Determination of Total Silicon and SiO2 Particles Using an ICP-MS Based Analytical Platform for Toxicokinetic Studies of Synthetic Amorphous Silica


 ,
,  ,
, 
 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Standards and Reagents
2.2. Instrumentation
2.3. Sample Preparation
2.3.1. Decontamination Procedures
2.3.2. Sample Collection
2.3.3. Microwave Digestion for Total Si Determination
2.4. Preparation and Characterization of the Quality Control Material (QCM)
2.5. Method Validation and Analytical Quality Control
2.6. Characterization of the Si Content of Maintenance Diets
2.7. Determination of SiO2 Particles by sp-ICP-MS and SEM-EDX
2.8. Statistics
3. Results
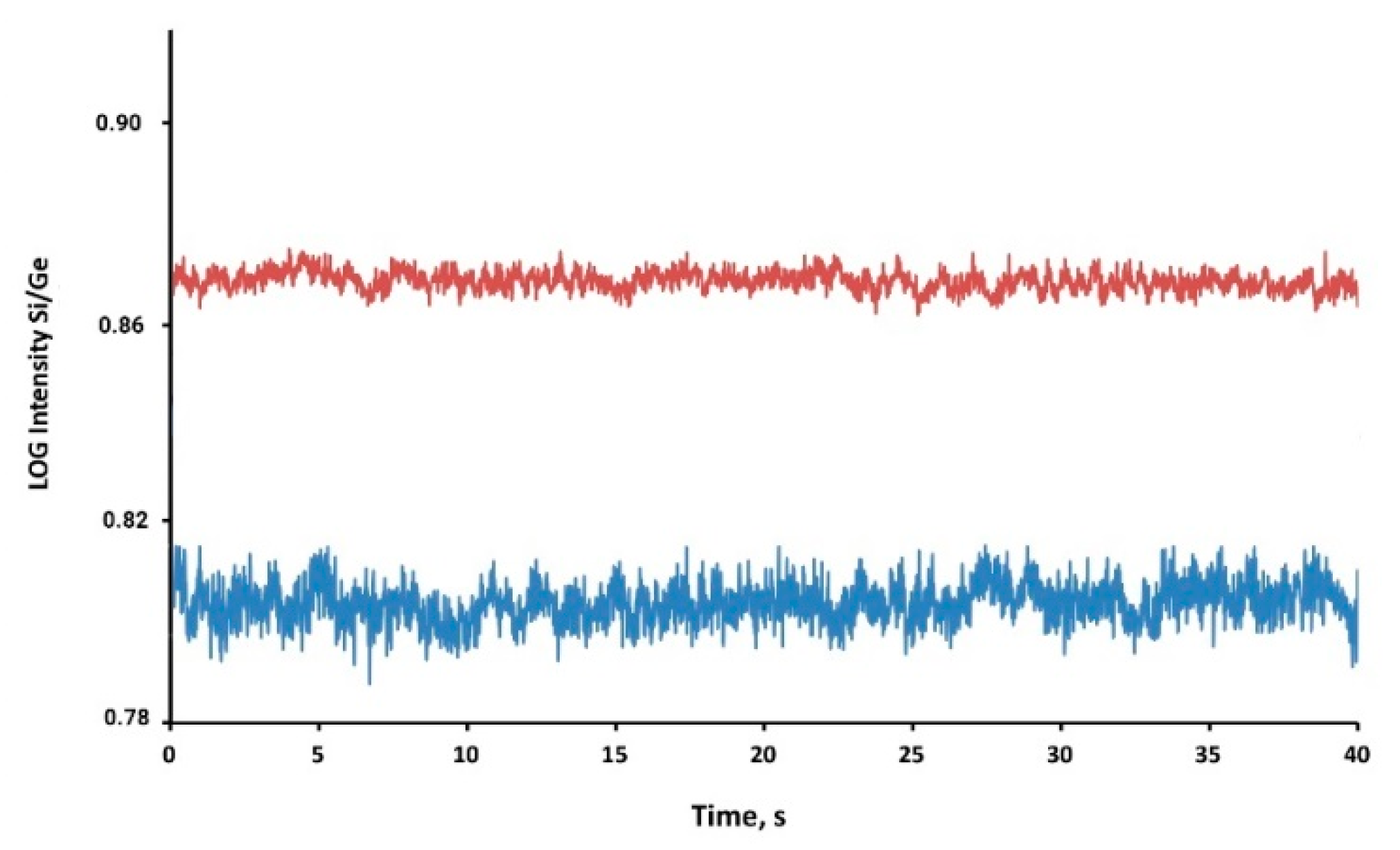
3.1. Interference-Free Detection of Total Si
3.2. Characterization of the QCM by a Multi-Technique Approach
3.3. Method Validation
3.3.1. Limits of Detection and Quantification
3.3.2. Trueness
3.3.3. Linearity and Precision
3.4. Applications to Real Samples
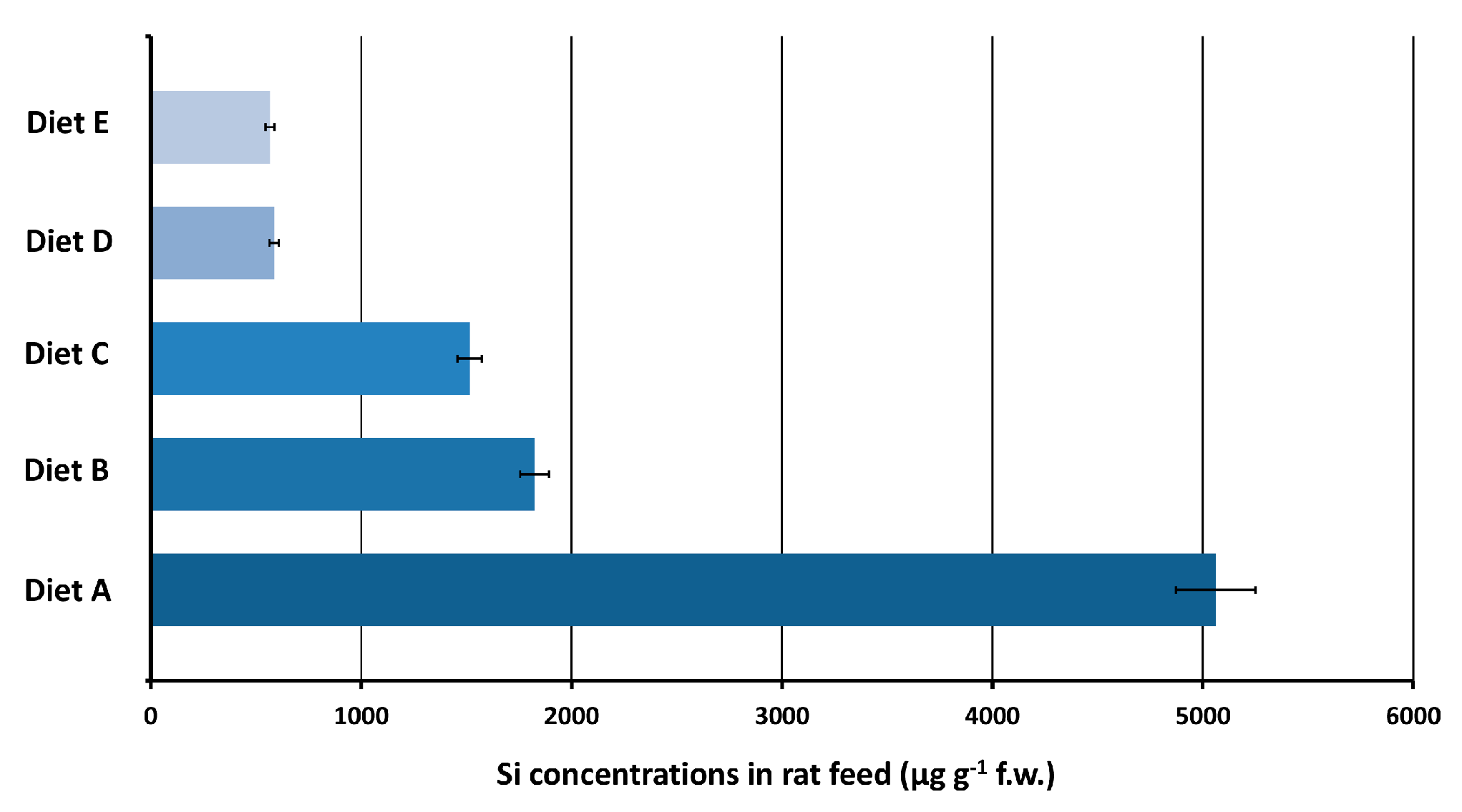
3.4.1. Maintenance Diet
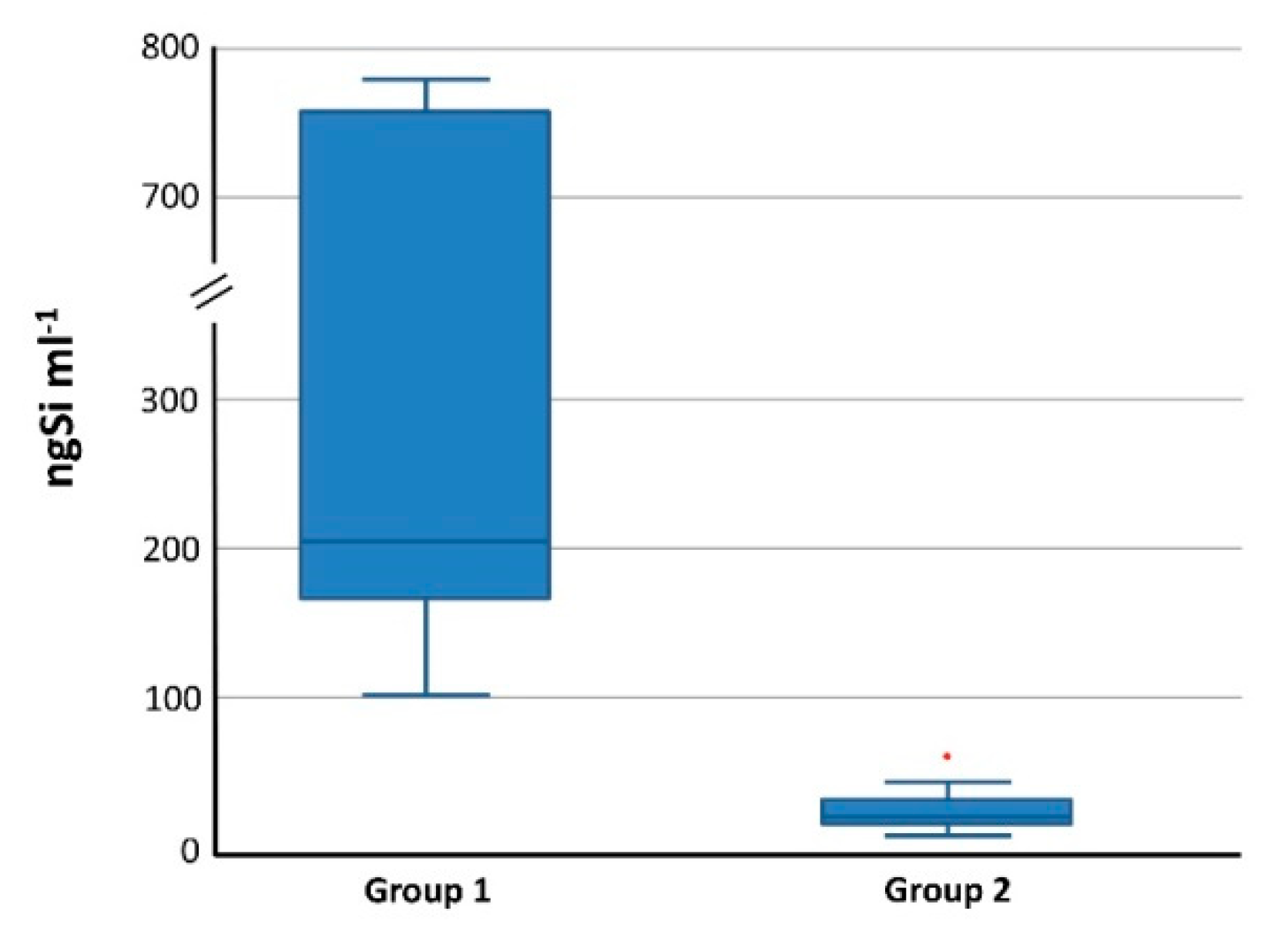
3.4.2. Total Si in Blood and Organs
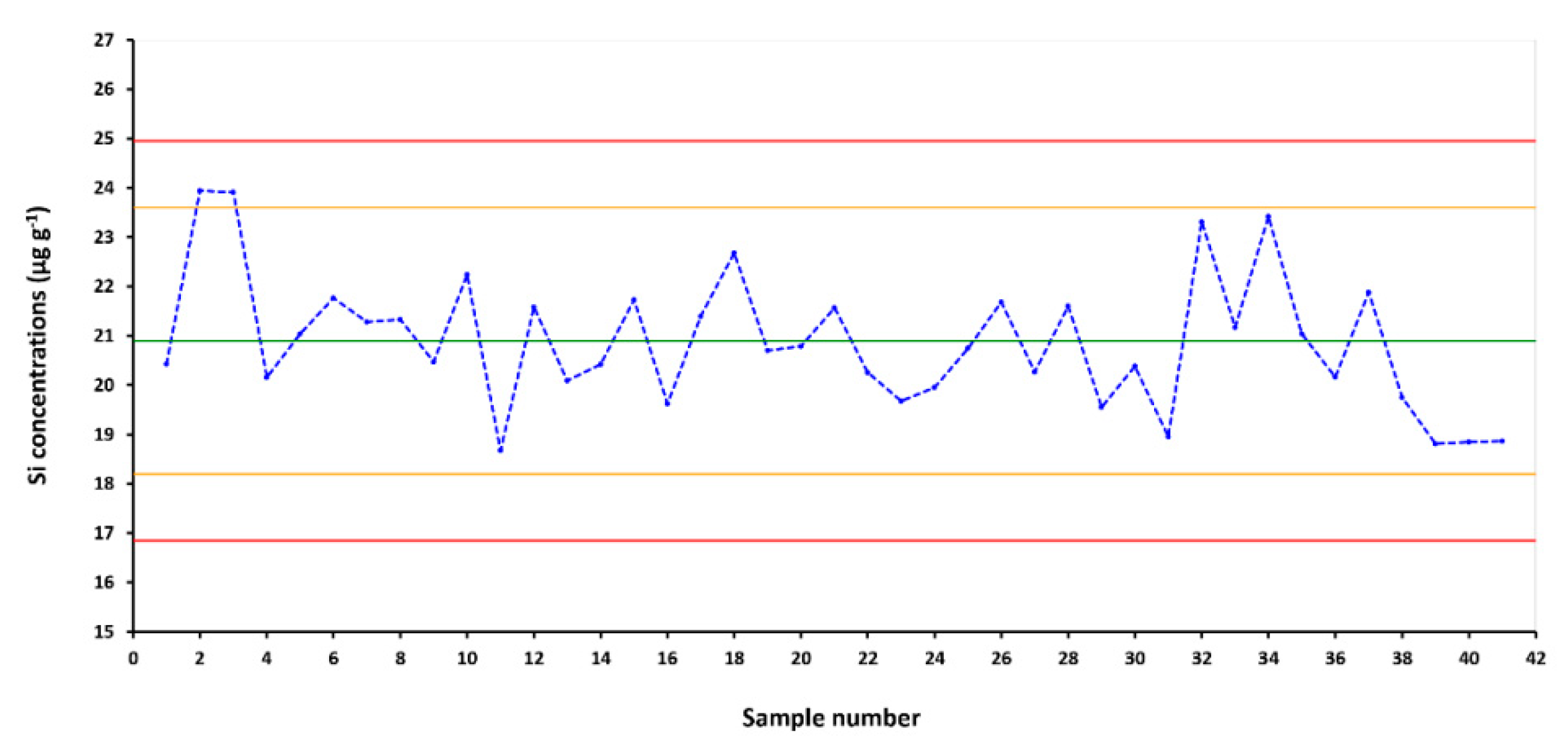
3.5. Analytical Quality Control
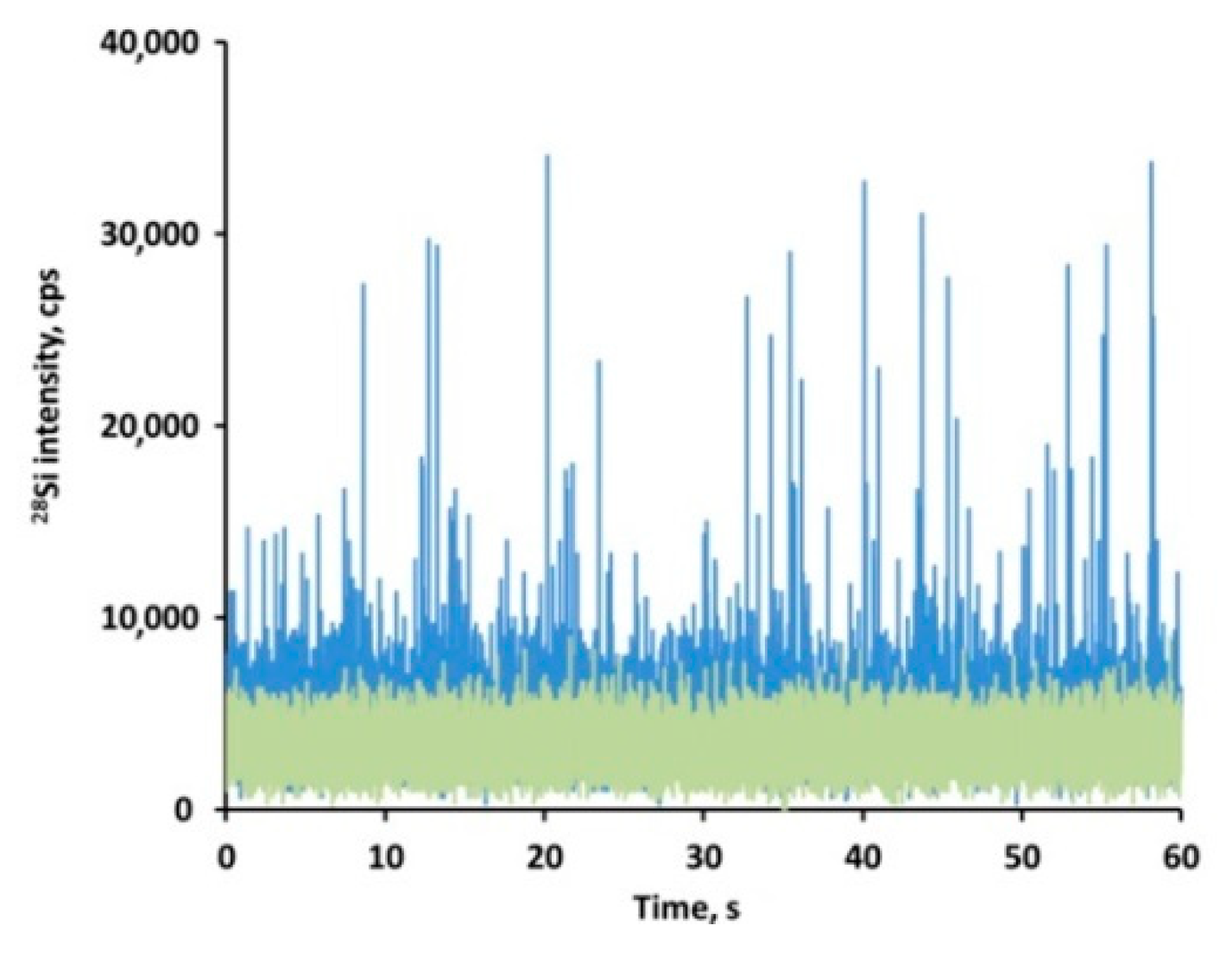
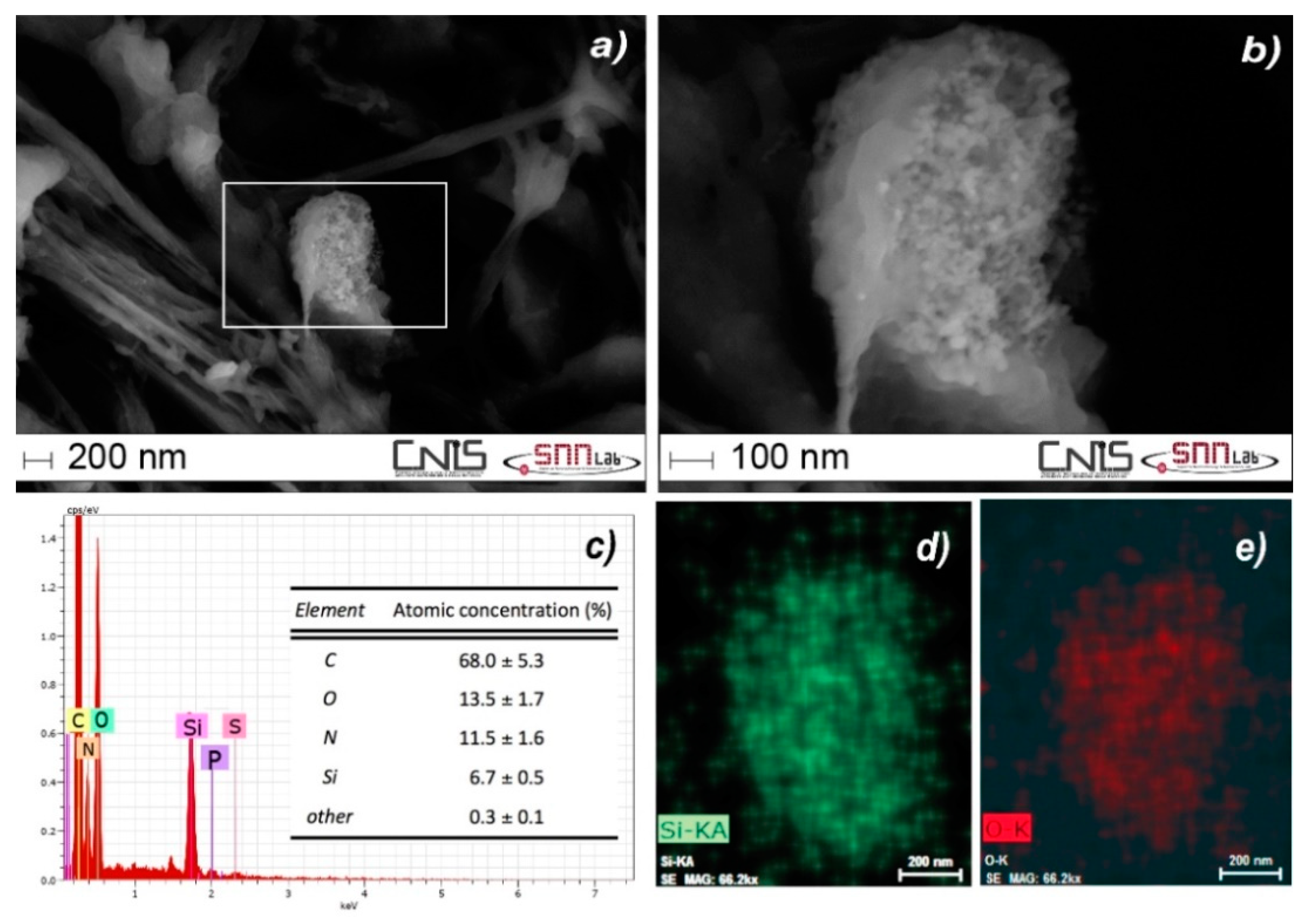
3.6. Screening the Presence of Particulate SiO2 in Tissues by spICP-MS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Barahona, F.; Ojea-Jimenez, I.; Geiss, O.; Gilliland, D.; Barrero-Moreno, J. Multimethod approach for the detection and characterisation of food-grade synthetic amorphous silica nanoparticles. J. Chromatogr. A 2016, 1432, 92–100. [Google Scholar] [CrossRef]
