
Nanoparticles for Targeted Drug Delivery to Cancer Stem Cells: A Review of Recent Advances





Abstract
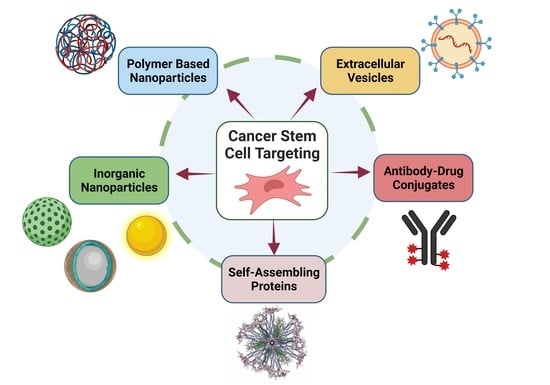
:
1. Introduction
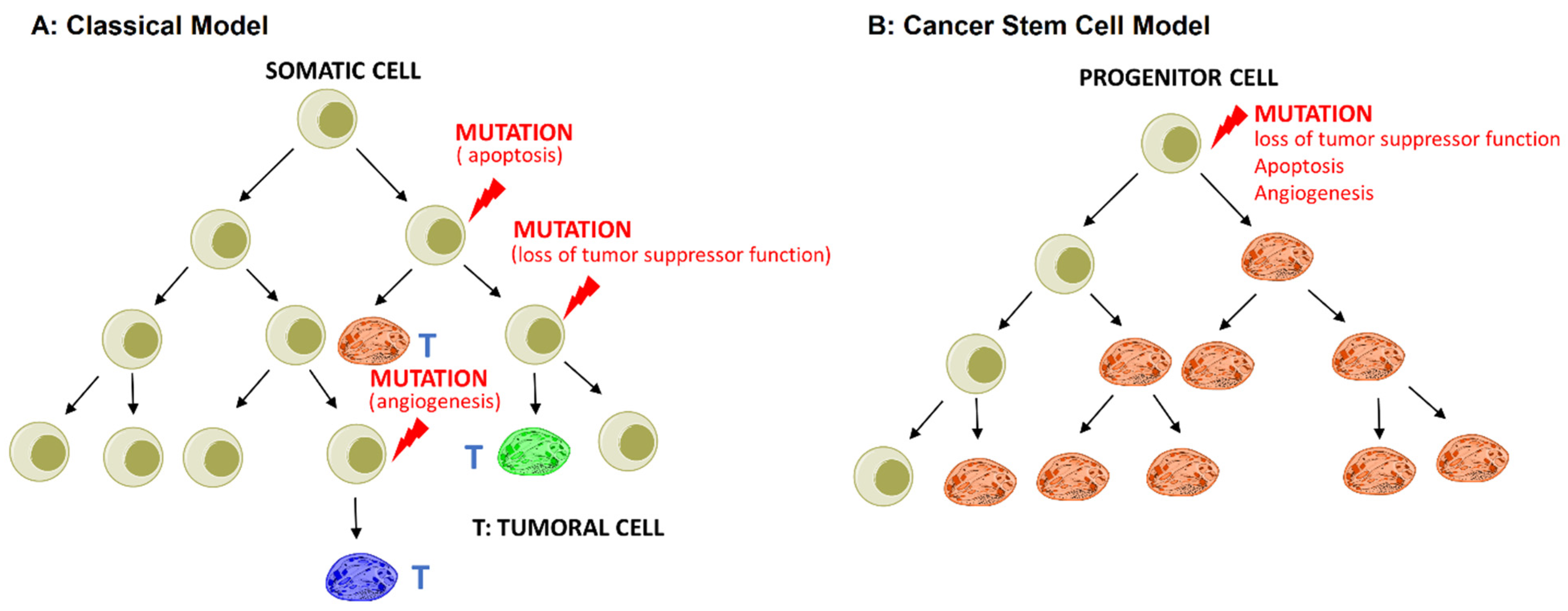
2. Cancer Stem Cell Biology
- Self-renewal and DNA repair: this extraordinary property of CSCs causes tumor relapse and radiation-resistance in tumors [22].
- Differentiation into multiple cell types: the pluripotency of CSCs causes heterogeneity in solid tumors [23].
- Ionizing radiation: this feature makes CSCs resistant to radiotherapy.
- Infinite proliferative potential: unlimited cell division, which leads to rapid tumor growth.
- Dormancy state: CSCs enter dormancy to evade the attack by the immune system, awaiting new signals from the environment to re-enter the cell cycle [22].
- Elevated expression of ATP-binding cassette (ABC) pumps and detoxifying enzymes to increase the drug’s efflux, which is considered to be an important mechanism for multi-drug resistance (MDR). Multi-drug resistance is either intrinsic and present before the start of treatment or acquired after exposure to treatment [25].
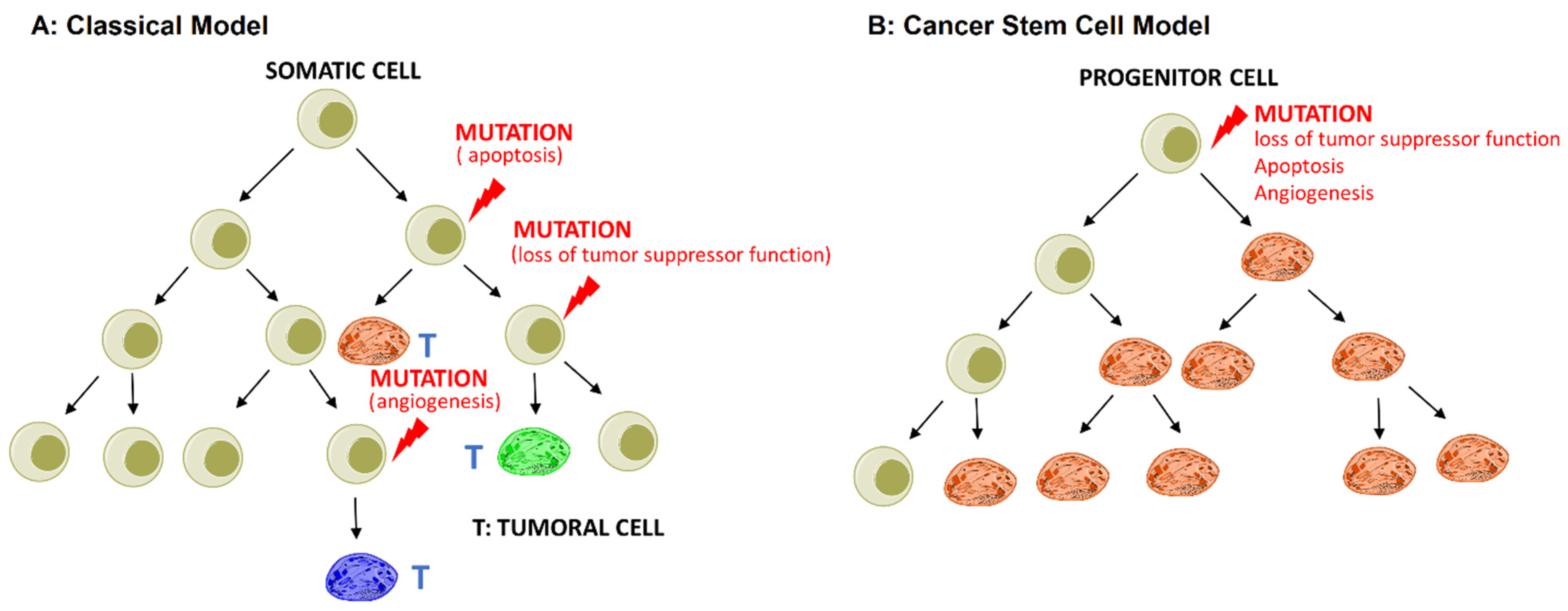
- The stochastic or classical model states that any somatic cell has the intrinsic ability to undergo mutation and transform into CSCs driven by genetic instability or environmental signals, as shown in Figure 1A;
2.1. Extracellular Matrix (ECM)
2.2. CSC Niche
2.3. Tumor Angiogenesis
2.4. Mitochondrial Activity of CSCs
2.5. Surface Biomarkers
2.6. Signaling Pathways
2.7. CYP Family of Enzymes
3. Polymer-Based NPs
- Passive targeting: Pathophysiological conditions, specifically impaired angiogenesis and a high demand for nutrients and oxygen by proliferating tumor cells, result in an overexpression of vascular endothelial growth factor (VEGF) and the formation of abnormal tumor vessels, with relatively large gaps between the endothelial cells’ lining lumen of the vessels. The large intercellular clefts and poor lymphatic drainage leads to an accumulation and retention of NPs, with a size range of 100–200 nm in the tumor tissue. This enhanced permeability and retention (EPR) effect allows for the passive targeting of drug-loaded NPs to the tumor vasculature. However, NPs with a short circulation time are rapidly taken up by the mononuclear phagocyte system (MPS), prior to uptake by the tumor vasculature. Therefore, NPs should be surface modified to prolong their residence time in circulation [32,33]. The surface modification of NPs with non-adhesive, highly water soluble polymers, like polyethylene glycol (PEG), polyacrylic acid (PAA), and dextran, has been shown to reduce the undesired uptake of NPs by MPS [1].
- Active targeting: Antibodies or ligands that interact specifically with one or multiple CSC surface biomarkers are used for targeting therapeutic agents to stem cells in the tumor tissue. This approach significantly reduces drug toxicity and undesirable uptake by normal cells [8].
3.1. PLGA NPs
3.2. PEG NPs
3.3. PLGA-PEG Copolymer NPs
3.4. Polylysine NPs
3.5. PLA-PEG NPs
3.6. Lipid-Polymer NPs
3.7. mPEG NPs
3.8. Hyaluronic Acid NPs
3.9. PLGA/TPGS NPs
3.10. Liposomes
3.11. Multi-Polymeric NPs
3.12. Other Polymeric NPs
4. Inorganic NPs
4.1. Gold NPs
4.2. Iron Oxide NPs
4.3. Silica NPs
5. Self-Assembling Protein NPs
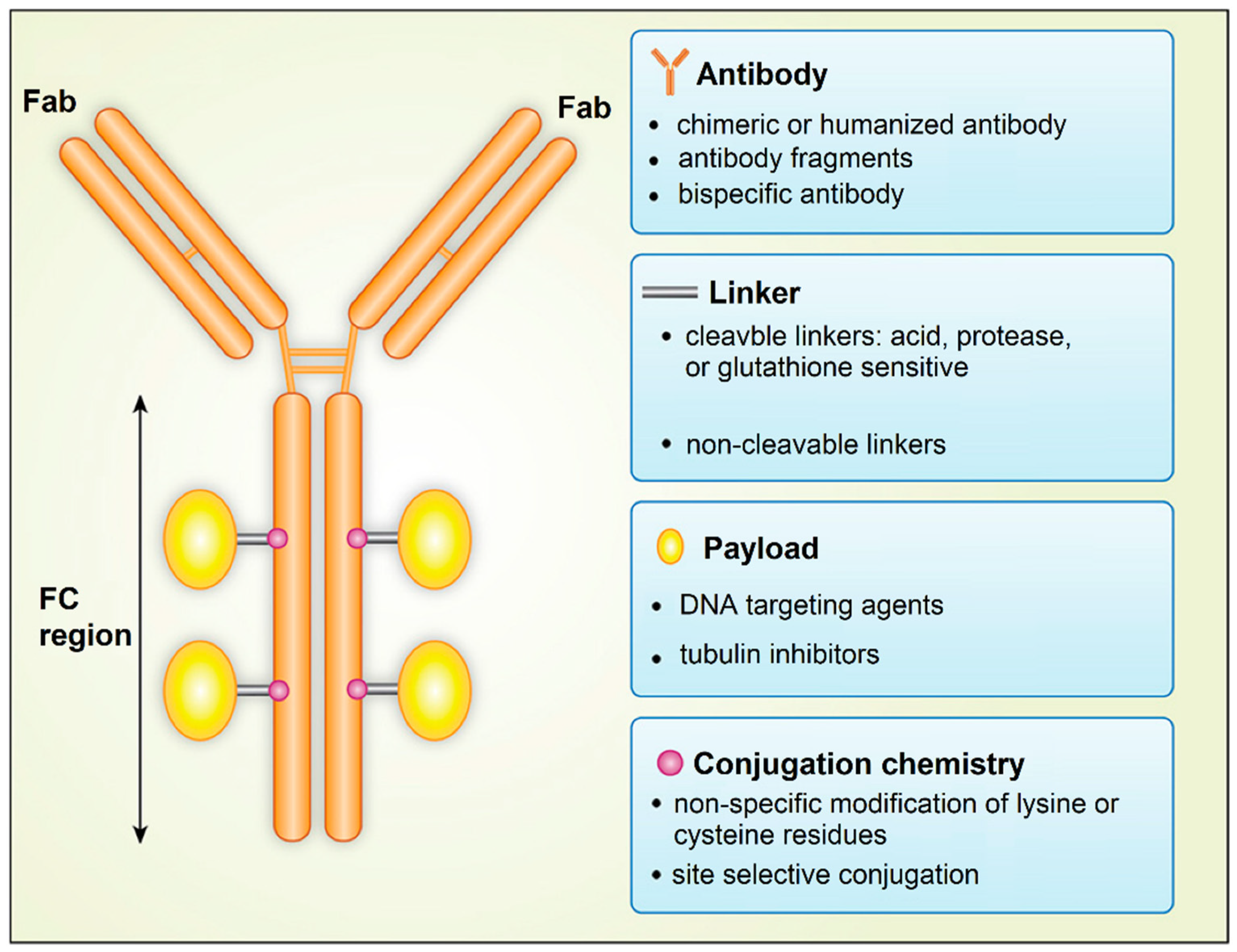
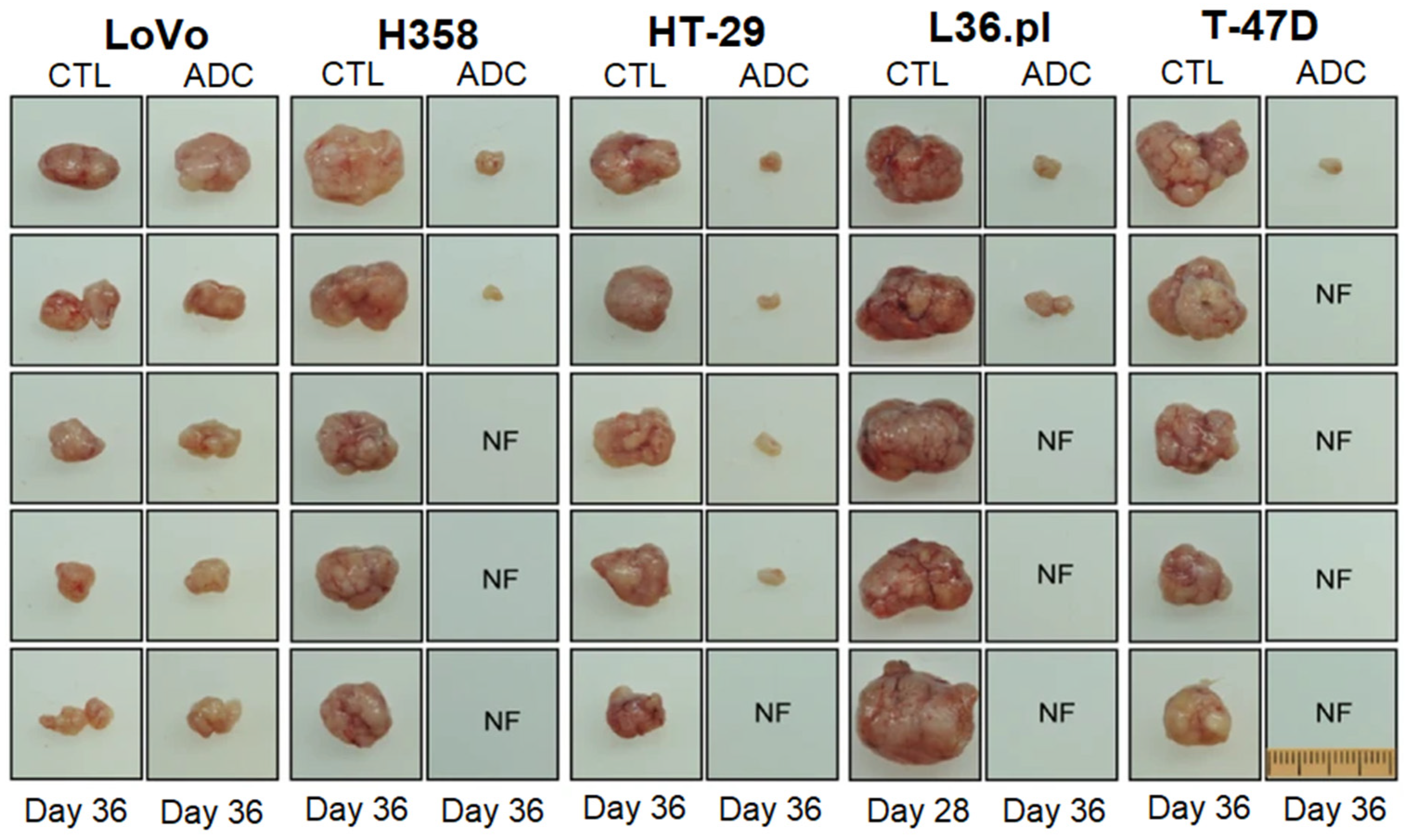
6. Antibody Drug Conjugates
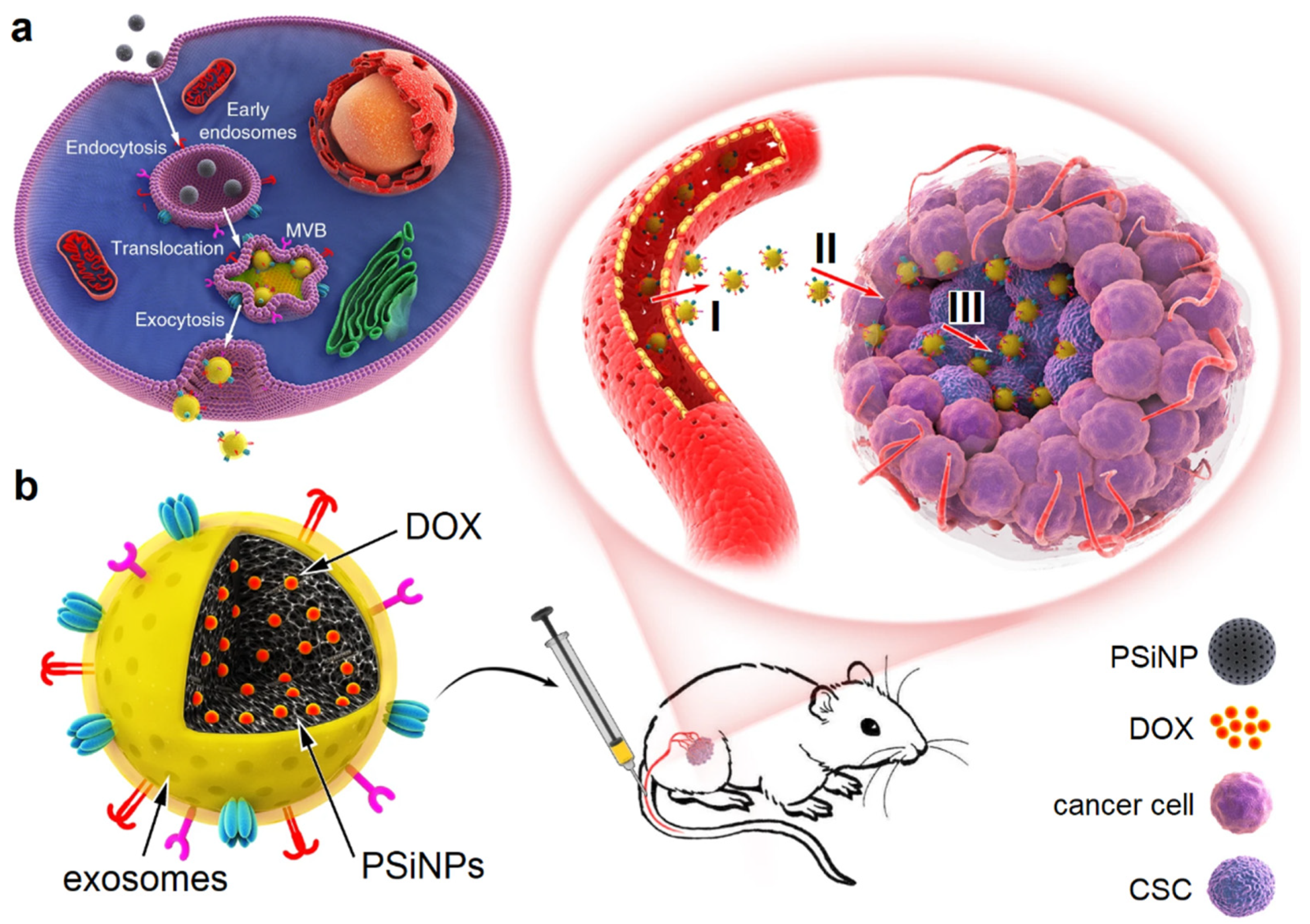
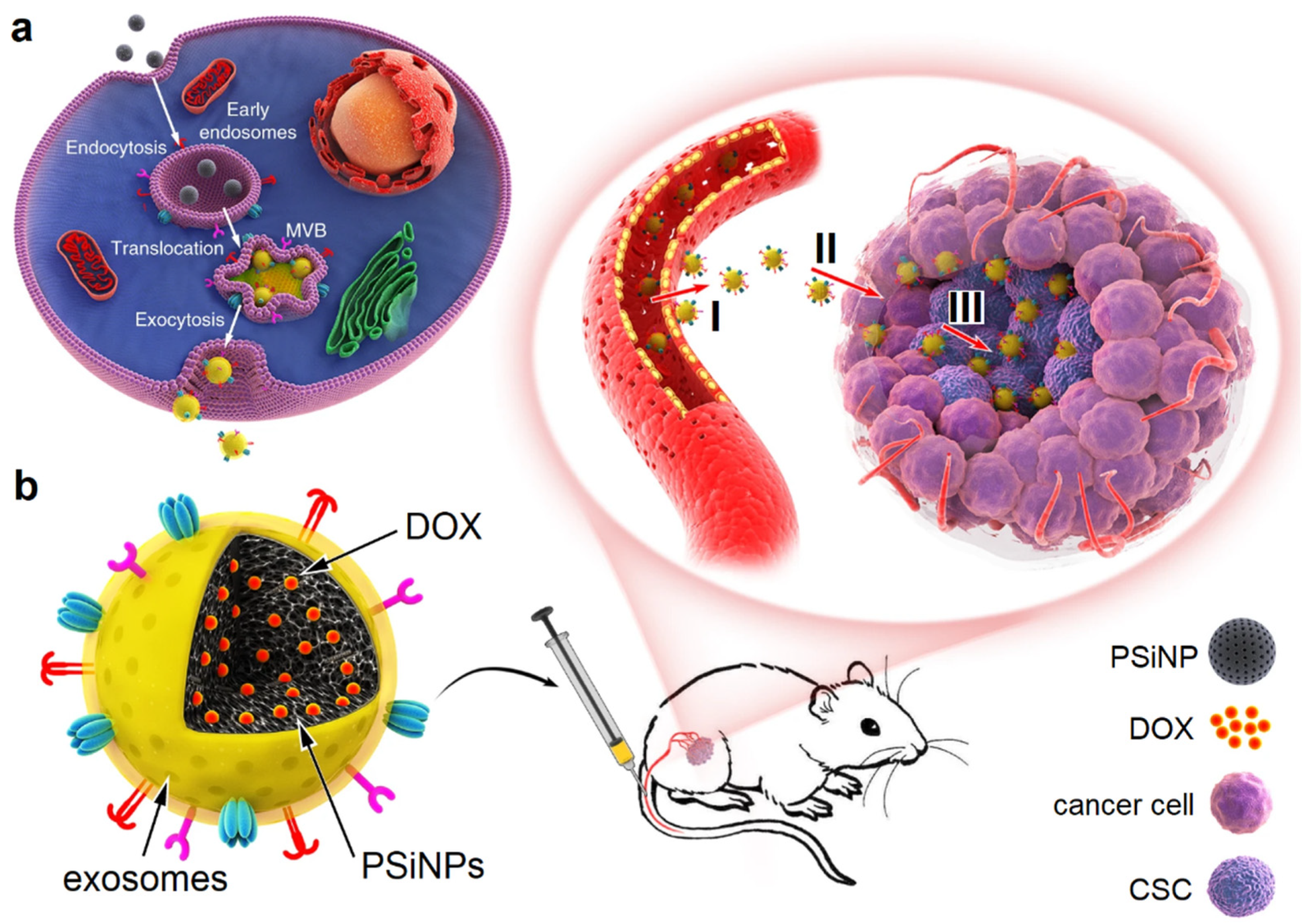
7. Extracellular Vesicles
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Duan, H.; Liu, Y.; Gao, Z.; Huang, W. Recent advances in drug delivery systems for targeting cancer stem cells. Acta Pharm. Sin. B 2021, 11, 55–70. [Google Scholar] [CrossRef]
- World Health Organization. WHO Report on Cancer: Setting Priorities, Investing Wisely and Providing Care for All; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Asghari, F.; Khademi, R.; Ranjbar, F.E.; Malekshahi, Z.V.; Majidi, R.F. Application of Nanotechnology in Targeting of Cancer Stem Cells: A Review. Int. J. Stem Cells 2019, 12, 227–239. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.F.B.; Jackson, E.L.; Woolfenden, A.E.; Lawrence, S.; Babar, I.; Vogel, S.; Crowley, D.; Bronson, R.T.; Jacks, T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005, 121, 823–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, A.T.; Maitland, N.J. Prostate cancer stem cells. Eur. J. Cancer 2006, 42, 1213–1218. [Google Scholar] [CrossRef]
- Yang, Z.F.; Ngai, P.; Ho, D.W.; Yu, W.C.; Ng, M.N.; Lau, C.K.; Li, M.L.; Tam, K.H.; Lam, C.T.; Poon, R.T.; et al. Identification of local and circulating cancer stem cells in human liver cancer. Hepatology 2008, 47, 919–928. [Google Scholar] [CrossRef]
