girard, O.M.; Hanahan, D.; Mattrey, R.F.; Ruoslahti, E. Tissue-Penetrating Delivery of Compounds and Nanoparticles into Tumors. Cancer Cell 2009, 16, 510–520. [Google Scholar] [CrossRef]
- Arap, W.; Pasqualini, R.; Ruoslahti, E. Cancer Treatment by Targeted Drug Delivery to Tumor Vasculature in a Mouse Model. Science 1998, 279, 377–380. [Google Scholar] [CrossRef]
- Hausner, S.H.; Bold, R.J.; Cheuy, L.Y.; Chew, H.K.; Daly, M.E.; Davis, R.A.; Foster, C.C.; Kim, E.J.; Sutcliffe, J.L. Preclinical Development and First-in-Human Imaging of the Integrin Avβ6 with [18F]Avβ6-Binding Peptide in Metastatic Carcinoma. Clin. Cancer Res. 2019, 25, 1206–1215. [Google Scholar] [CrossRef]
- Guo, Z.S.; Liu, Z.; Bartlett, D.L. Oncolytic Immunotherapy: Dying the Right Way Is a Key to Eliciting Potent Antitumor Immunity. Front. Oncol. 2014, 4, 74. [Google Scholar] [CrossRef]
- Tian, Q.; Bagheri, Y.; Keshri, P.; Wu, R.; Ren, K.; Yu, Q.; Zhao, B.; You, M. Efficient and Selective DNA Modification on Bacterial Membranes. Chem. Sci. 2021, 12, 2629–2634. [Google Scholar] [CrossRef]
- Orlando, I.; Basnett, P.; Nigmatullin, R.; Wang, W.; Knowles, J.C.; Roy, I. Chemical Modification of Bacterial Cellulose for the Development of an Antibacterial Wound Dressing. Front. Bioeng. Biotechnol. 2020, 8, 1072. [Google Scholar] [CrossRef]
- Filley, A.C.; Dey, M. Immune System, Friend or Foe of Oncolytic Virotherapy? Front. Oncol. 2017, 7, 106. [Google Scholar] [CrossRef]
- Neagu, M.; Piperigkou, Z.; Karamanou, K.; Engin, A.B.; Docea, A.O.; Constantin, C.; Negrei, C.; Nikitovic, D.; Tsatsakis, A. Protein Bio-Corona: Critical Issue in Immune Nanotoxicology. Arch. Toxicol. 2017, 91, 1031–1048. [Google Scholar] [CrossRef]
- Coffey, M.; Strong, J.; Forsyth, P.; Lee, P. Reovirus Therapy of Tumors with Activated Ras Pathway. Science 1998, 282, 1332–1334. [Google Scholar] [CrossRef]
- Annels, N.E.; Mansfield, D.; Arif, M.; Ballesteros-Merino, C.; Simpson, G.R.; Denyer, M.; Sandhu, S.S.; Melcher, A.A.; Harrington, K.J.; Davies, B.; et al. Phase I Trial of an ICAM-1-Targeted Immunotherapeutic-Coxsackievirus A21 (CVA21) as an Oncolytic Agent Against Non Muscle-Invasive Bladder Cancer. Clin. Cancer Res. 2019, 25, 5818–5831. [Google Scholar] [CrossRef]
- Delaunay, T.; Achard, C.; Boisgerault, N.; Grard, M.; Petithomme, T.; Chatelain, C.; Dutoit, S.; Blanquart, C.; Royer, P.-J.; Minvielle, S.; et al. Frequent Homozygous Deletions of Type I Interferon Genes in Pleural Mesothelioma Confer Sensitivity to Oncolytic Measles Virus. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2020, 15, 827–842. [Google Scholar] [CrossRef]
- Dirkx, A.E.M.; Oude Egbrink, M.G.A.; Wagstaff, J.; Griffioen, A.W. Monocyte/Macrophage Infiltration in Tumors: Modulators of Angiogenesis. J. Leukoc. Biol. 2006, 80, 1183–1196. [Google Scholar] [CrossRef] [PubMed]
- Fend, L.; Yamazaki, T.; Remy, C.; Fahrner, C.; Gantzer, M.; Nourtier, V.; Préville, X.; Quéméneur, E.; Kepp, O.; Adam, J.; et al. Immune Checkpoint Blockade, Immunogenic Chemotherapy or IFN-α Blockade Boost the Local and Abscopal Effects of Oncolytic Virotherapy. Cancer Res. 2017, 77, 4146–4157. [Google Scholar] [CrossRef] [PubMed]
- Sarinella, F.; Calistri, A.; Sette, P.; Palù, G.; Parolin, C. Oncolysis of Pancreatic Tumour Cells by a Gamma34.5-Deleted HSV-1 Does Not Rely upon Ras-Activation, but on the PI 3-Kinase Pathway. Gene Ther. 2006, 13, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.; Liu, Q.; Li, P.; Luo, H.; Wang, H.; Kong, Q. Genetically Engineered Salmonella Typhimurium: Recent Advances in Cancer Therapy. Cancer Lett. 2019, 448, 168–181. [Google Scholar] [CrossRef]
- Guo, S.; Huang, L. Nanoparticles Escaping RES and Endosome: Challenges for SiRNA Delivery for Cancer Therapy. J. Nanomater. 2011, 2011, 742895. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Cunningham, C.; Tong, A.W.; Post, L.; Netto, G.; Paulson, A.S.; Rich, D.; Blackburn, A.; Sands, B.; Gibson, B.; et al. Pilot Trial of Intravenous Infusion of a Replication-Selective Adenovirus (ONYX-015) in Combination with Chemotherapy or IL-2 Treatment in Refractory Cancer Patients. Cancer Gene Ther. 2003, 10, 341–352. [Google Scholar] [CrossRef]
- Post, D.E.; Sandberg, E.M.; Kyle, M.M.; Devi, N.S.; Brat, D.J.; Xu, Z.; Tighiouart, M.; Van Meir, E.G. Targeted Cancer Gene Therapy Using a Hypoxia Inducible Factor Dependent Oncolytic Adenovirus Armed with Interleukin-4. Cancer Res. 2007, 67, 6872–6881. [Google Scholar] [CrossRef]