- Younes, M.; Aggett, P.; Aguilar, F.; Crebelli, R.; Dusemund, B.; Filipič, M.; Frutos, M.J.; Galtier, P.; Gott, D.; Gundert-Remy, U.; et al. Re-evaluation of silicon dioxide (E 551) as a food additive. EFSA J. 2018, 16, e05088. [Google Scholar]
- Fruijtier-Pölloth, C. The safety of nanostructured synthetic amorphous silica (SAS) as a food additive (E 551). Arch. Toxicol. 2016, 90, 2885–2916. [Google Scholar] [CrossRef]
- Aureli, F.; D’Amato, M.; Berardis, B.D.; Raggi, A.; Turco, A.C.; Cubadda, F. Investigating agglomeration and dissolution of silica nanoparticles in aqueous suspensions by dynamic reaction cell inductively coupled plasma-mass spectrometry in time resolved mode. J. Anal. At. Spectrom. 2012, 27, 1540–1548. [Google Scholar] [CrossRef]
- Contado, C.; Mendoza, J.H.M.; García, O.L.; Piret, J.P.; Dumortier, E.; Toussaint, O.; Lucas, S. Physicochemical and toxicological evaluation of silica nanoparticles suitable for food and consumer products collected by following the EC recommendation. Anal. Bioanal. Chem. 2016, 408, 271–286. [Google Scholar] [CrossRef]
- De Temmerman, P.-J.; Van Doren, E.; Verleysen, E.; Van der Stede, Y.; Francisco, M.A.D.; Mast, J. Quantitative characterization of agglomerates and aggregates of pyrogenic and precipitated amorphous silica nanomaterials by transmission electron microscopy. J. Nanobiotechnol. 2012, 10, 24. [Google Scholar] [CrossRef]