- Fang, D.; Nguyen, T.K.; Leishear, K.; Finko, R.; Kulp, A.N.; Hotz, S.; Van Belle, P.A.; Xu, X.; Elder, D.E.; Herlyn, M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005, 65, 9328–9337. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Prince, M.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.; Kaplan, M.; Dalerba, P.; Weissman, I.; Clarke, M.; Ailles, L. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 973–978. [Google Scholar] [CrossRef] [Green Version]
- Szotek, P.P.; Pieretti-Vanmarcke, R.; Masiakos, P.T.; Dinulescu, D.M.; Connolly, D.; Foster, R.; Dombkowski, D.; Preffer, F.; MacLaughlin, D.T.; Donahoe, P.K. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc. Natl. Acad. Sci. USA 2006, 103, 11154–11159. [Google Scholar] [CrossRef] [Green Version]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [Green Version]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Masterman-Smith, M.; Geschwind, D.H.; Bronner-Fraser, M.; Kornblum, H.I. Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 15178–15183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.-Y.; Cheng, R.; Yang, Z.; Tian, Z.-M. Nanotechnology for cancer therapy based on chemotherapy. Molecules 2018, 23, 826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grodzinski, P.; Kircher, M.; Goldberg, M.; Gabizon, A. Integrating nanotechnology into cancer care. ACS Nano 2019, 13, 7370–7376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Li, Q.; Mo, J.; Dai, H. Drug-loaded polymeric nanoparticles for cancer stem cell targeting. Front. Pharmacol. 2017, 8, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, I.-S.; Jang, G.-B.; Lee, H.-Y.; Nam, J.-S. Targeting cancer stem cells by using the nanoparticles. Int. J. Nanomed. 2015, 10, 251. [Google Scholar]
- Tabassum, N.; Verma, V.; Kumar, M.; Kumar, A.; Singh, B. Nanomedicine in cancer stem cell therapy: From fringe to forefront. Cell Tissue Res. 2018, 374, 427–438. [Google Scholar] [CrossRef]
- Bhartiya, D.; Patel, H.; Ganguly, R.; Shaikh, A.; Shukla, Y.; Sharma, D.; Singh, P. Novel insights into adult and cancer stem cell biology. Stem Cells Dev. 2018, 27, 1527–1539. [Google Scholar] [CrossRef]
- Lau, E.Y.-T.; Ho, N.P.-Y.; Lee, T.K.-W. Cancer stem cells and their microenvironment: Biology and therapeutic implications. Stem Cells Int. 2017, 2017, 3714190. [Google Scholar] [CrossRef]
- Mokhtarzadeh, A.; Hassanpour, S.; Vahid, Z.F.; Hejazi, M.; Hashemi, M.; Ranjbari, J.; Tabarzad, M.; Noorolyai, S.; de la Guardia, M. Nano-delivery system targeting to cancer stem cell cluster of differentiation biomarkers. J. Control. Release 2017, 266, 166–186. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Huang, G.; Chen, Z.; Zhang, Y. Nanomaterials in targeting cancer stem cells for cancer therapy. Front. Pharmacol. 2017, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Soltani, F.; Ramezani, M.; Amel Farzad, S.; Mokhtarzadeh, A.; Hashemi, M. Comparison study of the effect of alkyl-modified and unmodified PAMAM and PPI dendrimers on solubility and antitumor activity of crocetin. Artif. Cells Nanomed. Biotechnol. 2017, 45, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- Dalpiaz, A.; Paganetto, G.; Botti, G.; Pavan, B. Cancer stem cells and nanomedicine: New opportunities to combat multidrug resistance? Drug Discov. Today 2020, 25, 1651–1667. [Google Scholar] [CrossRef]
- Nunes, T.; Hamdan, D.; Leboeuf, C.; El Bouchtaoui, M.; Gapihan, G.; Nguyen, T.T.; Meles, S.; Angeli, E.; Ratajczak, P.; Lu, H. Targeting cancer stem cells to overcome chemoresistance. Int. J. Mol. Sci. 2018, 19, 4036. [Google Scholar] [CrossRef] [Green Version]
- Dianat-Moghadam, H.; Heidarifard, M.; Jahanban-Esfahlan, R.; Panahi, Y.; Hamishehkar, H.; Pouremamali, F.; Rahbarghazi, R.; Nouri, M. Cancer stem cells-emanated therapy resistance: Implications for liposomal drug delivery systems. J. Control. Release 2018, 288, 62–83. [Google Scholar] [CrossRef]
- Loureiro, R.; Mesquita, K.A.; Magalhães-Novais, S.; Oliveira, P.J.; Vega-Naredo, I. Mitochondrial biology in cancer stem cells. Semin. Cancer Biol. 2017, 47, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Mokhtarzadeh, A.; Alibakhshi, A.; Hashemi, M.; Hejazi, M.; Hosseini, V.; de la Guardia, M.; Ramezani, M. Biodegradable nano-polymers as delivery vehicles for therapeutic small non-coding ribonucleic acids. J. Control. Release 2017, 245, 116–126. [Google Scholar] [CrossRef]
- Mokhtarzadeh, A.; Tabarzad, M.; Ranjbari, J.; de la Guardia, M.; Hejazi, M.; Ramezani, M. Aptamers as smart ligands for nano-carriers targeting. TrAC Trends Anal. Chem. 2016, 82, 316–327. [Google Scholar] [CrossRef]
- Mokhtarzadeh, A.; Alibakhshi, A.; Yaghoobi, H.; Hashemi, M.; Hejazi, M.; Ramezani, M. Recent advances on biocompatible and biodegradable nanoparticles as gene carriers. Expert Opin. Biol. Ther. 2016, 16, 771–785. [Google Scholar] [CrossRef]
- Iyer, A.K.; Singh, A.; Ganta, S.; Amiji, M.M. Role of integrated cancer nanomedicine in overcoming drug resistance. Adv. Drug Deliv. Rev. 2013, 65, 1784–1802. [Google Scholar] [CrossRef] [PubMed]