- Cattaneo, R.; Miest, T.; Shashkova, E.V.; Barry, M.A. Reprogrammed Viruses as Cancer Therapeutics: Targeted, Armed and Shielded. Nat. Rev. Microbiol. 2008, 6, 529–540. [Google Scholar] [CrossRef]
- Wang, Y.; Shang, W.; Niu, M.; Tian, J.; Xu, K. Hypoxia-Active Nanoparticles Used in Tumor Theranostic. Int. J. Nanomed. 2019, 14, 3705–3722. [Google Scholar] [CrossRef]
- Zhou, M.; Xie, Y.; Xu, S.; Xin, J.; Wang, J.; Han, T.; Ting, R.; Zhang, J.; An, F. Hypoxia-Activated Nanomedicines for Effective Cancer Therapy. Eur. J. Med. Chem. 2020, 195, 112274. [Google Scholar] [CrossRef]
- Erdogar, N.; İskit, A.B.; Eroglu, H.; Sargon, M.F.; Mungan, N.A.; Bilensoy, E. Cationic Core-Shell Nanoparticles for Intravesical Chemotherapy in Tumor-Induced Rat Model: Safety and Efficacy. Int. J. Pharm. 2014, 471, 1–9. [Google Scholar] [CrossRef]
- Caramés Masana, F.; de Reijke, T.M. The Efficacy of Apaziquone in the Treatment of Bladder Cancer. Expert Opin. Pharmacother. 2017, 18, 1781–1788. [Google Scholar] [CrossRef]
- Pierce, S.E.; Guziec, L.J.; Guziec, F.S.; Brodbelt, J.S. Characterization of aziridinylbenzoquinone DNA cross-links by liquid chromatography-infrared multiphoton dissociation-mass spectrometry. Chem. Res. Toxicol. 2010, 23, 1097–1104. [Google Scholar] [CrossRef]
- Qian, C.; Yu, J.; Chen, Y.; Hu, Q.; Xiao, X.; Sun, W.; Wang, C.; Feng, P.; Shen, Q.-D.; Gu, Z. Light-Activated Hypoxia-Responsive Nanocarriers for Enhanced Anticancer Therapy. Adv. Mater. 2016, 28, 3313–3320. [Google Scholar] [CrossRef]
- Nishida, C.R.; Ortiz de Montellano, P.R. Reductive Heme-Dependent Activation of the N-Oxide Prodrug AQ4N by Nitric Oxide Synthase. J. Med. Chem. 2008, 51, 5118–5120. [Google Scholar] [CrossRef]
- McCarthy, H.O.; Yakkundi, A.; McErlane, V.; Hughes, C.M.; Keilty, G.; Murray, M.; Patterson, L.H.; Hirst, D.G.; McKeown, S.R.; Robson, T. Bioreductive GDEPT Using Cytochrome P450 3A4 in Combination with AQ4N. Cancer Gene Ther. 2003, 10, 40–48. [Google Scholar] [CrossRef][Green Version]
- Knox, H.J.; Hedhli, J.; Kim, T.W.; Khalili, K.; Dobrucki, L.W.; Chan, J. A bioreproducible N-oxide-based probe for photoacoustic imaging of hypoxia. Nat. Commun. 2017, 8, 1794. [Google Scholar] [CrossRef]
- Peters, K.B.; Brown, J.M. Tirapazamine: A Hypoxia-Activated Topoisomerase II Poison. Cancer Res. 2002, 62, 5248–5253. [Google Scholar]
- Guo, D.; Xu, S.; Wang, N.; Jiang, H.; Huang, Y.; Jin, X.; Xue, B.; Zhang, C.; Zhu, X. Prodrug-Embedded Angiogenic Vessel-Targeting Nanoparticle: A Positive Feedback Amplifier in Hypoxia-Induced Chemo-Photo Therapy. Biomaterials 2017, 144, 188–198. [Google Scholar] [CrossRef]
- Wang, W.; Lin, L.; Ma, X.; Wang, B.; Liu, S.; Yan, X.; Li, S.; Tian, H.; Yu, X. Light-Induced Hypoxia-Triggered Living Nanocarriers for Synergistic Cancer Therapy. ACS Appl. Mater. Interfaces 2018, 10, 19398–19407. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, M.; Li, J.; Lan, S.; Zeng, Y.; Liu, X.; Liu, J. Light-Enhanced Hypoxia-Response of Conjugated Polymer Nanocarrier for Successive Synergistic Photodynamic and Chemo-Therapy. ACS Appl. Mater. Interfaces 2018, 10, 21909–21919. [Google Scholar] [CrossRef]
- Guo, D.; Xu, S.; Huang, Y.; Jiang, H.; Yasen, W.; Wang, N.; Su, Y.; Qian, J.; Li, J.; Zhang, C.; et al. Platinum(IV) complex-based two-in-one polyprodrug for a combinatorial chemo-photodynamic therapy. Biomaterials 2018, 177, 67–77. [Google Scholar] [CrossRef]
- Denny, W.A.; Wilson, W.R. Bioreducible Mustards: A Paradigm for Hypoxia-Selective Prodrugs of Diffusible Cytotoxins (HPDCs). Cancer Metastasis Rev. 1993, 12, 135–151. [Google Scholar] [CrossRef]
- Cheng, P.-H.; Wechman, S.L.; McMasters, K.M.; Zhou, H.S. Oncolytic Replication of E1b-Deleted Adenoviruses. Viruses 2015, 7, 5767–5779. [Google Scholar] [CrossRef]
- Connor, J.H.; Naczki, C.; Koumenis, C.; Lyles, D.S. Replication and Cytopathic Effect of Oncolytic Vesicular Stomatitis Virus in Hypoxic Tumor Cells in Vitro and in Vivo. J. Virol. 2004, 78, 8960–8970. [Google Scholar] [CrossRef]
- Dróżdż, M.; Makuch, S.; Cieniuch, G.; Woźniak, M.; Ziółkowski, P. Obligate and Facultative Anaerobic Bacteria in Targeted Cancer Therapy: Current Strategies and Clinical Applications. Life Sci. 2020, 261, 118296. [Google Scholar] [CrossRef]
- Dailey, K.M.; Allgood, J.E.; Johnson, P.R.; Ostlie, M.A.; Schaner, K.C.; Brooks, B.D.; Brooks, A.E. The next Frontier of Oncotherapy: Accomplishing Clinical Translation of Oncolytic Bacteria through Genetic Engineering. Future Microbiol. 2021, 16, 341–368. [Google Scholar] [CrossRef]