- Heroult, J.; Nischwitz, V.; Bartczak, D.; Goenaga Infante, H. The Potential of Asymmetric Flow Field-Flow Fractionation Hyphenated to Multiple Detectors for the Quantification and Size Estimation of Silica Nanoparticles in a Food Matrix. Anal. Bioanal. Chem. 2014, 406, 3919–3927. [Google Scholar] [CrossRef]
- Lim, J.-H.; Sisco, P.; Mudalige, T.K.; Sánchez-Pomales, G.; Howard, P.C.; Linder, S.W. Detection and characterization of SiO2 and TiO2 nanostructures in dietary supplements. J. Agric. Food Chem. 2015, 63, 3144–3152. [Google Scholar] [CrossRef]
- Peters, R.; Kramer, E.; Oomen, A.G.; Rivera, Z.E.H.; Oegema, G.; Tromp, P.C.; Fokkink, R.; Rietveld, A.; Marvin, H.J.P.; Weigel, S.; et al. Presence of nano-sized silica during in vitro digestion of foods containing silica as a food additive. ACS Nano 2012, 6, 2441–2451. [Google Scholar] [CrossRef]
- Rasmussen, K.; Mech, A.; Mast, J.; De Temmerman, P.-J.; Waegeneers, N.; Van Steen, F.; Pizzolon, J.C.; De Temmerman, L.; Van Doren, E.; Jensen, K.A.; et al. Synthetic Amorphous Silicon Dioxide (NM-200, NM-201, NM-202, NM-203, NM-204): Characterisation and Physico-Chemical Properties; JRC Scientific and Policy Reports; Publication Office of the European Union: Luxembourg, 2013. [Google Scholar]