- Pavan, B.; Paganetto, G.; Rossi, D.; Dalpiaz, A. Multidrug resistance in cancer or inefficacy of neuroactive agents: Innovative strategies to inhibit or circumvent the active efflux transporters selectively. Drug Discov. Today 2014, 19, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Sun, N.; Cheng, R.; Zhao, C.; Liu, J.; Tian, Z. Hybrid nanoparticles coated with hyaluronic acid lipoid for targeted co-delivery of paclitaxel and curcumin to synergistically eliminate breast cancer stem cells. J. Mater. Chem. B 2017, 5, 6762–6775. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xu, W.; Yuan, X.; Chen, H.; Song, H.; Wang, B.; Han, J. Polymer–lipid hybrid anti-HER2 nanoparticles for targeted salinomycin delivery to HER2-positive breast cancer stem cells and cancer cells. Int. J. Nanomed. 2017, 12, 6909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abou-ElNaga, A.; Mutawa, G.; El-Sherbiny, I.M.; Abd-ElGhaffar, H.; Allam, A.A.; Ajarem, J.; Mousa, S.A. Novel nano-therapeutic approach actively targets human ovarian cancer stem cells after xenograft into nude mice. Int. J. Mol. Sci. 2017, 18, 813. [Google Scholar] [CrossRef] [Green Version]
- Verma, R.K.; Yu, W.; Singh, S.P.; Shankar, S.; Srivastava, R.K. Anthothecol-encapsulated PLGA nanoparticles inhibit pancreatic cancer stem cell growth by modulating sonic hedgehog pathway. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 2061–2070. [Google Scholar] [CrossRef]
- Ni, M.; Xiong, M.; Zhang, X.; Cai, G.; Chen, H.; Zeng, Q.; Yu, Z. Poly (lactic-co-glycolic acid) nanoparticles conjugated with CD133 aptamers for targeted salinomycin delivery to CD133+ osteosarcoma cancer stem cells. Int. J. Nanomed. 2015, 10, 2537. [Google Scholar]
- Muntimadugu, E.; Kumar, R.; Saladi, S.; Rafeeqi, T.A.; Khan, W. CD44 targeted chemotherapy for co-eradication of breast cancer stem cells and cancer cells using polymeric nanoparticles of salinomycin and paclitaxel. Colloids Surf. B Biointerfaces 2016, 143, 532–546. [Google Scholar] [CrossRef]
- Sun, N.; Zhao, C.; Cheng, R.; Liu, Z.; Li, X.; Lu, A.; Tian, Z.; Yang, Z. Cargo-free nanomedicine with pH sensitivity for codelivery of DOX conjugated prodrug with SN38 to synergistically eradicate breast cancer stem cells. Mol. Pharm. 2018, 15, 3343–3355. [Google Scholar] [CrossRef]
- Wang, M.; Xie, F.; Wen, X.; Chen, H.; Zhang, H.; Liu, J.; Zhang, H.; Zou, H.; Yu, Y.; Chen, Y. Therapeutic PEG-ceramide nanomicelles synergize with salinomycin to target both liver cancer cells and cancer stem cells. Nanomedicine 2017, 12, 1025–1042. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.-Y.; Ng, V.W.L.; Gao, S.-J.; Tong, Y.W.; Hedrick, J.L.; Yang, Y.Y. Co-delivery of thioridazine and doxorubicin using polymeric micelles for targeting both cancer cells and cancer stem cells. Biomaterials 2014, 35, 1096–1108. [Google Scholar] [CrossRef]
- Mi, Y.; Huang, Y.; Deng, J. The enhanced delivery of salinomycin to CD133+ ovarian cancer stem cells through CD133 antibody conjugation with poly (lactic-co-glycolic acid)-poly (ethylene glycol) nanoparticles. Oncol. Lett. 2018, 15, 6611–6621. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Pan, X.; Xie, F.; Lu, Y.; Zou, H.; Yin, C.; Zhang, Y.; Gao, J. Codelivery of doxorubicin and elacridar to target both liver cancer cells and stem cells by polylactide-co-glycolide/d-alpha-tocopherol polyethylene glycol 1000 succinate nanoparticles. Int. J. Nanomed. 2018, 13, 6855–6870. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, Q.; Sun, J.; Liu, H.; Li, Q. The combination therapy of salinomycin and gefitinib using poly (d, l-lactic-co-glycolic acid)-poly (ethylene glycol) nanoparticles for targeting both lung cancer stem cells and cancer cells. OncoTargets Ther. 2017, 10, 5653. [Google Scholar] [CrossRef] [Green Version]
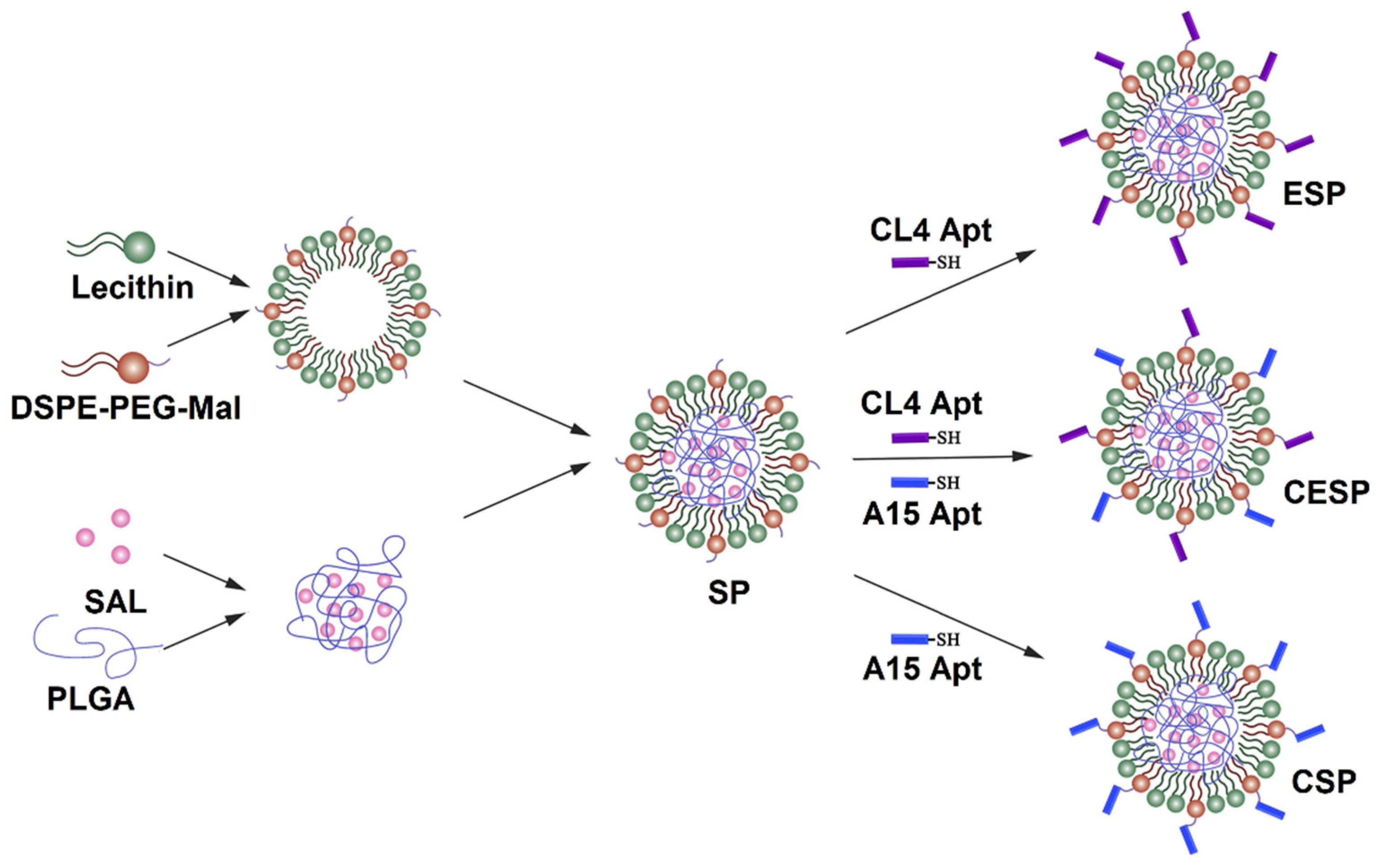
- Qiao, S.; Zhao, Y.; Geng, S.; Li, Y.; Hou, X.; Liu, Y.; Lin, F.-H.; Yao, L.; Tian, W. A novel double-targeted nondrug delivery system for targeting cancer stem cells. Int. J. Nanomed. 2016, 11, 6667–6678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Lin, J.; Shan, Y.; Zhengmao, L. The promotion of nanoparticle delivery to two populations of gastric cancer stem cells by CD133 and CD44 antibodies. Biomed. Pharmacother. 2019, 115, 108857. [Google Scholar] [CrossRef] [PubMed]
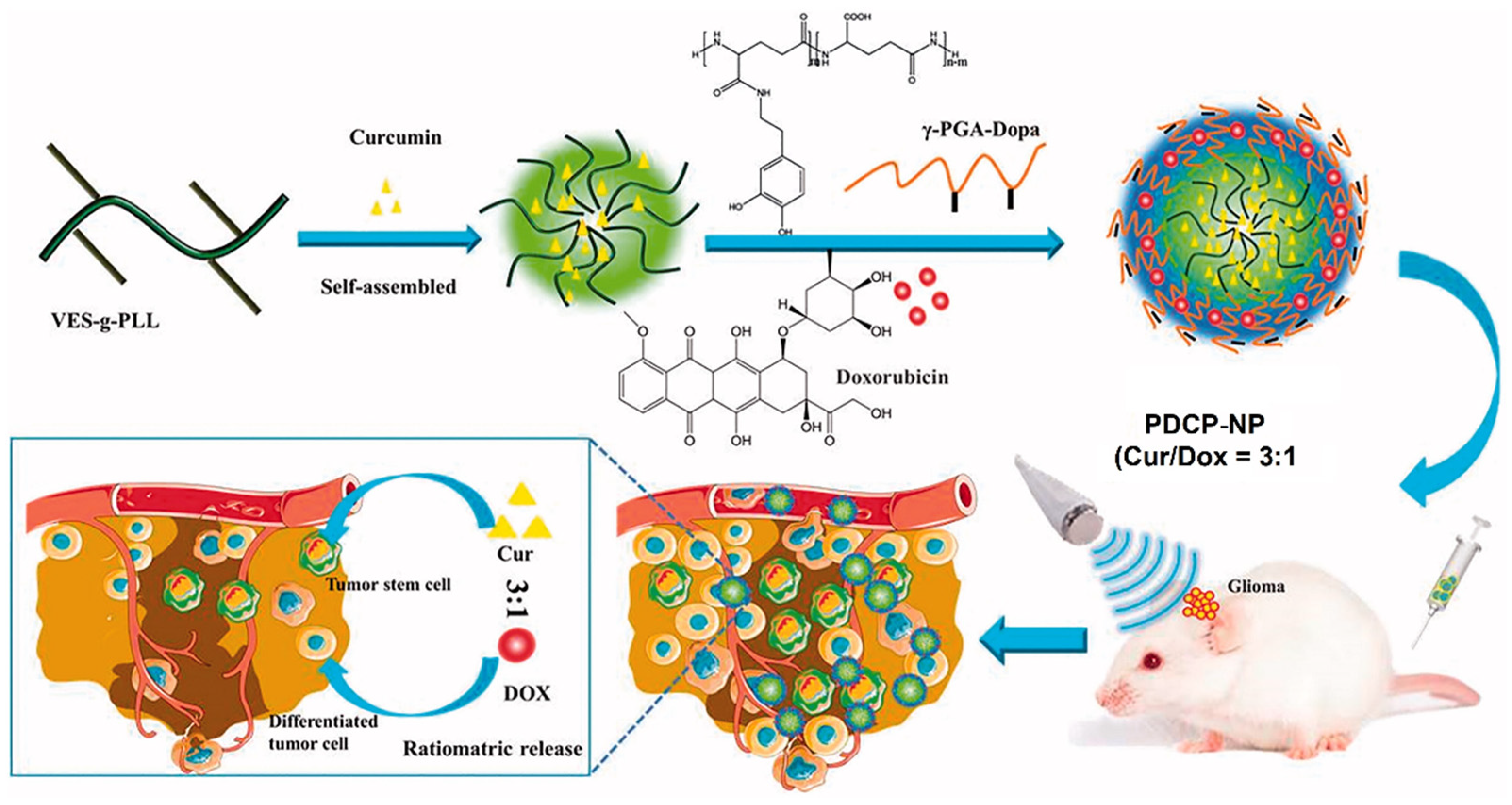
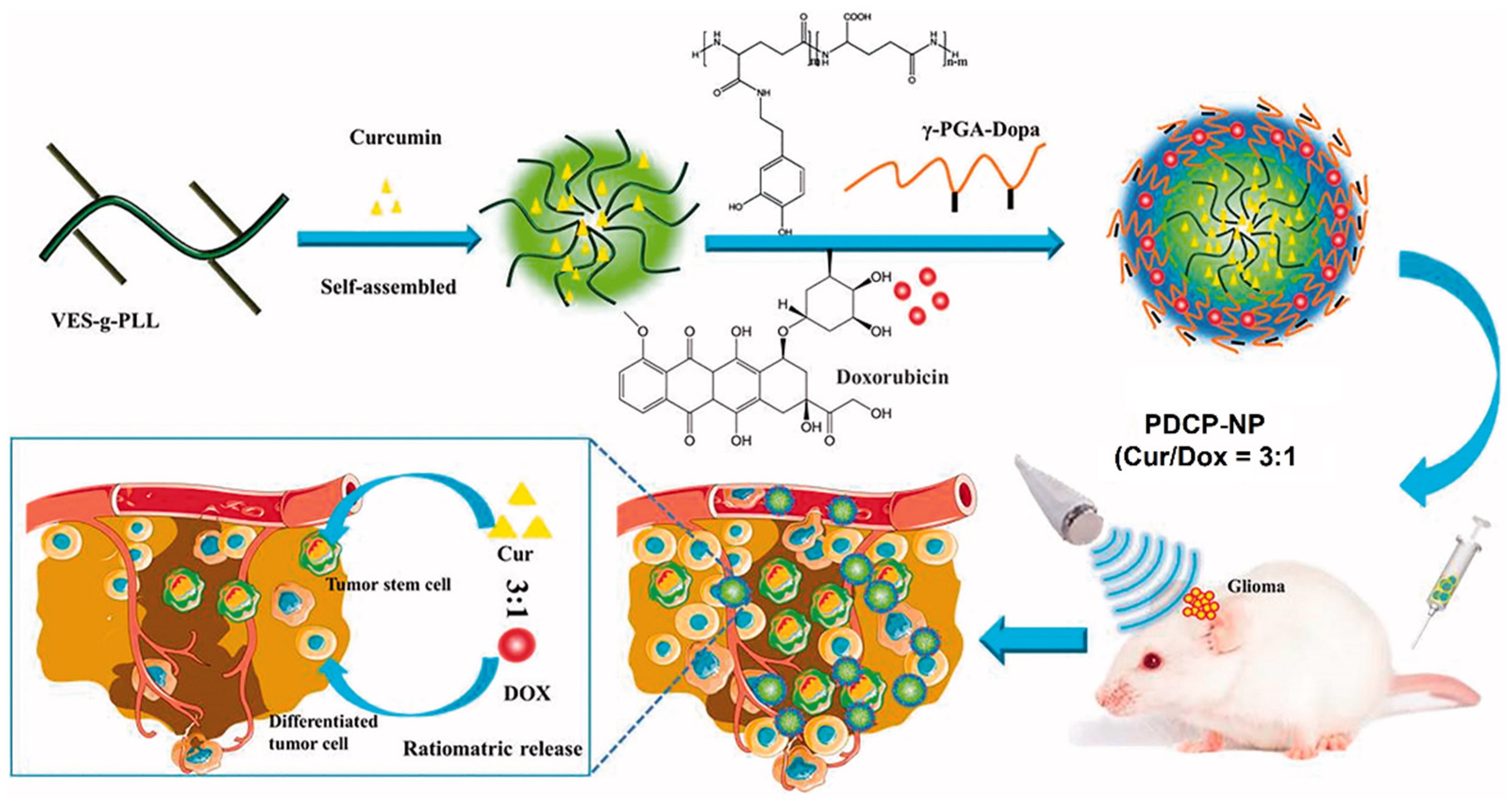
- Xu, H.-L.; Fan, Z.-L.; ZhuGe, D.-L.; Tong, M.-Q.; Shen, B.-X.; Lin, M.-T.; Zhu, Q.-Y.; Jin, B.-H.; Sohawon, Y.; Yao, Q. Ratiometric delivery of two therapeutic candidates with inherently dissimilar physicochemical property through pH-sensitive core–shell nanoparticles targeting the heterogeneous tumor cells of glioma. Drug Deliv. 2018, 25, 1302–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, E.; Kim, E.; Son, H.-Y.; Lim, E.-K.; Lee, H.; Choi, Y.; Park, K.; Han, S.; Suh, J.-S.; Huh, Y.-M. Nanovesicle-mediated systemic delivery of microRNA-34a for CD44 overexpressing gastric cancer stem cell therapy. Biomaterials 2016, 105, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Shen, S.; Zhang, Y.-J.; Xu, C.-F.; Cao, Z.-T.; Wen, L.-P.; Wang, J. Nanoparticle-facilitated autophagy inhibition promotes the efficacy of chemotherapeutics against breast cancer stem cells. Biomaterials 2016, 103, 44–55. [Google Scholar] [CrossRef]