- Du, J.; Lane, L.A.; Nie, S. Stimuli-Responsive Nanoparticles for Targeting the Tumor Microenvironment. J. Control. Release Off. J. Control. Release Soc. 2015, 219, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Wang, Y.; Huang, X.; Luby-Phelps, K.K.; Sumer, B.D.; Gao, J. Tunable, Ultra-Sensitive PH Responsive Nanoparticles Targeting Specific Endocytic Organelles in Living Cells. Angew. Chem. Int. Ed. Engl. 2011, 50, 6109–6114. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.; Bae, Y.H. Recent Progress in Tumor PH Targeting Nanotechnology. J. Control. Release Off. J. Control. Release Soc. 2008, 132, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Du, J.-Z.; Mao, C.-Q.; Yuan, Y.-Y.; Yang, X.-Z.; Wang, J. Tumor Extracellular Acidity-Activated Nanoparticles as Drug Delivery Systems for Enhanced Cancer Therapy. Biotechnol. Adv. 2014, 32, 789–803. [Google Scholar] [CrossRef]
- Andreev, O.A.; Engelman, D.M.; Reshetnyak, Y.K. Targeting Diseased Tissues by PHLIP Insertion at Low Cell Surface PH. Front. Physiol. 2014, 5, 97. [Google Scholar] [CrossRef]
- Choi, J.W.; Jung, S.J.; Kasala, D.; Hwang, J.K.; Hu, J.; Bae, Y.H.; Yun, C.O. pH-sensitive oncolytic adenovirus hybrid targeting acidic tumor microenvironment and angiogenesis. J Control Release. 2015, 205, 134–143. [Google Scholar] [CrossRef]
- Moon, C.Y.; Choi, J.-W.; Kasala, D.; Jung, S.-J.; Kim, S.W.; Yun, C.-O. Dual Tumor Targeting with PH-Sensitive and Bioreducible Polymer-Complexed Oncolytic Adenovirus. Biomaterials 2015, 41, 53–68. [Google Scholar] [CrossRef]
- Kim, C.S.; Duncan, B.; Creran, B.; Rotello, V.M. Triggered Nanoparticles as Therapeutics. Nano Today 2013, 8, 439–447. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering Precision Nanoparticles for Drug Delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Cherukuri, P.; Glazer, E.S.; Curley, S.A. Targeted Hyperthermia Using Metal Nanoparticles. Adv. Drug Deliv. Rev. 2010, 62, 339–345. [Google Scholar] [CrossRef]
- Eisenberg, D.P.; Carpenter, S.G.; Adusumilli, P.S.; Chan, M.-K.; Hendershott, K.J.; Yu, Z.; Fong, Y. Hyperthermia Potentiates Oncolytic Herpes Viral Killing of Pancreatic Cancer through a Heat Shock Protein Pathway. Surgery 2010, 148, 325–334. [Google Scholar] [CrossRef]
- Tian, X.-L.; Yan, Z.; Chen, J.; Zhao, W.-H.; Guo, W. Clinical Application of Highly Agglutinative Staphylococcin in Cancer Treatment Updates of the Literature. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2718–2725. [Google Scholar]
- Rius-Rocabert, S.; Llinares Pinel, F.; Pozuelo, M.J.; García, A.; Nistal-Villan, E. Oncolytic Bacteria: Past, Present and Future. FEMS Microbiol. Lett. 2019, 366, fnz136. [Google Scholar] [CrossRef]
- Hamada, K.; Desaki, J.; Nakagawa, K.; Zhang, T.; Shirakawa, T.; Gotoh, A.; Tagawa, M. Carrier cell-mediated delivery of a replication-competent adenovirus for cancer gene therapy. Mol. Ther. 2007, 15, 1121–1128. [Google Scholar] [CrossRef]
- Komarova, S.; Kawakami, Y.; Stoff-Khalili, M.A.; Curiel, D.T.; Pereboeva, L. Mesenchymal Progenitor Cells as Cellular Vehicles for Delivery of Oncolytic Adenoviruses. Mol. Cancer Ther. 2006, 5, 755–766. [Google Scholar] [CrossRef]
- Lang, S.I.; Kottke, T.; Thompson, J.; Vile, R.G. Unbiased Selection of Bone Marrow Derived Cells as Carriers for Cancer Gene Therapy. J. Gene Med. 2007, 9, 927–937. [Google Scholar] [CrossRef]
- Wei, J.; Wahl, J.; Nakamura, T.; Stiller, D.; Mertens, T.; Debatin, K.-M.; Beltinger, C. Targeted Release of Oncolytic Measles Virus by Blood Outgrowth Endothelial Cells in Situ Inhibits Orthotopic Gliomas. Gene Ther. 2007, 14, 1573–1586. [Google Scholar] [CrossRef]
- Raykov, Z.; Balboni, G.; Aprahamian, M.; Rommelaere, J. Carrier Cell-Mediated Delivery of Oncolytic Parvoviruses for Targeting Metastases. Int. J. Cancer 2004, 109, 742–749. [Google Scholar] [CrossRef]
- García-Castro, J.; Martínez-Palacio, J.; Lillo, R.; García-Sánchez, F.; Alemany, R.; Madero, L.; Bueren, J.A.; Ramírez, M. Tumor Cells as Cellular Vehicles to Deliver Gene Therapies to Metastatic Tumors. Cancer Gene Ther. 2005, 12, 341–349. [Google Scholar] [CrossRef]
- Santos, J.; Heiniö, C.; Quixabeira, D.; Zafar, S.; Clubb, J.; Pakola, S.; Cervera-Carrascon, V.; Havunen, R.; Kanerva, A.; Hemminki, A. Systemic Delivery of Oncolytic Adenovirus to Tumors Using Tumor-Infiltrating Lymphocytes as Carriers. Cells 2021, 10, 978. [Google Scholar] [CrossRef]
- Dailey, K.; Brooks, A.; Jacobson, R.; Kim, J.; Mallik, S. Probing Clinical Relevance: Establishing the Efficacy of C. Novyi against a Panel of 2D Cultured Pancreatic Cancer Cells. Biomed. Sci. Instrum. 2021, 57, 93–99. [Google Scholar] [CrossRef]