- Yang, Y.; Faust, J.J.; Schoepf, J.; Hristovski, K.; Capco, D.G.; Herckes, P.; Westerhoff, P. Survey of food-grade silica dioxide nanomaterial occurrence, characterization, human gut impacts and fate across its lifecycle. Sci. Total Environ. 2016, 565, 902–912. [Google Scholar] [CrossRef]
- Murugadoss, S.; Lison, D.; Godderis, L.; Van Den Brule, S.; Mast, J.; Brassinne, F.; Sebaihi, N.; Hoet, P.H. Toxicology of silica nanoparticles: An update. Arch. Toxicol. 2017, 91, 2967–3010. [Google Scholar] [CrossRef] [PubMed]
- Boudard, D.; Aureli, F.; Laurent, B.; Sturm, N.; Raggi, A.; Antier, E.; Lakhdar, L.; Marche, P.N.; Cottier, M.; Cubadda, F.; et al. Chronic Oral Exposure to Synthetic Amorphous Silica (NM-200) Results in Renal and Liver Lesions in Mice. Kidney Int. Rep. 2019, 4, 1463–1471. [Google Scholar] [CrossRef] [PubMed]
- Boudard, D.; Aureli, F.; Laurent, B.; Sturm, N.; Raggi, A.; Antier, E.; Lakhdar, L.; Marche, P.N.; Cottier, M.; Cubadda, F.; et al. The Authors Reply. Kidney Int. Rep. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Tassinari, R.; Di Felice, G.; Butteroni, C.; Barletta, B.; Corinti, S.; Cubadda, F.; Aureli, F.; Raggi, A.; Narciso, L.; Tait, S.; et al. Hazard identification of pyrogenic synthetic amorphous silica (NM-203) after sub-chronic oral exposure in rat: A multitarget approach. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2020, 137, 111168. [Google Scholar] [CrossRef]