- Sun, R.; Liu, Y.; Li, S.-Y.; Shen, S.; Du, X.-J.; Xu, C.-F.; Cao, Z.-T.; Bao, Y.; Zhu, Y.-H.; Li, Y.-P. Co-delivery of all-trans-retinoic acid and doxorubicin for cancer therapy with synergistic inhibition of cancer stem cells. Biomaterials 2015, 37, 405–414. [Google Scholar] [CrossRef]
- Li, S.-Y.; Sun, R.; Wang, H.-X.; Shen, S.; Liu, Y.; Du, X.-J.; Zhu, Y.-H.; Jun, W. Combination therapy with epigenetic-targeted and chemotherapeutic drugs delivered by nanoparticles to enhance the chemotherapy response and overcome resistance by breast cancer stem cells. J. Control. Release 2015, 205, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Waters, A.K.; Kalyan, P.; Achrol, A.S.; Kesari, S.; Yenugonda, V.M. Lipid–polymer hybrid nanoparticles as a next-generation drug delivery platform: State of the art, emerging technologies, and perspectives. Int. J. Nanomed. 2019, 14, 1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
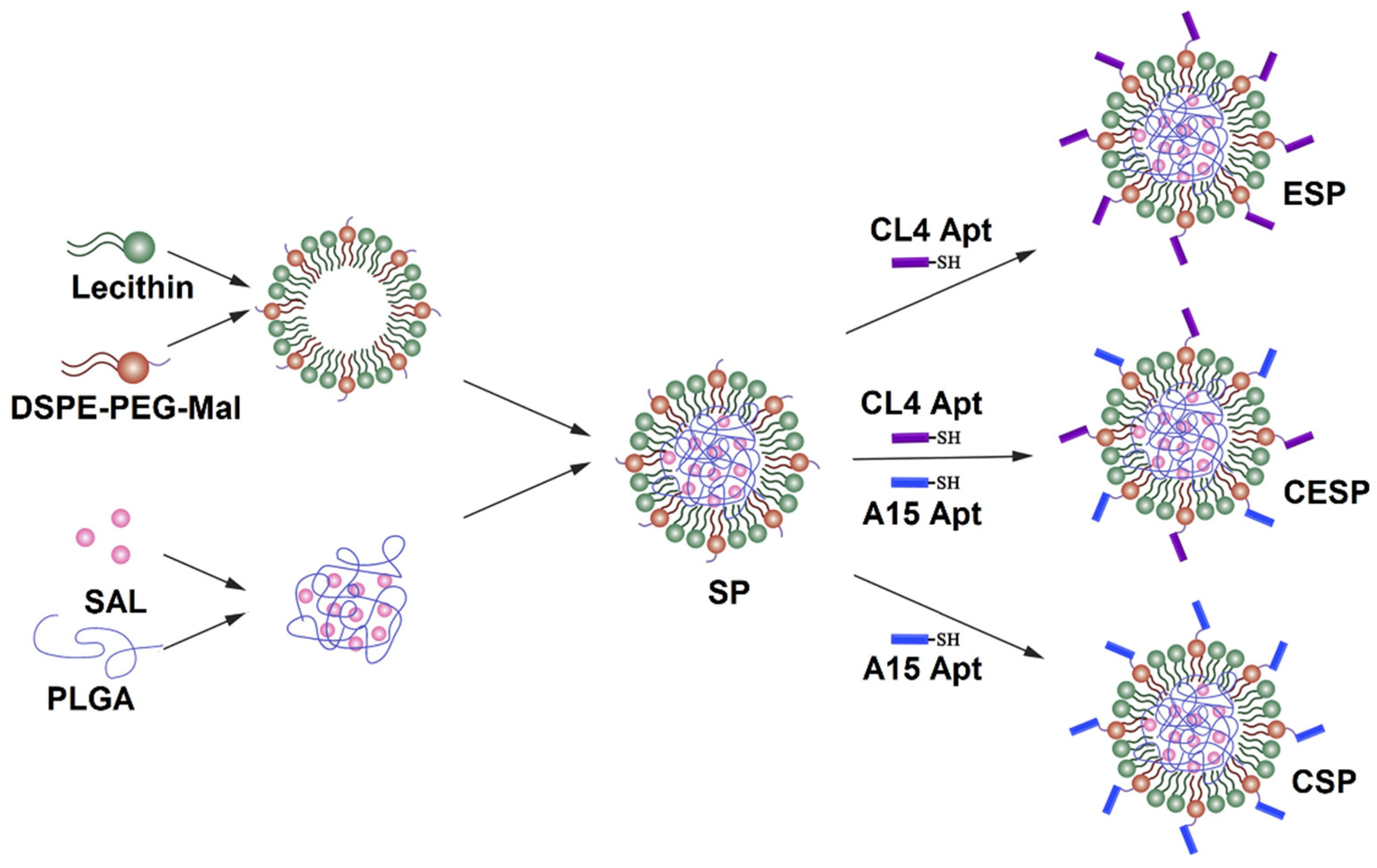
- Zeng, Y.-B.; Yu, Z.-C.; He, Y.-N.; Zhang, T.; Du, L.-B.; Dong, Y.-M.; Chen, H.-W.; Zhang, Y.-Y.; Wang, W.-Q. Salinomycin-loaded lipid-polymer nanoparticles with anti-CD20 aptamers selectively suppress human CD20+ melanoma stem cells. Acta Pharmacol. Sin. 2018, 39, 261–274. [Google Scholar] [CrossRef]
- Chen, F.; Zeng, Y.; Qi, X.; Chen, Y.; Ge, Z.; Jiang, Z.; Zhang, X.; Dong, Y.; Chen, H.; Yu, Z. Targeted salinomycin delivery with EGFR and CD133 aptamers based dual-ligand lipid-polymer nanoparticles to both osteosarcoma cells and cancer stem cells. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 2115–2127. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.-Y.; Shen, Y.-A.; Chen, M.-H.; Wen, Y.-H.; Hsieh, P.-I.; Lo, C.-L. The feasibility of ROS-and GSH-responsive micelles for treating tumor-initiating and metastatic cancer stem cells. J. Mater. Chem. B 2019, 7, 3109–3118. [Google Scholar] [CrossRef]
- Yang, R.; Mondal, G.; Wen, D.; Mahato, R.I. Combination therapy of paclitaxel and cyclopamine polymer-drug conjugates to treat advanced prostate cancer. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 391–401. [Google Scholar] [CrossRef] [PubMed]
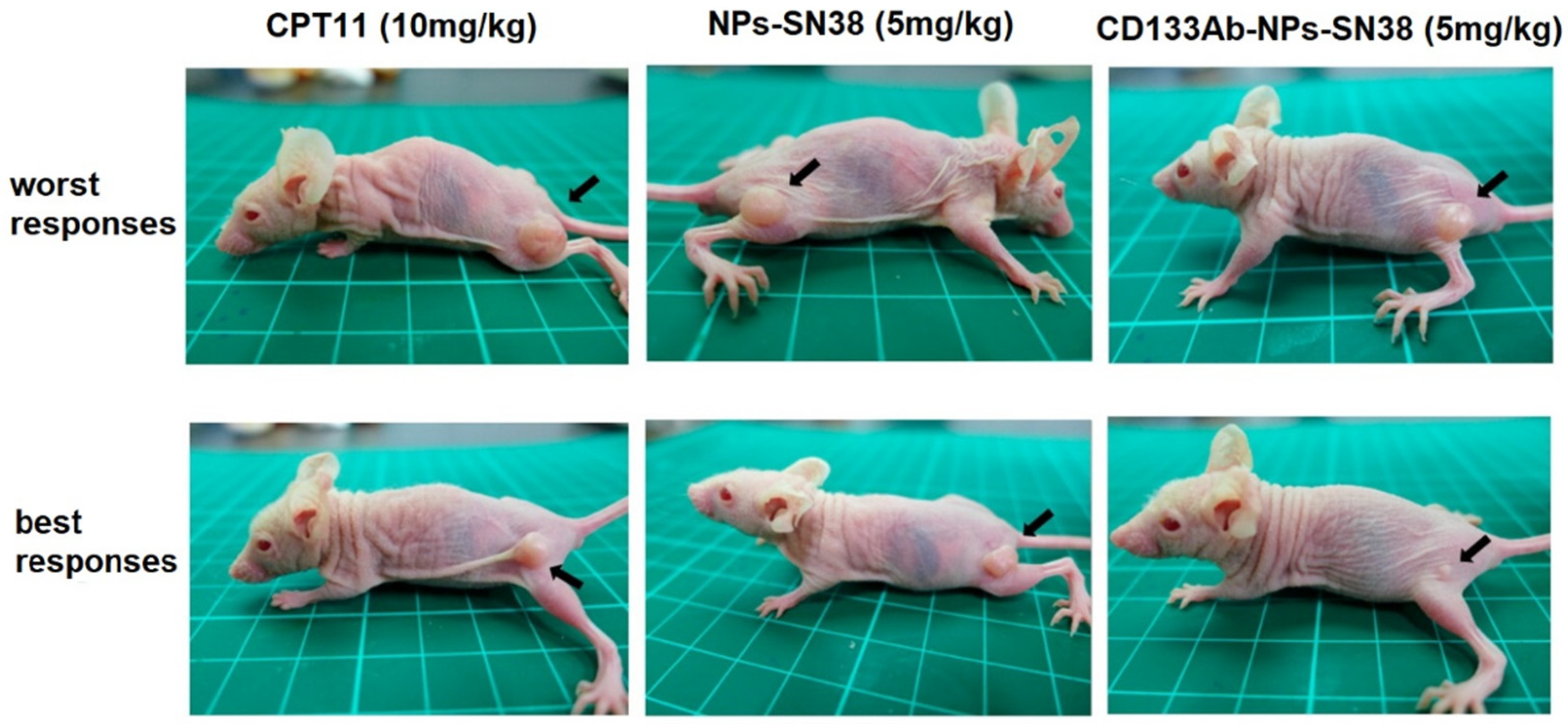
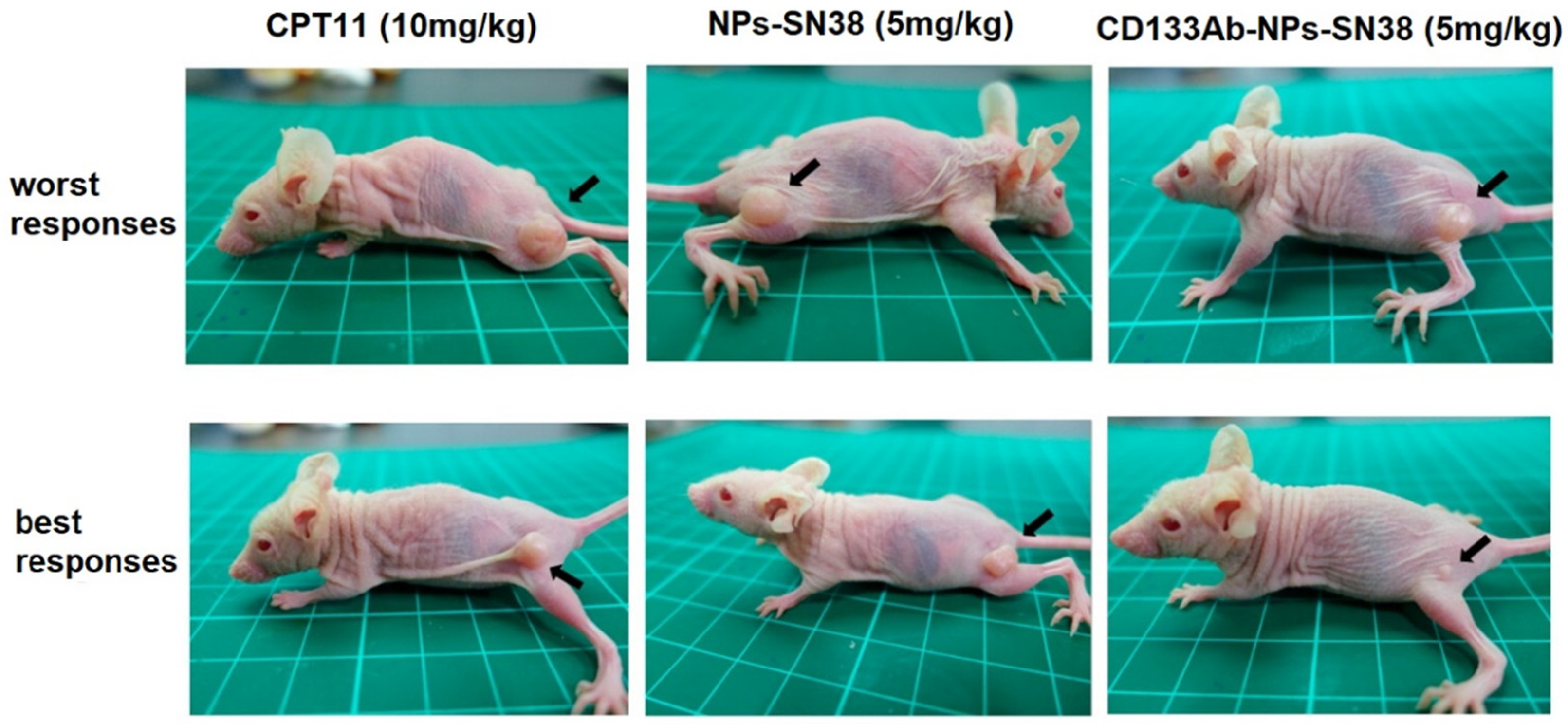
- Ning, S.-T.; Lee, S.-Y.; Wei, M.-F.; Peng, C.-L.; Lin, S.Y.-F.; Tsai, M.-H.; Lee, P.-C.; Shih, Y.-H.; Lin, C.-Y.; Luo, T.-Y.; et al. Targeting colorectal cancer stem-like cells with anti-CD133 antibody-conjugated SN-38 nanoparticles. ACS Appl. Mater. Interfaces 2016, 8, 17793–17804. [Google Scholar] [CrossRef] [PubMed]
- Debele, T.A.; Yu, L.-Y.; Yang, C.-S.; Shen, Y.-A.; Lo, C.-L. pH-and GSH-sensitive hyaluronic acid-MP conjugate micelles for intracellular delivery of doxorubicin to colon cancer cells and cancer stem cells. Biomacromolecules 2018, 19, 3725–3737. [Google Scholar] [CrossRef]
- Gaio, E.; Conte, C.; Esposito, D.; Reddi, E.; Quaglia, F.; Moret, F. CD44 Targeting Mediated by Polymeric Nanoparticles and Combination of Chlorine TPCS2a-PDT and Docetaxel-Chemotherapy for Efficient Killing of Breast Differentiated and Stem Cancer Cells In Vitro. Cancers 2020, 12, 278. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Liu, J.; Xie, F.; Lu, Y.; Yin, C.; Shen, X. Co-Delivery of Docetaxel and Salinomycin to Target Both Breast Cancer Cells and Stem Cells by PLGA/TPGS Nanoparticles. Int. J. Nanomed. 2019, 14, 9199. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sun, M.; Li, W.; Fan, L.; Zhou, Y.; Hu, Z. A novel CD133-and EpCAM-targeted liposome with redox-responsive properties capable of synergistically eliminating liver cancer stem cells. Front. Chem. 2020, 8, 649. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Chen, Y.; Zhao, H.; Qiao, G.; Liu, M.; Zhang, C.; Cui, D.; Ma, L. Dual-modified cationic liposomes loaded with paclitaxel and survivin siRNA for targeted imaging and therapy of cancer stem cells in brain glioma. Drug Deliv. 2018, 25, 1718–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.; Lu, M.; Ming, L.; Chen, Y.; Cheng, K.; Zhou, J.; Jiang, S.; Lin, Z.; Chen, D. 89Zr-Labeled Multifunctional Liposomes Conjugate Chitosan for PET-Trackable Triple-Negative Breast Cancer Stem Cell Targeted Therapy. Int. J. Nanomed. 2020, 15, 9061. [Google Scholar] [CrossRef]
- Yang, Z.; Sun, N.; Cheng, R.; Zhao, C.; Liu, Z.; Li, X.; Liu, J.; Tian, Z. pH multistage responsive micellar system with charge-switch and PEG layer detachment for co-delivery of paclitaxel and curcumin to synergistically eliminate breast cancer stem cells. Biomaterials 2017, 147, 53–67. [Google Scholar] [CrossRef]