- Kai, M.P.; Brighton, H.E.; Fromen, C.A.; Shen, T.W.; Luft, J.C.; Luft, Y.E.; Keeler, A.W.; Robbins, G.R.; Ting, J.P.Y.; Zamboni, W.C.; et al. Tumor Presence Induces Global Immune Changes and Enhances Nanoparticle Clearance. ACS Nano 2016, 10, 861–870. [Google Scholar] [CrossRef]
- Jones, S.W.; Roberts, R.A.; Robbins, G.R.; Perry, J.L.; Kai, M.P.; Chen, K.; Bo, T.; Napier, M.E.; Ting, J.P.Y.; DeSimone, J.M.; et al. Nanoparticle Clearance Is Governed by Th1/Th2 Immunity and Strain Background. J. Clin. Investig. 2013, 123, 3061–3073. [Google Scholar] [CrossRef]
- Müller, L.K.; Simon, J.; Rosenauer, C.; Mailänder, V.; Morsbach, S.; Landfester, K. The Transferability from Animal Models to Humans: Challenges Regarding Aggregation and Protein Corona Formation of Nanoparticles. Biomacromolecules 2018, 19, 374–385. [Google Scholar] [CrossRef]
- Cai, R.; Chen, C. The Crown and the Scepter: Roles of the Protein Corona in Nanomedicine. Adv. Mater. 2019, 31, 1805740. [Google Scholar] [CrossRef]
- Lundqvist, M.; Stigler, J.; Cedervall, T.; Berggård, T.; Flanagan, M.B.; Lynch, I.; Elia, G.; Dawson, K. The Evolution of the Protein Corona around Nanoparticles: A Test Study. ACS Nano 2011, 5, 7503–7509. [Google Scholar] [CrossRef]
- Corbo, C.; Molinaro, R.; Parodi, A.; Toledano Furman, N.E.; Salvatore, F.; Tasciotti, E. The Impact of Nanoparticle Protein Corona on Cytotoxicity, Immunotoxicity and Target Drug Delivery. Nanomedicines 2016, 11, 81–100. [Google Scholar] [CrossRef]
- Salvati, A.; Pitek, A.S.; Monopoli, M.P.; Prapainop, K.; Bombelli, F.B.; Hristov, D.R.; Kelly, P.M.; Åberg, C.; Mahon, E.; Dawson, K.A. Transferrin-Functionalized Nanoparticles Lose Their Targeting Capabilities When a Biomolecule Corona Adsorbs on the Surface. Nat. Nanotechnol. 2013, 8, 137–143. [Google Scholar] [CrossRef]
- Gräfe, C.; Weidner, A.; von der Lühe, M.; Bergemann, C.; Schacher, F.; Clement, J.; Dutz, S. Intentional Formation of a Protein Corona on Nanoparticles—Serum Concentration Affects Protein Corona Mass, Surface Charge, and Nanoparticle-Cell Interaction. Int. J. Biochem. Cell Biol. 2016, 75, 196–202. [Google Scholar] [CrossRef]
- Partikel, K.; Korte, R.; Mulac, D.; Humpf, H.-U.; Langer, K. Serum Type and Concentration Both Affect the Protein-Corona Composition of PLGA Nanoparticles. Beilstein J. Nanotechnol. 2019, 10, 1002–1015. [Google Scholar] [CrossRef]
- Mendt, M.; Kamerkar, S.; Sugimoto, H.; McAndrews, K.M.; Wu, C.-C.; Gagea, M.; Yang, S.; Blanko, E.V.R.; Peng, Q.; Ma, X.; et al. Generation and Testing of Clinical-Grade Exosomes for Pancreatic Cancer. JCI Insight 2018, 3, e99263. [Google Scholar] [CrossRef] [PubMed]
- Grenier, P.; de Viana, I.M.O.; Lima, E.M.; Bertrand, N. Anti-Polyethylene Glycol Antibodies Alter the Protein Corona Deposited on Nanoparticles and the Physiological Pathways Regulating Their Fate in Vivo. J. Control. Release 2018, 287, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Innate Immunity. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Mues, N.; Chu, H.W. Out-Smarting the Host: Bacteria Maneuvering the Immune Response to Favor Their Survival. Front. Immunol. 2020, 11, 819. [Google Scholar] [CrossRef] [PubMed]
- Minasyan, H. Mechanisms and Pathways for the Clearance of Bacteria from Blood Circulation in Health and Disease. Pathophysiology 2016, 23, 61–66. [Google Scholar] [CrossRef]
- Kell, A.M.; Gale, M. RIG-I in RNA Virus Recognition. Virology 2015, 479–480, 110–121. [Google Scholar] [CrossRef]
- Loo, Y.M.; Fornek, J.; Crochet, N.; Bajwa, G.; Perwitasari, O.; Martinez-Sobrido, L.; Akira, S.; Gill, M.A.; García-Sastre, A.; Katze, M.G.; et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 2008, 82, 335–45. [Google Scholar] [CrossRef]
- Liu, Y.; Cai, J.; Liu, W.; Lin, Y.; Guo, L.; Liu, X.; Qin, Z.; Xu, C.; Zhang, Y.; Su, X.; et al. Intravenous Injection of the Oncolytic Virus M1 Awakens Antitumor T Cells and Overcomes Resistance to Checkpoint Blockade. Cell Death Dis. 2020, 11, 1062. [Google Scholar] [CrossRef]
- Puzanov, I.; Milhem, M.M.; Minor, D.; Hamid, O.; Li, A.; Chen, L.; Chastain, M.; Gorski, K.S.; Anderson, A.; Chou, J.; et al. Talimogene Laherparepvec in Combination with Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J. Clin. Oncol. 2016, 34, 2619–2626. [Google Scholar] [CrossRef]
- Melcher, A.; Parato, K.; Rooney, C.M.; Bell, J.C. Thunder and Lightning: Immunotherapy and Oncolytic Viruses Collide. Mol. Ther. 2011, 19, 1008–1016. [Google Scholar] [CrossRef]