- Van Kesteren, P.C.E.; Cubadda, F.; Bouwmeester, H.; van Eijkeren, J.C.H.; Dekkers, S.; de Jong, W.H.; Oomen, A.G. Novel insights into the risk assessment of the nanomaterial synthetic amorphous silica, additive E551, in food. Nanotoxicology 2015, 9, 442–452. [Google Scholar] [CrossRef]
- Hardy, A.; Benford, D.; Halldorsson, T.; Jeger, M.J.; Knutsen, H.K.; More, S.; Naegeli, H.; Noteborn, H.; Ockleford, C.; Ricci, A.; et al. Guidance on risk assessment of the application of nanoscience and nanotechnologies in the food and feed chain: Part 1, human and animal health. EFSA J. 2018, 16, e05327. [Google Scholar]
- Tassinari, R.; Cubadda, F.; Moracci, G.; Aureli, F.; D’Amato, M.; Valeri, M.; De Berardis, B.; Raggi, A.; Mantovani, A.; Passeri, D.; et al. Oral, short-term exposure to titanium dioxide nanoparticles in Sprague-Dawley rat: Focus on reproductive and endocrine systems and spleen. Nanotoxicology 2014, 8, 654–662. [Google Scholar] [CrossRef]
- Aureli, F.; D’Amato, M.; Raggi, A.; Cubadda, F. Quantitative characterization of silica nanoparticles by asymmetric flow field flow fractionation coupled with online multiangle light scattering and ICP-MS/MS detection. J. Anal. At. Spectrom. 2015, 30, 1266–1273. [Google Scholar] [CrossRef]
- ANSES. French Agency for Food, Environmental and Occupational Health & Safety Nanogenotox: The Project. Available online: https://www.anses.fr/en/content/nanogenotox-project (accessed on 23 March 2020).