- Wang, H.; Agarwal, P.; Zhao, S.; Xu, R.X.; Yu, J.; Lu, X.; He, X. Hyaluronic acid-decorated dual responsive nanoparticles of Pluronic F127, PLGA, and chitosan for targeted co-delivery of doxorubicin and irinotecan to eliminate cancer stem-like cells. Biomaterials 2015, 72, 74–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Shi, S.; Ming, Y.; Wang, L.; Li, C.; Luo, M.; Li, Z.; Li, B.; Chen, J. Specific cancer stem cell-therapy by albumin nanoparticles functionalized with CD44-mediated targeting. J. Nanobiotechnol. 2018, 16, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Usacheva, M.; Swaminathan, S.K.; Kirtane, A.R.; Panyam, J. Enhanced photodynamic therapy and effective elimination of cancer stem cells using surfactant–polymer nanoparticles. Mol. Pharm. 2014, 11, 3186–3195. [Google Scholar] [CrossRef]
- Rao, W.; Wang, H.; Han, J.; Zhao, S.; Dumbleton, J.; Agarwal, P.; Zhang, W.; Zhao, G.; Yu, J.; Zynger, D.L. Chitosan-decorated doxorubicin-encapsulated nanoparticle targets and eliminates tumor reinitiating cancer stem-like cells. ACS Nano 2015, 9, 5725–5740. [Google Scholar] [CrossRef]
- Wu, P.; Liu, Q.; Wang, Q.; Qian, H.; Yu, L.; Liu, B.; Li, R. Novel silk fibroin nanoparticles incorporated silk fibroin hydrogel for inhibition of cancer stem cells and tumor growth. Int. J. Nanomed. 2018, 13, 5405. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Dong, S.; Bhattacharyya, J.; Chen, M. iTEP nanoparticle-delivered salinomycin displays an enhanced toxicity to cancer stem cells in orthotopic breast tumors. Mol. Pharm. 2014, 11, 2703–2712. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug. Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Sharma, A.; Goyal, A.K.; Rath, G. Recent advances in metal nanoparticles in cancer therapy. J. Drug Target. 2018, 26, 617–632. [Google Scholar] [CrossRef]
- McNeil, S.E. Evaluation of nanomedicines: Stick to the basics. Nat. Rev. Mater. 2016, 1, 1–2. [Google Scholar] [CrossRef]
- Mohd-Zahid, M.H.; Mohamud, R.; Abdullah, C.A.C.; Lim, J.; Alem, H.; Hanaffi, W.N.W.; Iskandar, Z. Colorectal cancer stem cells: A review of targeted drug delivery by gold nanoparticles. RSC Adv. 2020, 10, 973–985. [Google Scholar] [CrossRef]
- Murphy, C.J.; Gole, A.M.; Stone, J.W.; Sisco, P.N.; Alkilany, A.M.; Goldsmith, E.C.; Baxter, S.C. Gold nanoparticles in biology: Beyond toxicity to cellular imaging. Acc. Chem. Res. 2008, 41, 1721–1730. [Google Scholar] [CrossRef]
- Safwat, M.A.; Soliman, G.M.; Sayed, D.; Attia, M.A. Gold nanoparticles enhance 5-fluorouracil anticancer efficacy against colorectal cancer cells. Int. J. Pharm. 2016, 513, 648–658. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, W.; Lim, Y.C.; Liu, T. Salinomycin-loaded gold nanoparticles for treating cancer stem cells by ferroptosis-induced cell death. Mol. Pharm. 2019, 16, 2532–2539. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Niestroj, M.; Yuan, D.; Chang, S.; Chen, J. Treating cancer stem cells and cancer metastasis using glucose-coated gold nanoparticles. Int. J. Nanomed. 2015, 10, 2065–2077. [Google Scholar]
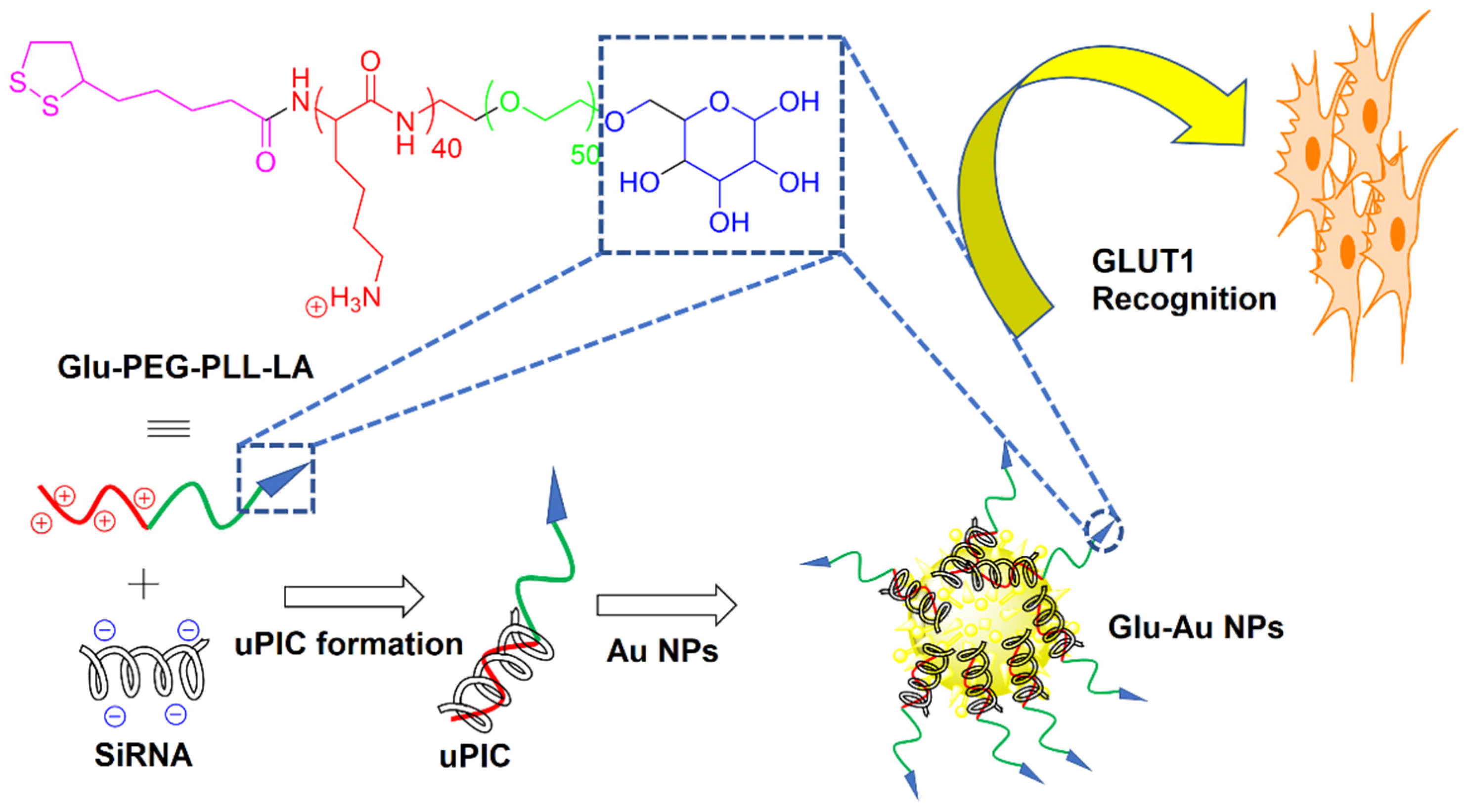
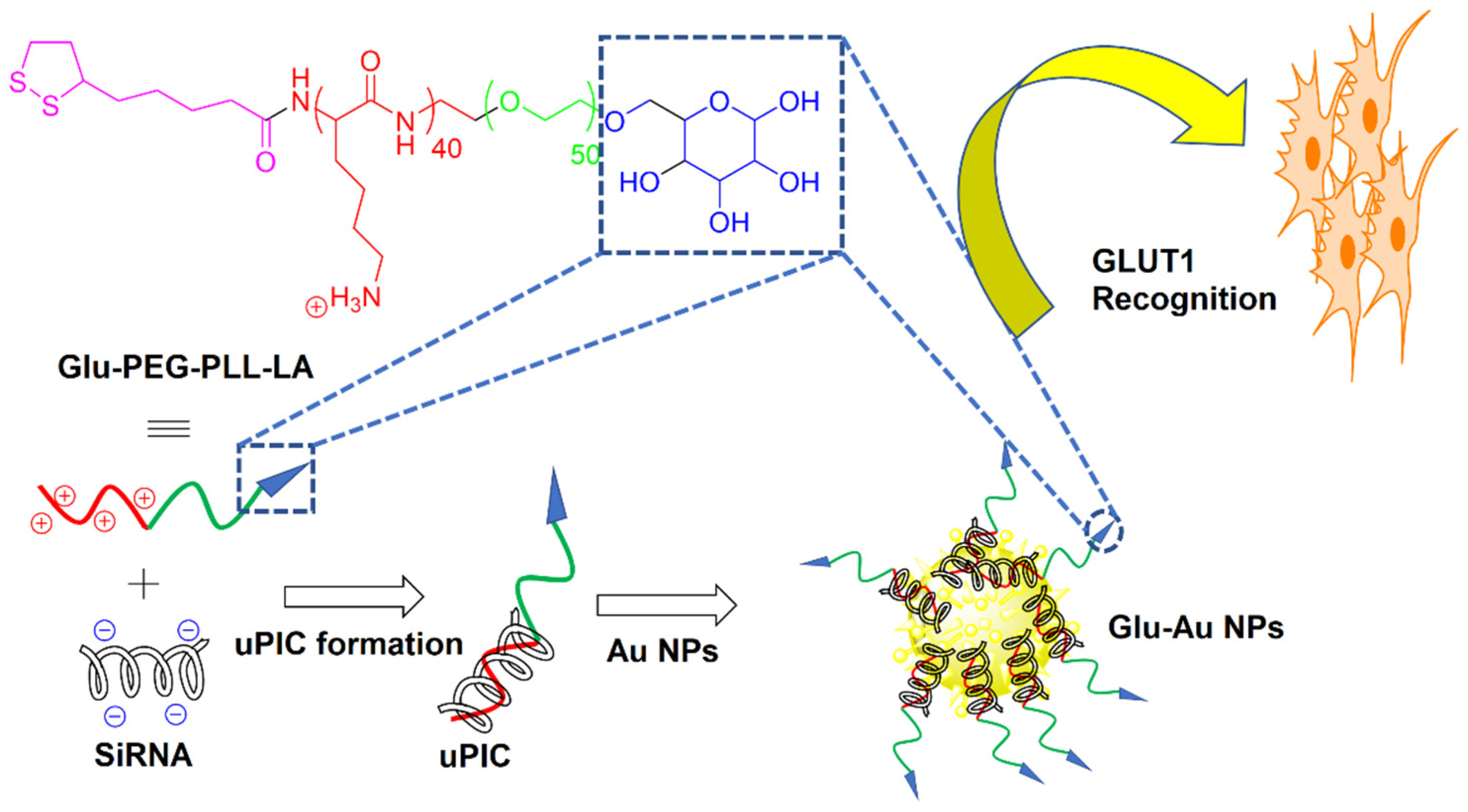
- Yi, Y.; Kim, H.J.; Zheng, M.; Mi, P.; Naito, M.; Kim, B.S.; Min, H.S.; Hayashi, K.; Perche, F.; Toh, K. Glucose-linked sub-50-nm unimer polyion complex-assembled gold nanoparticles for targeted siRNA delivery to glucose transporter 1-overexpressing breast cancer stem-like cells. J. Control. Release 2019, 295, 268–277. [Google Scholar] [CrossRef]
- Kim, H.J.; Takemoto, H.; Yi, Y.; Zheng, M.; Maeda, Y.; Chaya, H.; Hayashi, K.; Mi, P.; Pittella, F.; Christie, R.J.; et al. Precise Engineering of siRNA Delivery Vehicles to Tumors Using Polyion Complexes and Gold Nanoparticles. ACS Nano 2014, 8, 8979–8991. [Google Scholar] [CrossRef]
- Yi, Y.; Kim, H.J.; Mi, P.; Zheng, M.; Takemoto, H.; Toh, K.; Kim, B.S.; Hayashi, K.; Naito, M.; Matsumoto, Y.; et al. Targeted systemic delivery of siRNA to cervical cancer model using cyclic RGD-installed unimer polyion complex-assembled gold nanoparticles. J. Control. Release 2016, 244, 247–256. [Google Scholar] [CrossRef]
- Patra, C.R.; Bhattacharya, R.; Mukherjee, P. Fabrication and functional characterization of goldnanoconjugates for potential application in ovarian cancer. J. Mater. Chem. 2009, 20, 547–554. [Google Scholar] [CrossRef]
- Chanda, N.; Kattumuri, V.; Shukla, R.; Zambre, A.; Katti, K.; Upendran, A.; Kulkarni, R.R.; Kan, P.; Fent, G.M.; Casteel, S.W. Bombesin functionalized gold nanoparticles show in vitro and in vivo cancer receptor specificity. Proc. Natl. Acad. Sci. USA 2010, 107, 8760–8765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.-L.; Wang, L.; Liu, X.-Y.; Zhang, Z.-P.; Guo, H.-C.; Liu, W.-M.; Tang, S.-H. In vitro cancer cell imaging and therapy using transferrin-conjugated gold nanoparticles. Cancer Lett. 2009, 274, 319–326. [Google Scholar] [CrossRef]