- Sloot, S.; Rashid, O.M.; Zager, J.S. Intralesional Therapy for Metastatic Melanoma. Expert Opin. Pharmacother. 2014, 15, 2629–2639. [Google Scholar] [CrossRef]
- Hamid, O.; Ismail, R.; Puzanov, I. Intratumoral Immunotherapy—Update 2019. Oncologist 2020, 25, e423–e438. [Google Scholar] [CrossRef]
- Tang, B.; Guo, Z.S.; Bartlett, D.L.; Liu, J.; McFadden, G.; Shisler, J.L.; Roy, E.J. A Cautionary Note on the Selectivity of Oncolytic Poxviruses. Oncolytic Virother. 2019, 8, 3–8. [Google Scholar] [CrossRef]
- Marabelle, A.; Andtbacka, R.; Harrington, K.; Melero, I.; Leidner, R.; de Baere, T.; Robert, C.; Ascierto, P.A.; Baurain, J.-F.; Imperiale, M.; et al. Starting the Fight in the Tumor: Expert Recommendations for the Development of Human Intratumoral Immunotherapy (HIT-IT). Ann. Oncol. 2018, 29, 2163–2174. [Google Scholar] [CrossRef]
- Na, Y.; Nam, J.-P.; Hong, J.; Oh, E.; Shin, H.C.; Kim, H.S.; Kim, S.W.; Yun, C.-O. Systemic Administration of Human Mesenchymal Stromal Cells Infected with Polymer-Coated Oncolytic Adenovirus Induces Efficient Pancreatic Tumor Homing and Infiltration. J. Control. Release 2019, 305, 75–88. [Google Scholar] [CrossRef]
- Foreman, P.M.; Friedman, G.K.; Cassady, K.A.; Markert, J.M. Oncolytic Virotherapy for the Treatment of Malignant Glioma. Neurotherapeutics 2017, 14, 333–344. [Google Scholar] [CrossRef]
- Jaime-Ramirez, A.C.; Dmitrieva, N.; Yoo, J.Y.; Banasavadi-Siddegowda, Y.; Zhang, J.; Relation, T.; Bolyard-Blessing, C.; Wojton, J.; Kaur, B. Humanized Chondroitinase ABC Sensitizes Glioblastoma Cells to Temozolomide. J. Gene Med. 2017, 19, 2942. [Google Scholar] [CrossRef]
- Eble, J.A.; Niland, S. The Extracellular Matrix in Tumor Progression and Metastasis. Clin. Exp. Metastasis 2019, 36, 171–198. [Google Scholar] [CrossRef]
- Buijs, P.R.; Verhagen, J.H.; van Eijck, C.H.; van den Hoogen, B.G. Oncolytic Viruses: From Bench to Bedside with a Focus on Safety. Hum. Vaccines Immunother. 2015, 11, 1573–1584. [Google Scholar] [CrossRef]
- Duggan, S.T.; Keating, G.M. Pegylated Liposomal Doxorubicin. Drugs 2011, 71, 2531–2558. [Google Scholar] [CrossRef]
- Veronese, F.M.; Mero, A. The Impact of PEGylation on Biological Therapies. BioDrugs 2008, 22, 315–329. [Google Scholar] [CrossRef]
- Bando, H.; Shimodaira, H.; Fujitani, K.; Takashima, A.; Yamaguchi, K.; Nakayama, N.; Takahashi, T.; Oki, E.; Azuma, M.; Nishina, T.; et al. A Phase II Study of Nab-Paclitaxel in Combination with Ramucirumab in Patients with Previously Treated Advanced Gastric Cancer. Eur. J. Cancer 2018, 91, 86–91. [Google Scholar] [CrossRef]
- Guo, S.; Zhang, Y.; Wu, Z.; Zhang, L.; He, D.; Li, X.; Wang, Z. Synergistic Combination Therapy of Lung Cancer: Cetuximab Functionalized Nanostructured Lipid Carriers for the Co-Delivery of Paclitaxel and 5-Demethylnobiletin. Biomed. Pharmacother. 2019, 118, 109225. [Google Scholar] [CrossRef]
- Ota, D.; Akatsuka, S.; Nishi, T.; Kato, T.; Takeuchi, M.; Tsuji, M.; Fukuuchi, A. Phase I Study of Combination Therapy with Weekly Nanoparticle Albumin-Bound Paclitaxel and Cyclophosphamide in Metastatic Breast Cancer Patients. Anticancer Res. 2019, 39, 6903–6907. [Google Scholar] [CrossRef]
- Tian, Z.; Zhang, F.; Li, P.; Wang, J.; Yang, J.; Zhang, P.; Yao, W.; Wang, X. Albumin-Bound Paclitaxel and Gemcitabine Combination Therapy in Soft Tissue Sarcoma. BMC Cancer 2020, 20, 698. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the Clinic. Bioeng. Transl. Med. 2016, 1, 10–29. [Google Scholar] [CrossRef]
- Rezvantalab, S.; Drude, N.I.; Moraveji, M.K.; Güvener, N.; Koons, E.K.; Shi, Y.; Lammers, T.; Kiessling, F. PLGA-Based Nanoparticles in Cancer Treatment. Front. Pharmacol. 2018, 9, 1260. [Google Scholar] [CrossRef]
- Ito, I.; Ji, L.; Tanaka, F.; Saito, Y.; Gopalan, B.; Branch, C.D.; Xu, K.; Atkinson, E.N.; Bekele, B.N.; Stephens, L.C.; et al. Liposomal Vector Mediated Delivery of the 3p FUS1 Gene Demonstrates Potent Antitumor Activity against Human Lung Cancer in Vivo. Cancer Gene Ther. 2004, 11, 733–739. [Google Scholar] [CrossRef][Green Version]
- Kim, S.-S.; Harford, J.B.; Moghe, M.; Rait, A.; Chang, E.H. Combination with SGT-53 Overcomes Tumor Resistance to a Checkpoint Inhibitor. Oncoimmunology 2018, 7, e1484982. [Google Scholar] [CrossRef]
- Patel, M.R.; Bauer, T.M.; Jimeno, A.; Wang, D.; LoRusso, P.; Do, K.T.; Stemmer, S.M.; Maurice-Dror, C.; Geva, R.; Zacharek, S.; et al. A Phase I Study of MRNA-2752, a Lipid Nanoparticle Encapsulating MRNAs Encoding Human OX40L, IL-23, and IL-36γ, for Intratumoral (ITu) Injection Alone and in Combination with Durvalumab. J. Clin. Oncol. 2020, 38, 3092–3092. [Google Scholar] [CrossRef]