- European Commission Joint Research Center. JRC NANOMATERIALS REPOSITORY. Available online: https://ec.europa.eu/jrc/sites/jrcsh/files/JRC%20Nanomaterials%20Repository-List%20of%20Representative%20Nanomaterials-201606.pdf (accessed on 24 March 2020).
- Cubadda, F.; Oomen, A.G.; Laurentie, M.; Aureli, F.; D’Amato, M.; Maranghi, F.; Moracci, G.; Raggi, A.; Tassinari, R.; de Jong, W.H.; et al. Toxicokinetics of synthetic amorphous silica after oral and intravenous administration in rats. Part. Fibre Toxicol. 2020. manuscript in preparation. [Google Scholar]
- Arslan, Z.; Tyson, J.F. Determination of trace elements in siliceous samples by ICP-MS after precipitation of silicon as sodium fluorosilicate. Microchim. Acta 2008, 160, 219–225. [Google Scholar] [CrossRef][Green Version]
- Schramel, P.; Wendler, I.; Knapp, G. Total digestion of silicate containing matrices (plants, soil, sludges) using a pressure ashing device with PFA-vessels. Fresenius J. Anal. Chem. 1996, 356, 512–514. [Google Scholar] [CrossRef]
- Carter, R.H. Solubilities of Some Inorganic Flurides in Water at 25° C.1. Ind. Eng. Chem. 1928, 20, 1195. [Google Scholar] [CrossRef]
- Tanner, S.D.; Baranov, V.I.; Bandura, D.R. Reaction cells and collision cells for ICP-MS: A tutorial review. Spectrochim. Acta Part B At. Spectrosc. 2002, 57, 1361–1452. [Google Scholar] [CrossRef]
- Practical Guide to ICP-MS: A Tutorial for Beginners, Third Edition. Available online: https://www.crcpress.com/Practical-Guide-to-ICP-MS-A-Tutorial-for-Beginners-Third-Edition/Thomas/p/book/9781466555433 (accessed on 23 March 2020).