- Latorre, A.; Latorre, A.; Castellanos, M.; Rodriguez Diaz, C.; Lazaro-Carrillo, A.; Aguado, T.; Lecea, M.; Romero-Pérez, S.; Calero, M.; Sanchez-Puelles, J.M. Multifunctional albumin-stabilized gold nanoclusters for the reduction of cancer stem cells. Cancers 2019, 11, 969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
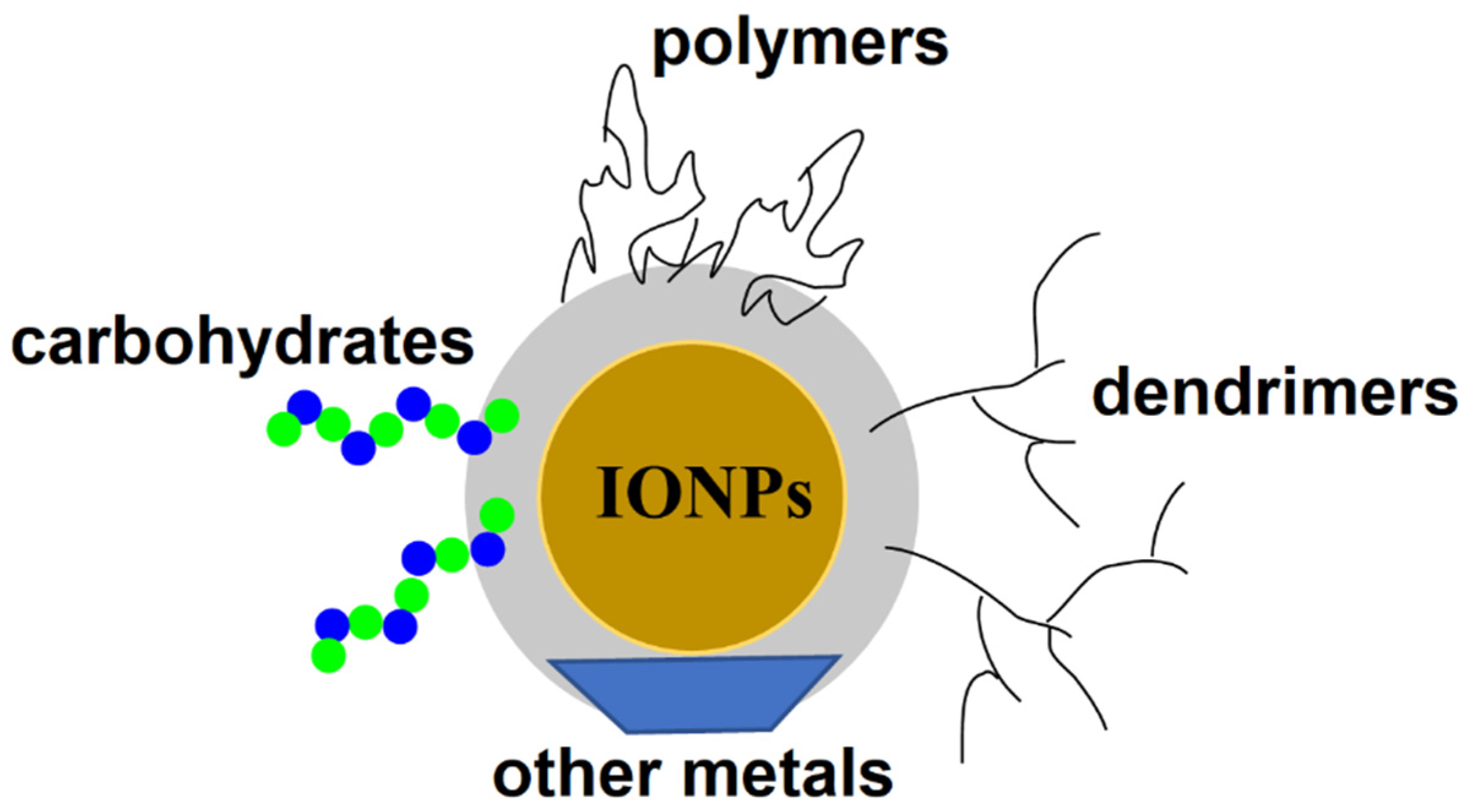
- El-Boubbou, K. Magnetic iron oxide nanoparticles as drug carriers: Preparation, conjugation and delivery. Nanomedicine 2018, 13, 929–952. [Google Scholar] [CrossRef]
- Su, Z.; Liu, D.; Chen, L.; Zhang, J.; Ru, L.; Chen, Z.; Gao, Z.; Wang, X. CD44-targeted magnetic nanoparticles kill head and neck squamous cell carcinoma stem cells in an alternating magnetic field. Int. J. Nanomed. 2019, 14, 7549. [Google Scholar] [CrossRef] [Green Version]
- Miller-Kleinhenz, J.; Guo, X.; Qian, W.; Zhou, H.; Bozeman, E.N.; Zhu, L.; Ji, X.; Wang, Y.A.; Styblo, T.; O’Regan, R. Dual-targeting Wnt and uPA receptors using peptide conjugated ultra-small nanoparticle drug carriers inhibited cancer stem-cell phenotype in chemo-resistant breast cancer. Biomaterials 2018, 152, 47–62. [Google Scholar] [CrossRef]
- Chen, L.; Liu, M.; Zhou, Q.; Li, X. Recent developments of mesoporous silica nanoparticles in biomedicine. Emergent Mater. 2020, 3, 381–405. [Google Scholar] [CrossRef]
- Zhou, Y.; Quan, G.; Wu, Q.; Zhang, X.; Niu, B.; Wu, B.; Huang, Y.; Pan, X.; Wu, C. Mesoporous silica nanoparticles for drug and gene delivery. Acta Pharm. Sin. B 2018, 8, 165–177. [Google Scholar] [CrossRef]
- Pan, Y.; Zhou, S.; Li, Y.; Parshad, B.; Li, W.; Haag, R. Novel dendritic polyglycerol-conjugated, mesoporous silica-based targeting nanocarriers for co-delivery of doxorubicin and tariquidar to overcome multidrug resistance in breast cancer stem cells. J. Control. Release 2021, 330, 1106–1117. [Google Scholar] [CrossRef]
- Tsai, P.-H.; Wang, M.-L.; Chang, J.-H.; Yarmishyn, A.A.; Nhi Nguyen, P.N.; Chen, W.; Chien, Y.; Huo, T.-I.; Mou, C.-Y.; Chiou, S.-H. Dual delivery of HNF4α and cisplatin by mesoporous silica nanoparticles inhibits cancer pluripotency and tumorigenicity in hepatoma-derived CD133-expressing stem cells. ACS Appl. Mater. Interfaces 2019, 11, 19808–19818. [Google Scholar] [CrossRef]
- Shen, J.L.; Wolfram, J.; Ferrari, M.; Shen, H.F. Taking the vehicle out of drug delivery. Mater. Today 2017, 20, 95–97. [Google Scholar] [CrossRef] [Green Version]
- Volta-Duran, E.; Serna, N.; Sanchez-Garcia, L.; Avino, A.; Sanchez, J.M.; Lopez-Laguna, H.; Cano-Garrido, O.; Casanova, I.; Mangues, R.; Eritja, R.; et al. Design and engineering of tumor-targeted, dual-acting cytotoxic nanoparticles. Acta Biomater. 2021, 119, 312–322. [Google Scholar] [CrossRef]
- Casanova, S.; Unzueta, U.; Arroyo-Solera, I.; Cespedes, M.V.; Villaverde, A.; Mangues, R.; Vazquez, E. Protein-driven nanomedicines in oncotherapy. Curr. Opin. Pharmacol. 2019, 47, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Deci, M.B.; Liu, M.X.; Gonya, J.; Lee, C.J.; Li, T.Y.; Ferguson, S.W.; Bonacquisti, E.E.; Wang, J.L.; Nguyen, J. Carrier-Free CXCR4-Targeted Nanoplexes Designed for Polarizing Macrophages to Suppress Tumor Growth. Cell Mol. Bioeng. 2019, 12, 375–388. [Google Scholar] [CrossRef]
- Volta-Duran, E.; Cano-Garrido, O.; Serna, N.; Lopez-Laguna, H.; Sanchez-Garcia, L.; Pesarrodona, M.; Sanchez-Chardi, A.; Mangues, R.; Villaverde, A.; Vazquez, E.; et al. Controlling self-assembling and tumor cell-targeting of protein-only nanoparticles through modular protein engineering. Sci. China Mater. 2020, 63, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Shipunova, V.O.; Kotelnikova, P.A.; Aghayeva, U.F.; Stremovskiy, O.A.; Novikov, I.A.; Schulga, A.A.; Nikitin, M.P.; Deyev, S.M. Self-assembling nanoparticles biofunctionalized with magnetite-binding protein for the targeted delivery to HER2/neu overexpressing cancer cells. J. Magn. Mater. 2019, 469, 450–455. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, L.; Ji, X.R.; Shin, M.C.; Xie, S.P.; Pan, B.Y.; Yu, F.; Zhao, J.W.; Yang, V.C. A self-assembly and stimuli-responsive fusion gelonin delivery system for tumor treatment. J. Ind. Eng. Chem. 2020, 89, 409–415. [Google Scholar] [CrossRef]
- Serna, N.; Sanchez-Garcia, L.; Unzueta, U.; Diaz, R.; Vazquez, E.; Mangues, R.; Villaverde, A. Protein-Based Therapeutic Killing for Cancer Therapies. Trends Biotechnol. 2018, 36, 318–335. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.C.; Zhi, K.K.; Ding, Z.Y.; Sun, Y.; Li, S.; Li, M.Y.; Pu, K.F.; Zou, J. Emergence in protein derived nanomedicine as anticancer therapeutics: More than a tour de force. Semin. Cancer Biol. 2021, 69, 77–90. [Google Scholar] [CrossRef]
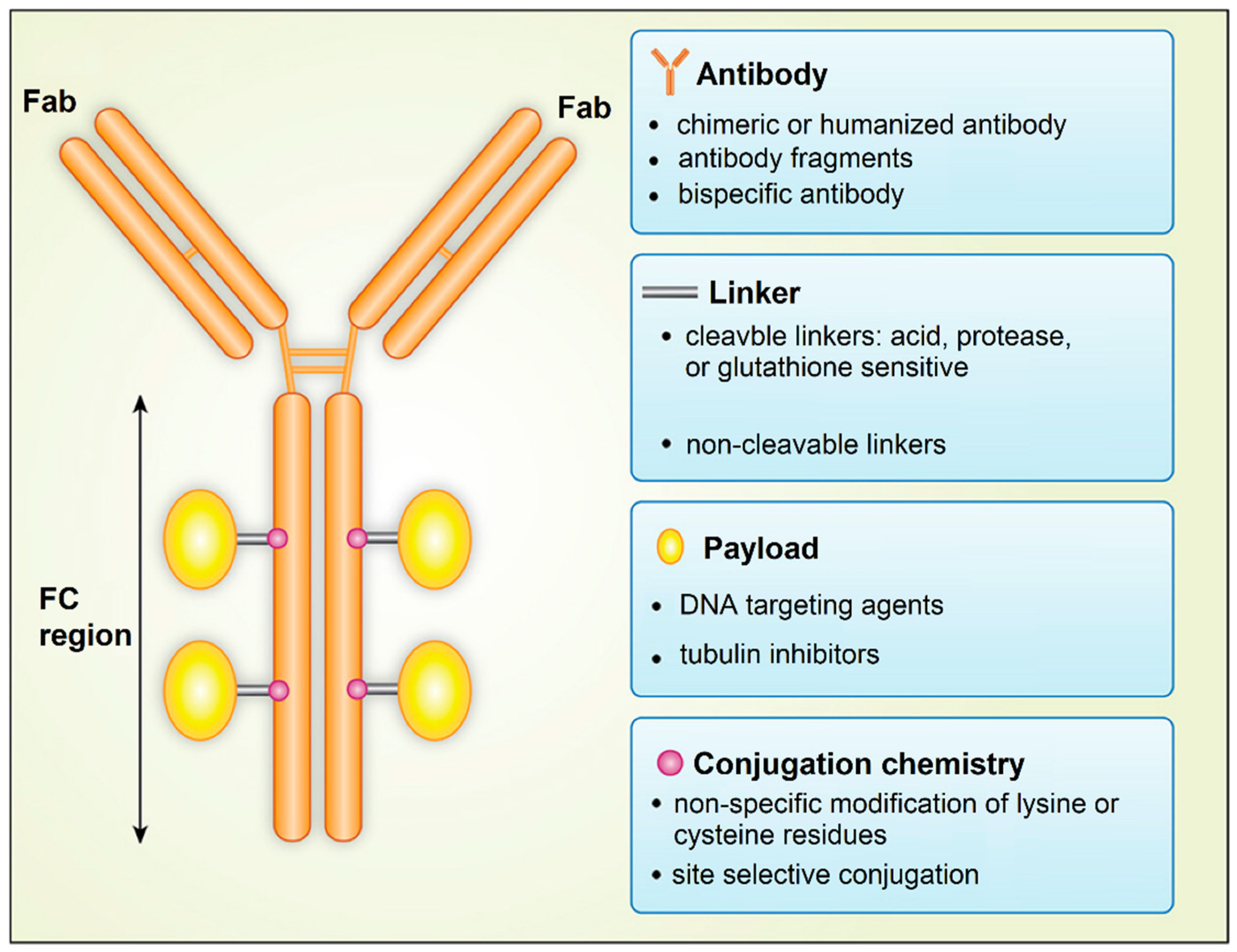
- Villela-Martinez, L.M.; Velez-Ayala, A.K.; Lopez-Sanchez, R.D.; Martinez-Cardona, J.A.; Hernandez-Hernandez, J.A. Advantages of Drug Selective Distribution in Cancer Treatment: Brentuximab Vedotin. Int J. Pharmacol. 2017, 13, 785–807. [Google Scholar] [CrossRef]