- Sarker, D.; Plummer, R.; Meyer, T.; Sodergren, M.H.; Basu, B.; Chee, C.E.; Huang, K.-W.; Palmer, D.H.; Ma, Y.T.; Evans, T.R.J.; et al. MTL-CEBPA, a Small Activating RNA Therapeutic Upregulating C/EBP-α, in Patients with Advanced Liver Cancer: A First-in-Human, Multicenter, Open-Label, Phase I Trial. Clin. Cancer Res. 2020, 26, 3936–3946. [Google Scholar] [CrossRef]
- Bader, A.G. MiR-34—A MicroRNA Replacement Therapy Is Headed to the Clinic. Front. Genet. 2012, 3, 120. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro Story and the Clinical Translation of Nanomedicines Containing Nucleic Acid-Based Drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.J.; Chang, A.; Cunningham, P.N. Chronic Microangiopathy Due to DCR-MYC, a Myc-Targeted Short Interfering RNA. Am. J. Kidney Dis. 2020, 75, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Schiffelers, R.M.; Ansari, A.; Xu, J.; Zhou, Q.; Tang, Q.; Storm, G.; Molema, G.; Lu, P.Y.; Scaria, P.V.; Woodle, M.C. Cancer SiRNA Therapy by Tumor Selective Delivery with Ligand-Targeted Sterically Stabilized Nanoparticle. Nucleic Acids Res. 2004, 32, e149. [Google Scholar] [CrossRef]
- Dawidczyk, C.M.; Kim, C.; Park, J.H.; Russell, L.M.; Lee, K.H.; Pomper, M.G.; Searson, P.C. State-of-the-Art in Design Rules for Drug Delivery Platforms: Lessons from FDA-Approved Nanomedicines. J. Control. Release Off. J. Control. Release Soc. 2014, 187, 133–144. [Google Scholar] [CrossRef]
- Passero, F.C.; Grapsa, D.; Syrigos, K.N.; Saif, M.W. The Safety and Efficacy of Onivyde (Irinotecan Liposome Injection) for the Treatment of Metastatic Pancreatic Cancer Following Gemcitabine-Based Therapy. Expert Rev. Anticancer Ther. 2016, 16, 697–703. [Google Scholar] [CrossRef]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef]
- Current Nanotechnology Treatments—National Cancer Institute. Available online: https://www.cancer.gov/nano/cancer-nanotechnology/current-treatments (accessed on 23 September 2021).
- Mamot, C.; Ritschard, R.; Wicki, A.; Stehle, G.; Dieterle, T.; Bubendorf, L.; Hilker, C.; Deuster, S.; Herrmann, R.; Rochlitz, C. Tolerability, Safety, Pharmacokinetics, and Efficacy of Doxorubicin-Loaded Anti-EGFR Immunoliposomes in Advanced Solid Tumours: A Phase 1 Dose-Escalation Study. Lancet Oncol. 2012, 13, 1234–1241. [Google Scholar] [CrossRef]
- Leighl, N.B.; Page, R.D.; Raymond, V.M.; Daniel, D.B.; Divers, S.G.; Reckamp, K.L.; Villalona-Calero, M.A.; Dix, D.; Odegaard, J.I.; Lanman, R.B.; et al. Clinical Utility of Comprehensive Cell-Free DNA Analysis to Identify Genomic Biomarkers in Patients with Newly Diagnosed Metastatic Non-Small Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 4691–4700. [Google Scholar] [CrossRef]
- Soung, Y.H.; Ford, S.; Zhang, V.; Chung, J. Exosomes in Cancer Diagnostics. Cancers 2017, 9, 8. [Google Scholar] [CrossRef]
- Zhou, S.; Yang, Y.; Wu, Y.; Liu, S. Review: Multiplexed Profiling of Biomarkers in Extracellular Vesicles for Cancer Diagnosis and Therapy Monitoring. Anal. Chim. Acta 2021, 1175, 338633. [Google Scholar] [CrossRef]
- Besse, B.; Charrier, M.; Lapierre, V.; Dansin, E.; Lantz, O.; Planchard, D.; Le Chevalier, T.; Livartoski, A.; Barlesi, F.; Laplanche, A.; et al. Dendritic Cell-Derived Exosomes as Maintenance Immunotherapy after First Line Chemotherapy in NSCLC. Oncoimmunology 2015, 5, e1071008. [Google Scholar] [CrossRef]
- Cheng, Q.; Shi, X.; Han, M.; Smbatyan, G.; Lenz, H.-J.; Zhang, Y. Reprogramming Exosomes as Nanoscale Controllers of Cellular Immunity. J. Am. Chem. Soc. 2018, 140, 16413–16417. [Google Scholar] [CrossRef]
- Fu, W.; Lei, C.; Liu, S.; Cui, Y.; Wang, C.; Qian, K.; Li, T.; Shen, Y.; Fan, X.; Lin, F.; et al. CAR Exosomes Derived from Effector CAR-T Cells Have Potent Antitumour Effects and Low Toxicity. Nat. Commun. 2019, 10, 4355. [Google Scholar] [CrossRef]
- Whiteside, T.L. Stimulatory Role of Exosomes in the Context of Therapeutic Anti-Cancer Vaccines. Biotarget 2017, 1, 5. [Google Scholar] [CrossRef]
- Gu, X.; Erb, U.; Büchler, M.W.; Zöller, M. Improved Vaccine Efficacy of Tumor Exosome Compared to Tumor Lysate Loaded Dendritic Cells in Mice. Int. J. Cancer 2015, 136, E74–E84. [Google Scholar] [CrossRef]
- Anderson, M.D. Cancer Center Phase I Study of Mesenchymal Stromal Cells-Derived Exosomes with KrasG12D SiRNA for Metastatic Pancreas Cancer Patients Harboring KrasG12D Mutation. Available online: clinicaltrials.gov (accessed on 28 July 2021).