- International Organization for Standardization. ISO Guide 35:2017 Reference Materials—Guidance for Characterization and Assessment of Homogeneity and Stability; ISO: Geneva, Switzerland, 2017. [Google Scholar]
- International Organization for Standardization. ISO Guide 30:2015 Reference Materials—Selected Terms and Definitions; ISO: Geneva, Switzerland, 2015. [Google Scholar]
- International Atomic Energy Agency. IAEA TECDOC No. 1350 Development and Use of Reference Materials and Quality Control Materials; IAEA: Vienna, Austria, 2003. [Google Scholar]
- Inorganic Mass Spectrometry. Inorganic Mass Spectrometry. F. Adams, R. Gijbels, and R. Van Grieken, Eds. Wiley-Interscience, New York, 1988. xii, 404 pp., illus. $65. Chemical Analysis, vol. 95. Science 1989, 243, 1499. [Google Scholar] [CrossRef]
- Wollenweber, D.; Straßburg, S.; Wünsch, G. Determination of Li, Na, Mg, K, Ca and Fe with ICP-MS using cold plasma conditions. Fresenius J. Anal. Chem. 1999, 364, 433–437. [Google Scholar] [CrossRef]
- Balcaen, L.; Geuens, I.; Moens, L.; Vanhaecke, F. Determination of ultra-trace amounts of Fe in AgNO3 solutions by means of isotope dilution analysis applying an inductively coupled plasma mass spectrometer equipped with a dynamic reaction cell. Anal. Bioanal. Chem. 2003, 377, 1020–1025. [Google Scholar] [CrossRef]
- Van der Zande, M.; Vandebriel, R.J.; Groot, M.J.; Kramer, E.; Herrera Rivera, Z.E.; Rasmussen, K.; Ossenkoppele, J.S.; Tromp, P.; Gremmer, E.R.; Peters, R.J.; et al. Sub-chronic toxicity study in rats orally exposed to nanostructured silica. Part. Fibre Toxicol. 2014, 11, 8. [Google Scholar] [CrossRef]
- Paek, H.-J.; Chung, H.-E.; Lee, J.-A.; Kim, M.-K.; Lee, Y.-J.; Kim, M.-S.; Kim, S.-H.; Maeng, E.-H.; Lee, J.; Jeong, J.; et al. Quantitative Determination of Silica Nanoparticles in Biological Matrices and Their Pharmacokinetics and Toxicokinetics in Rats. Sci. Adv. Mater. 2014, 6, 1605–1610. [Google Scholar] [CrossRef]
- Waegeneers, N.; Brasseur, A.; Van Doren, E.; Van der Heyden, S.; Serreyn, P.-J.; Pussemier, L.; Mast, J.; Schneider, Y.-J.; Ruttens, A.; Roels, S. Short-term biodistribution and clearance of intravenously administered silica nanoparticles. Toxicol. Rep. 2018, 5, 632–638. [Google Scholar] [CrossRef]
- Peters, R.J.B.; Oomen, A.G.; Bemmel, G.; van Vliet, L.; van Undas, A.K.; Munniks, S.; Bleys, R.L.A.W.; Tromp, P.C.; Brand, W.; van der Lee, M. Silicon dioxide and titanium dioxide particles found in human tissues. Nanotoxicology 2020, 14, 420–432. [Google Scholar] [CrossRef]
- Capiau, S.; Bolea-Fernandez, E.; Balcaen, L.; Van Der Straeten, C.; Verstraete, A.G.; Vanhaecke, F.; Stove, C.P. Development, validation and application of an inductively coupled plasma–mass spectrometry method to determine cobalt in metal-on-metal prosthesis patients using volumetric absorptive microsampling. Talanta 2020, 208, 120055. [Google Scholar] [CrossRef]
- Jugdaohsingh, R.; Anderson, S.H.; Tucker, K.L.; Elliott, H.; Kiel, D.P.; Thompson, R.P.; Powell, J.J. Dietary silicon intake and absorption. Am. J. Clin. Nutr. 2002, 75, 887–893. [Google Scholar] [CrossRef]
- Powell, J.J.; McNaughton, S.A.; Jugdaohsingh, R.; Anderson, S.H.C.; Dear, J.; Khot, F.; Mowatt, L.; Gleason, K.L.; Sykes, M.; Thompson, R.P.H.; et al. A provisional database for the silicon content of foods in the United Kingdom. Br. J. Nutr. 2005, 94, 804–812. [Google Scholar] [CrossRef]
- Pennington, J.A. Silicon in foods and diets. Food Addit. Contam. 1991, 8, 97–118. [Google Scholar] [CrossRef]
- Kim, Y.-Y.; Kim, M.-H.; Choi, M.-K. Relationship between dietary intake and urinary excretion of silicon in free-living Korean adult men and women. Biol. Trace Elem. Res. 2019, 191, 286–293. [Google Scholar] [CrossRef]
- Prescha, A.; Zabłocka-Słowińska, K.; Grajeta, H. Dietary silicon and its impact on plasma silicon levels in the Polish population. Nutrients 2019, 11, 980. [Google Scholar] [CrossRef]