- Walko, C.M.; West, H. Antibody Drug Conjugates for Cancer Treatment. Jama Oncol. 2019, 5, 1648. [Google Scholar] [CrossRef] [Green Version]
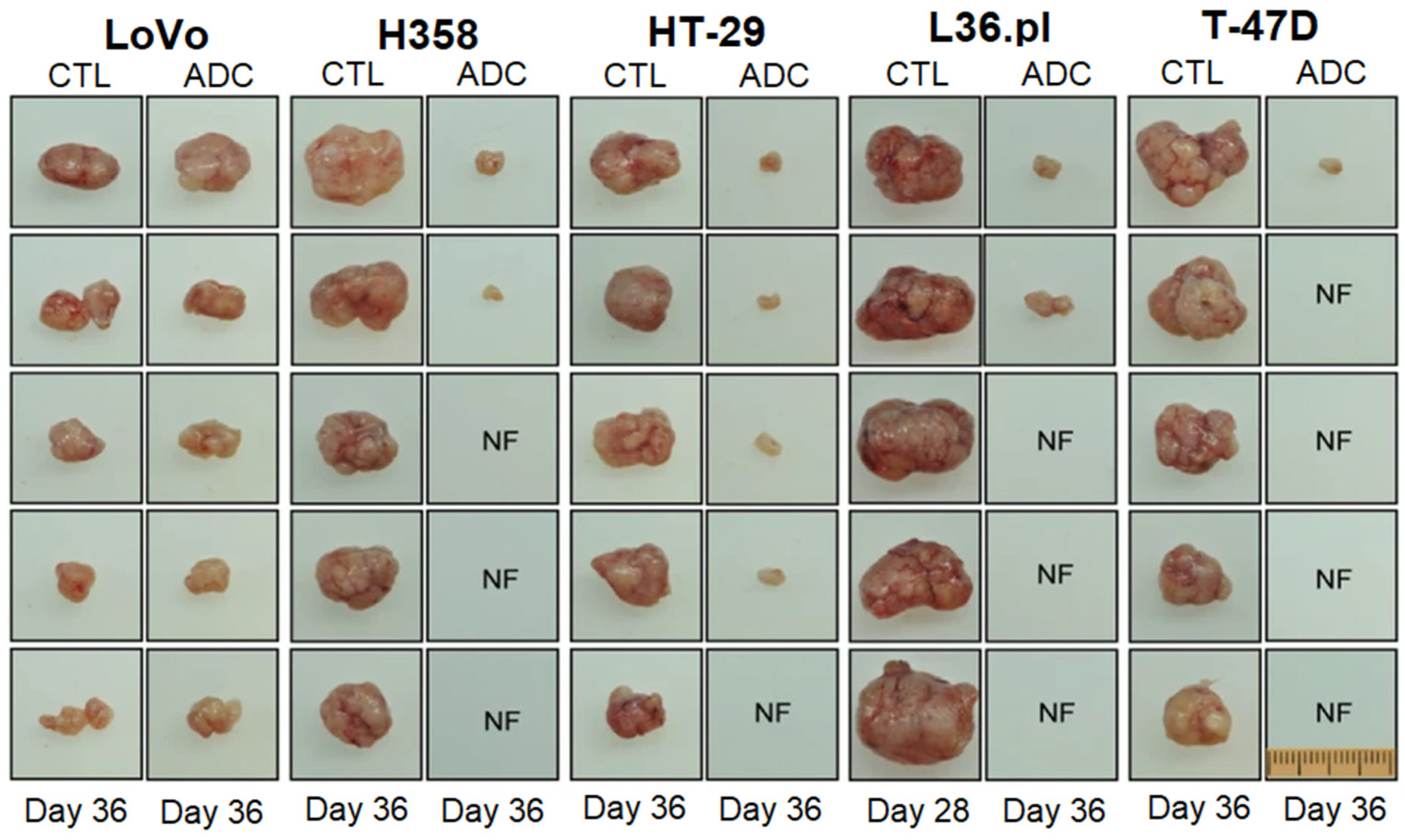
- Fernandes, E.; Ferreira, J.A.; Peixoto, A.; Lima, L.; Barroso, S.; Sarmento, B.; Santos, L.L. New trends in guided nanotherapies for digestive cancers: A systematic review. J. Control. Release 2015, 209, 288–307. [Google Scholar] [CrossRef]
- Hafeez, U.; Parakh, S.; Gan, H.K.; Scott, A.M. Antibody-Drug Conjugates for Cancer Therapy. Molecules 2020, 25, 4764. [Google Scholar] [CrossRef]
- Chari, R.V.J.; Miller, M.L.; Widdison, W.C. Antibody-Drug Conjugates: An Emerging Concept in Cancer Therapy. Angew. Chem. Int. Ed. 2014, 53, 3796–3827. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Bertozzi, C.R. Site-Specific Antibody-Drug Conjugates: The Nexus of Biciorthogonal Chemistry, Protein Engineering, and Drug Development. Bioconjugate Chem. 2015, 26, 176–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanaswamy, R.; Torchilin, V.P. Targeted Delivery of Combination Therapeutics Using Monoclonal Antibody 2C5-Modified Immunoliposomes for Cancer Therapy. Pharm Res. Dordr. 2021, 38, 429–450. [Google Scholar] [CrossRef]
- Masoumi, E.; Jafarzadeh, L.; Mirzaei, H.R.; Alishah, K.; Fallah-Mehrjardi, K.; Rostamian, H.; Khakpoor-Koosheh, M.; Meshkani, R.; Noorbakhsh, F.; Hadjati, J. Genetic and pharmacological targeting of A2a receptor improves function of anti-mesothelin CAR T cells. J. Exp. Clin. Cancer Res. 2020, 39, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Golfier, S.; Kopitz, C.; Kahnert, A.; Heisler, I.; Schatz, C.A.; Stelte-Ludwig, B.; Mayer-Bartschmid, A.; Unterschemmann, K.; Bruder, S.; Linden, L.; et al. Anetumab Ravtansine: A Novel Mesothelin-Targeting Antibody-Drug Conjugate Cures Tumors with Heterogeneous Target Expression Favored by Bystander Effect. Mol. Cancer Ther. 2014, 13, 1537–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. DS-8201a, A Novel HER2-Targeting ADC with a Novel DNA Topoisomerase I Inhibitor, Demonstrates a Promising Antitumor Efficacy with Differentiation from T-DM1. Clin. Cancer Res. 2016, 22, 5097–5108. [Google Scholar] [CrossRef] [Green Version]
- Millar, R.; Kilbey, A.; Remak, S.J.; Severson, T.M.; Dhayade, S.; Sandilands, E.; Foster, K.; Bryant, D.M.; Blyth, K.; Coffelt, S.B. The MSP-RON axis stimulates cancer cell growth in models of triple negative breast cancer. Mol. Oncol. 2020, 14, 1868–1880. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.M.; Feng, L.; Suthe, S.R.; Weng, T.H.; Hu, C.Y.; Liu, Y.Z.; Wu, Z.G.; Wang, M.H.; Yao, H.P. Therapeutic efficacy of a novel humanized antibody-drug conjugate recognizing plexin-semaphorin-integrin domain in the RON receptor for targeted cancer therapy. J. Immunother. Cancer 2019, 7, 250. [Google Scholar] [CrossRef]
- Godwin, C.D.; Laszlo, G.S.; Wood, B.L.; Correnti, C.E.; Bates, O.M.; Garling, E.E.; Mao, Z.J.; Beddoe, M.E.; Lunn, M.C.; Humbert, O.; et al. The CD33 splice isoform lacking exon 2 as therapeutic target in human acute myeloid leukemia. Leukemia 2020, 34, 2479–2483. [Google Scholar] [CrossRef]
- Kovtun, Y.; Noordhuis, P.; Whiteman, K.R.; Watkins, K.; Jones, G.E.; Harvey, L.; Lai, K.C.; Portwood, S.; Adams, S.; Sloss, C.M.; et al. IMGN779, a Novel CD33-Targeting Antibody-Drug Conjugate with DNA-Alkylating Activity, Exhibits Potent Antitumor Activity in Models of AML. Mol. Cancer Ther. 2018, 17, 1271–1279. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Leon, F.; Rauth, S.; Batra, S.K.; Ponnusamy, M.P. A Systematic Review on the Implications of O-linked Glycan Branching and Truncating Enzymes on Cancer Progression and Metastasis. Cells 2020, 9, 446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starbuck, K.; Al-Alem, L.; Eavarone, D.A.; Hernandez, S.F.; Bellio, C.; Prendergast, J.M.; Stein, J.; Dransfield, D.T.; Zarrella, B.; Growdon, W.B.; et al. Treatment of ovarian cancer by targeting the tumor stem cell-associated carbohydrate antigen, Sialyl-Thomsen-nouveau. Oncotarget 2018, 9, 23289–23305. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Oller, L.; Seras-Franzoso, J.; Andrade, F.; Rafael, D.; Abasolo, I.; Gener, P.; Schwartz, S. Extracellular Vesicles as Drug Delivery Systems in Cancer. Pharmaceutics 2020, 12, 1146. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.W.; Zheng, L.; Zou, X.F.; Wang, J.G.; Zhong, J.N.; Zhong, T.Y. Extracellular vesicles in type 2 diabetes mellitus: Key roles in pathogenesis, complications, and therapy. J. Extracell Vesicles 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Gener, P.; Callejo, P.G.; Seras-Franzoso, J.; Andrade, F.; Rafael, D.; Abasolo, I.; Schwartz, S.J. The potential of nanomedicine to alter cancer stem cell dynamics: The impact of extracellular vesicles. Nanomedicine 2020, 15, 2785–2800. [Google Scholar] [CrossRef]