- Rosenblum, D.; Gutkin, A.; Kedmi, R.; Ramishetti, S.; Veiga, N.; Jacobi, A.M.; Schubert, M.S.; Friedmann-Morvinski, D.; Cohen, Z.R.; Behlke, M.A.; et al. CRISPR-Cas9 Genome Editing Using Targeted Lipid Nanoparticles for Cancer Therapy. Sci. Adv. 2020, 6, eabc9450. [Google Scholar] [CrossRef]
- Beal, J.M.; Payne, M.A.; Gilder, H.; Johnson, G.; Craver, W.L. Experience with Administration of an Intravenous Fat Emulsion to Surgical Patients. Metabolism 1957, 6, 673–681. [Google Scholar]
- Lin, C.; Li, H.; Hao, M.; Xiong, D.; Luo, Y.; Huang, C.; Yuan, Q.; Zhang, J.; Xia, N. Increasing the Efficiency of CRISPR/Cas9-Mediated Precise Genome Editing of HSV-1 Virus in Human Cells. Sci. Rep. 2016, 6, 34531. [Google Scholar] [CrossRef]
- FDA Approves T-VEC to Treat Metastatic Melanoma—National Cancer Institute. Available online: https://www.cancer.gov/news-events/cancer-currents-blog/2015/t-vec-melanoma (accessed on 28 September 2021).
- Anesti, A.-M.; Simpson, G.R.; Price, T.; Pandha, H.S.; Coffin, R.S. Expression of RNA Interference Triggers from an Oncolytic Herpes Simplex Virus Results in Specific Silencing in Tumour Cells in Vitro and Tumours in Vivo. BMC Cancer 2010, 10, 486. [Google Scholar] [CrossRef]
- Herr, H.W.; Morales, A. History of bacillus Calmette-Guerin and bladder cancer: an immunotherapy success story. J. Urol. 2008, 179, 53–56. [Google Scholar] [CrossRef]
- Gardlik, R.; Behuliak, M.; Palffy, R.; Celec, P.; Li, C.J. Gene Therapy for Cancer: Bacteria-Mediated Anti-Angiogenesis Therapy. Gene Ther. 2011, 18, 425–431. [Google Scholar] [CrossRef]
- Basu, P.; Mehta, A.; Jain, M.; Gupta, S.; Nagarkar, R.V.; John, S.; Petit, R. A Randomized Phase 2 Study of ADXS11-001 Listeria Monocytogenes–Listeriolysin O Immunotherapy with or Without Cisplatin in Treatment of Advanced Cervical Cancer. Int. J. Gynecol. Cancer 2018, 28, 764–772. [Google Scholar] [CrossRef]
- Conry, R.M.; Westbrook, B.; McKee, S.; Norwood, T.G. Talimogene Laherparepvec: First in Class Oncolytic Virotherapy. Hum. Vaccines Immunother. 2018, 14, 839–846. [Google Scholar] [CrossRef]
- Kohlhapp, F.J.; Kaufman, H.L. Molecular Pathways: Mechanism of Action for Talimogene Laherparepvec, a New Oncolytic Virus Immunotherapy. Clin. Cancer Res. 2016, 22, 1048–1054. [Google Scholar] [CrossRef]
- Farassati, F.; Yang, A.D.; Lee, P.W. Oncogenes in Ras Signalling Pathway Dictate Host-Cell Permissiveness to Herpes Simplex Virus 1. Nat. Cell Biol. 2001, 3, 745–750. [Google Scholar] [CrossRef]
- Kemp, V.; van den Wollenberg, D.J.M.; Camps, M.G.M.; van Hall, T.; Kinderman, P.; Pronk-van Montfoort, N.; Hoeben, R.C. Arming Oncolytic Reovirus with GM-CSF Gene to Enhance Immunity. Cancer Gene Ther. 2019, 26, 268–281. [Google Scholar] [CrossRef]
- Nguyen, A.; Ho, L.; Wan, Y. Chemotherapy and Oncolytic Virotherapy: Advanced Tactics in the War against Cancer. Front. Oncol. 2014, 4, 145. [Google Scholar] [CrossRef] [PubMed]
- Middleton, M.R.; Hoeller, C.; Michielin, O.; Robert, C.; Caramella, C.; Öhrling, K.; Hauschild, A. Intratumoural Immunotherapies for Unresectable and Metastatic Melanoma: Current Status and Future Perspectives. Br. J. Cancer 2020, 123, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.; Curiel, D.; Cotten, M. Delivery of Drugs, Proteins and Genes into Cells Using Transferrin as a Ligand for Receptor-Mediated Endocytosis. Adv. Drug Deliv. Rev. 1994, 14, 113–135. [Google Scholar] [CrossRef]
- Ottolino-Perry, K.; Diallo, J.-S.; Lichty, B.D.; Bell, J.C.; McCart, J.A. Intelligent Design: Combination Therapy With Oncolytic Viruses. Mol. Ther. 2010, 18, 251–263. [Google Scholar] [CrossRef]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.-C. Optimizing Oncolytic Virotherapy in Cancer Treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef]
- Duong, M.T.-Q.; Qin, Y.; You, S.-H.; Min, J.-J. Bacteria-Cancer Interactions: Bacteria-Based Cancer Therapy. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, W.; Wu, X.; Zhang, Y.; Mannion, C.; Brouchkov, A.; Man, Y.-G.; Chen, T. Oncolytic Bacteria and Their Potential Role in Bacterium-Mediated Tumour Therapy: A Conceptual Analysis. J. Cancer 2019, 10, 4442–4454. [Google Scholar] [CrossRef]
- Pryor, K.; Goddard, J.; Goldstein, D.; Stricker, P.; Russell, P.; Golovsky, D.; Penny, R. Bacillus Calmette-Guerin (BCG) Enhances Monocyte- and Lymphocyte-Mediated Bladder Tumour Cell Killing. Br. J. Cancer 1995, 71, 801–807. [Google Scholar] [CrossRef]
- Janku, F.; Fu, S.; Murthy, R.; Karp, D.; Hong, D.; Tsimberidou, A.; Gillison, M.; Adat, A.; Raina, A.; Call, G.; et al. 383 First-in-Man Clinical Trial of Intratumoral Injection of Clostridium Novyi-NT Spores in Combination with Pembrolizumab in Patients with Treatment-Refractory Advanced Solid Tumors. J. Immunother. Cancer 2020, 8, 383. [Google Scholar] [CrossRef]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and Survival with GVAX Pancreas Prime and Listeria Monocytogenes–Expressing Mesothelin (CRS-207) Boost Vaccines for Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef]
- Kienle, G.S. Fever in Cancer Treatment: Coley’s Therapy and Epidemiologic Observations. Glob. Adv. Health Med. 2012, 1, 92–100. [Google Scholar] [CrossRef]
- Campisi, L.; Soudja, S.M.; Cazareth, J.; Bassand, D.; Lazzari, A.; Brau, F.; Narni-Mancinelli, E.; Glaichenhaus, N.; Geissmann, F.; Lauvau, G. Splenic CD8α+ Dendritic Cells Undergo Rapid Programming by Cytosolic Bacteria and Inflammation to Induce Protective CD8+ T-Cell Memory. Eur. J. Immunol. 2011, 41, 1594–1605. [Google Scholar] [CrossRef]
- Wallecha, A.; French, C.; Petit, R.; Singh, R.; Amin, A.; Rothman, J. Lm-LLO-Based Immunotherapies and HPV-Associated Disease. J. Oncol. 2012, 2012, 542851. [Google Scholar] [CrossRef]
- Janku, F.; Zhang, H.H.; Pezeshki, A.; Goel, S.; Murthy, R.; Wang-Gillam, A.; Shepard, D.R.; Helgason, T.; Masters, T.; Hong, D.S.; et al. Intratumoral Injection of Clostridium Novyi-NT Spores in Patients with Treatment-Refractory Advanced Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 96–106. [Google Scholar] [CrossRef]
- Masonic Cancer Center, University of Minnesota. A Phase 1 Study of an IL-2 Expressing, Attenuated Salmonella Typhimurium in Patients with Unresectable Hepatic Spread from Any Non-Hematologic Primary. Available online: clinicaltrials.gov (accessed on 28 July 2020).









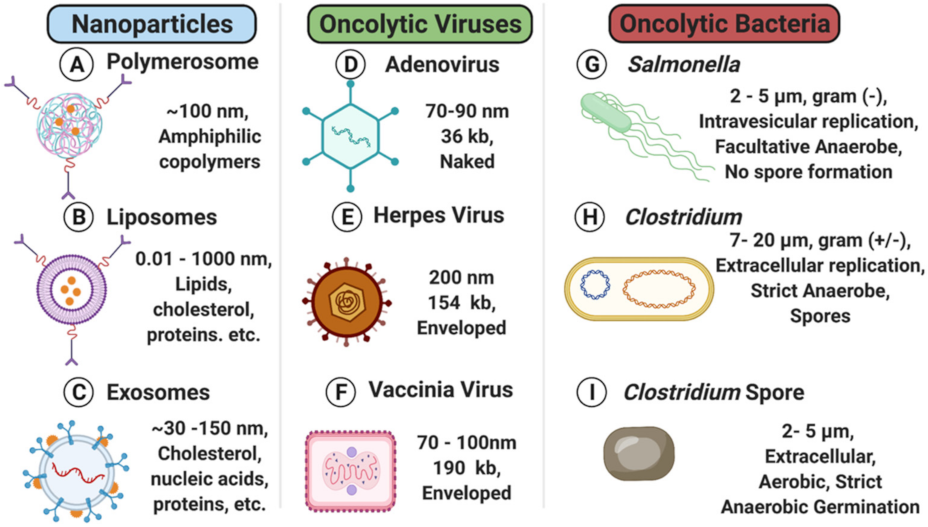{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Aspects | Liposomes | Polymersomes | Exosomes | Oncolytic Virus | Oncolytic Bacteria |
|---|---|---|---|---|---|
| Structure | Lipid bilayer membrane | Lipid bilayer membrane | Lipid bilayer membrane | Nucleocapsid | Cellular |
| Proliferation in tumors | No | No | No | Yes | Yes |
| Genetic Modification | N/A | N/A | N/A | Good | Good |
| Tumor Targeting | Specific and modifiable | Specific and modifiable | Specific and modifiable | Intratumor injection preferred to increase efficacy | Specific with systemic injection |
| Drug Delivery capacity | Contained within an aqueous core | Contained within an aqueous core | Contained within an aqueous core | Limited capacity of continuous expression | Continuous drug expression with termination control mechanisms |
| Immunomodulation | Low-Mild | Low-Mild | Low-Mild | Mild-Mod | Strong |
| Anticancer Effects | Drug delivery | Drug delivery | Drug delivery | Direct: cellular lysis Indirect: gene delivery and drug delivery | Direct: exotoxin and nutrient competition Indirect: unlimited delivery options |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pierce, K.M.; Miklavcic, W.R.; Cook, K.P.; Hennen, M.S.; Bayles, K.W.; Hollingsworth, M.A.; Brooks, A.E.; Pullan, J.E.; Dailey, K.M. The Evolution and Future of Targeted Cancer Therapy: From Nanoparticles, Oncolytic Viruses, and Oncolytic Bacteria to the Treatment of Solid Tumors. Nanomaterials 2021, 11, 3018. https://doi.org/10.3390/nano11113018
Pierce KM, Miklavcic WR, Cook KP, Hennen MS, Bayles KW, Hollingsworth MA, Brooks AE, Pullan JE, Dailey KM. The Evolution and Future of Targeted Cancer Therapy: From Nanoparticles, Oncolytic Viruses, and Oncolytic Bacteria to the Treatment of Solid Tumors. Nanomaterials. 2021; 11(11):3018. https://doi.org/10.3390/nano11113018
Chicago/Turabian StylePierce, Kyle M., William R. Miklavcic, Kyle P. Cook, Mikayla Sweitzer Hennen, Kenneth W. Bayles, Michael A. Hollingsworth, Amanda E. Brooks, Jessica E. Pullan, and Kaitlin M. Dailey. 2021. "The Evolution and Future of Targeted Cancer Therapy: From Nanoparticles, Oncolytic Viruses, and Oncolytic Bacteria to the Treatment of Solid Tumors" Nanomaterials 11, no. 11: 3018. https://doi.org/10.3390/nano11113018
APA StylePierce, K. M., Miklavcic, W. R., Cook, K. P., Hennen, M. S., Bayles, K. W., Hollingsworth, M. A., Brooks, A. E., Pullan, J. E., & Dailey, K. M. (2021). The Evolution and Future of Targeted Cancer Therapy: From Nanoparticles, Oncolytic Viruses, and Oncolytic Bacteria to the Treatment of Solid Tumors. Nanomaterials, 11(11), 3018. https://doi.org/10.3390/nano11113018