- Wang, J.H.; Zheng, Y.J.; Zhao, M. Exosome-Based Cancer Therapy: Implication for Targeting Cancer Stem Cells. Front. Pharmacol. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Smyth, T.J.; Redzic, J.S.; Michael, W.B.; Anchordoquy, T.J. Examination of the specificity of tumor cell derived exosomes with tumor cells in vitro. Biochim. Biophys. Acta Biomembr. 2014, 1838, 2954–2965. [Google Scholar] [CrossRef] [Green Version]
- Morishita, H.; Mizushima, N. Diverse Cellular Roles of Autophagy. Annu Rev. Cell Dev. Biol 2019, 35, 453–475. [Google Scholar] [CrossRef]
- Yong, T.Y.; Zhang, X.Q.; Bie, N.N.; Zhang, H.B.; Zhang, X.T.; Li, F.Y.; Hakeem, A.; Hu, J.; Gan, L.; Santos, H.A.; et al. Tumor exosome-based nanoparticles are efficient drug carriers for chemotherapy. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.Y.; Deng, T.; Liu, R.; Bai, M.; Zhou, L.K.; Wang, X.; Li, S.; Wang, X.Y.; Yang, H.; Li, J.L.; et al. Exosome-delivered EGFR regulates liver microenvironment to promote gastric cancer liver metastasis. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.J.; Lei, Z.Q.; Yang, P.H.; Si, A.F.; Xiang, D.M.; Tang, X.W.; Guo, G.M.; Zhou, J.H.; Huser, N. Exosome-transmitted p120-catenin suppresses hepatocellular carcinoma progression via STAT3 pathways. Mol. Carcinog. 2019, 58, 1389–1399. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Yin, H.B.; Li, X.Y.; Zhu, G.M.; He, W.Y.; Gou, X. Bladder cancer cellsecreted exosomal miR21 activates the PI3K/AKT pathway in macrophages to promote cancer progression. Int J. Oncol. 2019, 56, 151–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.; Li, H.J.; Zhu, Z.C.; Mei, P.; Hu, W.M.; Xiong, X.C.; Tao, J. microRNA-21-5p from M2 macrophage-derived extracellular vesicles promotes the differentiation and activity of pancreatic cancer stem cells by mediating KLF3. Cell Biol. Toxicol. 2021. [Google Scholar] [CrossRef]
- Ma, J.W.; Zhang, Y.; Tang, K.; Zhang, H.F.; Yin, X.N.; Li, Y.; Xu, P.W.; Sun, Y.L.; Ma, R.H.; Ji, T.T.; et al. Reversing drug resistance of soft tumor-repopulating cells by tumor cell-derived chemotherapeutic microparticles. Cell Res. 2016, 26, 713–727. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Indication | Targets |
|---|---|
| Acute myeloid leukaemia | CD25, CD33, CD123 (IL-3Rα), FLT3 |
| Breast cancer | CD25, CD174, CD197 (CCR7), CD205 (Ly75), CD228 (P79, SEMF), c-MET, CRIPTO, ErbB2 (HER2), ErbB3 (HER3), FLOR1 (FRα), Globo H, GPNMB, IGF-1R, integrin β-6, PTK7 (CCK4), nectin-4 (PVRL4), ROR2, SLC39A6 (LIV1A ZIP6) |
| Bladder cancer | CD25, CD205(Ly75) |
| Colorectal cancer | CD74, CD174, CD166, CD227 (MUC-1), CD326 (Epcam), CEACAM5, CRIPTO, FAP, ED-B, ErbB3 (HER3) |
| Gastric cancer | CD25, CD197 (CCR7), CD228 (P79, SEMF), FLOR1(FRα), Globo H, GRP20, GCC, SLC39A6 (LIV1A ZIP6) |
| Gliomas GIII and GIV | CD25, EGFR |
| Head and neck cancer | CD71 (transferrin R), CD197 (CCR7), EGFR, SLC39A6 (LIV1A ZIP6) |
| Hodgkin’s lymphoma | CD25, CD30, CD197 (CCR7) |
| Lung cancer | Axl, alpha v beta6, CD25, CD56, CD71 (transferrin R), CD228 (P79, SEMF), CD326, CRIPTO, EGFR, ErbB3 (HER3), FAP, Globo H, GD2, IGF-1R, integrin β-6, mesothelin, PTK7 (CCK4), ROR2, SLC34A2 (NaPi2b), SLC39A6 (LIV1A ZIP6) |
| Liver cancer | CD276 (B7-H3), c-MET |
| Melanoma | CD276 (B7-H3), GD2, GPNMB, ED-B, PMEL 17, endothelin B receptor |
| Mesothelioma | Mesothelin, CD228 (P79, SEMF) |
| Multiple Myeloma | CD38, CD46 (MCP), CD56, CD74, CD138, CD269 (BCMA), endothelin B receptor |
| Non-Hodgkin Lymphoma | CD19, CD20, CD22, CD25, CD30, CD37, CD70, CD71 (transferrin R), CD72, CD79, CD180, CD205 (Ly75), ROR1 |
| Ovarian cancer | CA125(MUC16), CD142 (TF), CD205 (Ly75), FLOR1(FRα), Globo H, mesothelin, PTK7 (CCK4) |
| Pancreatic cancer | CD25, CD71 (transferrin R), CD74, CD227 (MUC1), CD228 (P79, SEMF), GRP20, GCC, IGF-1R, integrin β-6, nectin-4 (PVRL4), SLC34A2 (NaPi2b), SLC44A4, alpha v beta6, mesothelin |
| Prostate cancer | CD46 (MCP), PSMA, STEAP-1, SLC44A4, TENB2 |
| Renal cancer | AGS-16, EGFR, c-MET, CAIX, CD70, FLOR1 (FRα) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ertas, Y.N.; Abedi Dorcheh, K.; Akbari, A.; Jabbari, E. Nanoparticles for Targeted Drug Delivery to Cancer Stem Cells: A Review of Recent Advances. Nanomaterials 2021, 11, 1755. https://doi.org/10.3390/nano11071755
Ertas YN, Abedi Dorcheh K, Akbari A, Jabbari E. Nanoparticles for Targeted Drug Delivery to Cancer Stem Cells: A Review of Recent Advances. Nanomaterials. 2021; 11(7):1755. https://doi.org/10.3390/nano11071755
Chicago/Turabian StyleErtas, Yavuz Nuri, Keyvan Abedi Dorcheh, Ali Akbari, and Esmaiel Jabbari. 2021. "Nanoparticles for Targeted Drug Delivery to Cancer Stem Cells: A Review of Recent Advances" Nanomaterials 11, no. 7: 1755. https://doi.org/10.3390/nano11071755
APA StyleErtas, Y. N., Abedi Dorcheh, K., Akbari, A., & Jabbari, E. (2021). Nanoparticles for Targeted Drug Delivery to Cancer Stem Cells: A Review of Recent Advances. Nanomaterials, 11(7), 1755. https://doi.org/10.3390/nano11071755