- Popplewell, J.F.; King, S.J.; Day, J.P.; Ackrill, P.; Fifield, L.K.; Cresswell, R.G.; di Tada, M.L.; Liu, K. Kinetics of uptake and elimination of silicic acid by a human subject: A novel application of 32Si and accelerator mass spectrometry. J. Inorg. Biochem. 1998, 69, 177–180. [Google Scholar] [CrossRef]
- Marcowycz, A.; Housez, B.; Maudet, C.; Cazaubiel, M.; Rinaldi, G.; Croizet, K. Digestive absorption of silicon, supplemented as orthosilicic acid-vanillin complex. Mol. Nutr. Food Res. 2015, 59, 1584–1589. [Google Scholar] [CrossRef]
- Sripanyakorn, S.; Jugdaohsingh, R.; Dissayabutr, W.; Anderson, S.H.C.; Thompson, R.P.H.; Powell, J.J. The comparative absorption of silicon from different foods and food supplements. Br. J. Nutr. 2009, 102, 825–834. [Google Scholar] [CrossRef]
- Pruksa, S.; Siripinyanond, A.; Powell, J.J.; Jugdaohsingh, R. Silicon balance in human volunteers; a pilot study to establish the variance in silicon excretion versus intake. Nutr. Metab. 2014, 11, 4. [Google Scholar] [CrossRef]
- Magnusson, B.; Hovind, H.; Krysell, M.; Lund, U.; Mäkinen, I. Handbook—InterQuality Control, Nordtest Report TR 569; 5.1.; Nordtest: Taastrup, Denmark, 2018. [Google Scholar]
- Westgard, J.O.; Barry, P.L.; Hunt, M.R.; Groth, T. A multi-rule Shewhart chart for quality control in clinical chemistry. Clin. Chem. 1981, 27, 493–501. [Google Scholar] [CrossRef]
- Westgard, J.O. Basic QC Practices: Training in Statistical Quality Control for Healthcare Laboratories; Westgard Quality Corp: Madison, WI, USA, 2002; Chapter 6; pp. 77–88. ISBN 978-1-886958-17-3. [Google Scholar]
- Westgard, J.O.; Klee, G.G. Quality Management. In Fundamentals of Clinical Chemistry; Burtis, C., Ed.; WB Saunders Company: Philadelphia, PA, USA, 1996; Chapter 16; pp. 211–223. [Google Scholar]
- Cembrowski, G.S.; Carey, R.N. Quality Control Procedures. In Laboratory Quality Management; ASCP Press: Chicago, IL, USA, 1989; Chapter 4; pp. 59–79. [Google Scholar]
- International Organization for Standardization. ISO/TS 19590:2017 Nanotechnologies—Size Distribution and Concentration of Inorganic Nanoparticles in Aqueous Media via Single Particle Inductively Coupled Plasma Mass Spectrometry; ISO: Geneva, Switzerland, 2017. [Google Scholar]
- Laborda, F.; Bolea, E.; Jiménez-Lamana, J. Single Particle Inductively Coupled Plasma Mass Spectrometry: A Powerful Tool for Nanoanalysis. Anal. Chem. 2014, 86, 2270–2278. [Google Scholar] [CrossRef]
- Degueldre, C.; Favarger, P.-Y.; Wold, S. Gold colloid analysis by inductively coupled plasma-mass spectrometry in a single particle mode. Anal. Chim. Acta 2006, 555, 263–268. [Google Scholar] [CrossRef]
- Mitrano, D.M.; Lesher, E.K.; Bednar, A.; Monserud, J.; Higgins, C.P.; Ranville, J.F. Detecting nanoparticulate silver using single-particle inductively coupled plasma–mass spectrometry. Environ. Toxicol. Chem. 2012, 31, 115–121. [Google Scholar] [CrossRef]
- Montaño, M.D.; Olesik, J.W.; Barber, A.G.; Challis, K.; Ranville, J.F. Single Particle ICP-MS: Advances toward routine analysis of nanomaterials. Anal. Bioanal. Chem. 2016, 408, 5053–5074. [Google Scholar] [CrossRef]
- Dan, Y.; Shi, H.; Stephan, C.; Liang, X. Rapid Analysis of Titanium Dioxide Nanoparticles in Sunscreens Using Single Particle Inductively Coupled Plasma-Mass Spectrometry. Microchem. J. 2015, 122, 119–126. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Instrument Parameter | Value |
|---|---|
| RF Power | 1.3 kV |
| Plasma Gas Flow | 15.5 l min−1 |
| Aux Gas Flow | 1.2 l min−1 |
| Carrier gas | 1.00 l min−1 |
| DRC | CH4 |
| Cell gas flow 0.8 mL min−1 | |
| RPq 0.55 | |
| CPV −17 V | |
| QRO −6 V | |
| CRO −8 V | |
| Internal standard | 74Ge |
| Analytical mass | 28Si |
| Sample flow rate | 1 mL min−1 |
| Sweeps | 20 |
| Dwell time | 200 ms |
| Reading | 1 |
| Replicates | 3 |
| Integration time | 4 s |
| Tissue | LoD | LoQ |
|---|---|---|
| Liver | 0.3 | 0.9 |
| Spleen | 0.4 | 1.5 |
| Lungs | 0.5 | 1.8 |
| Heart | 0.4 | 1.2 |
| Brain | 0.2 | 0.8 |
| Kidneys | 0.3 | 0.9 |
| Ovaries | 1.8 | 6.1 |
| Testis | 0.2 | 0.8 |
| GI tract | 0.3 | 1.1 |
| Mesenteric lymph nodes | 0.4 | 1.5 |
| Blood | 0.2 | 0.7 |
| Sample | NM-203 | NM-200 | ||
|---|---|---|---|---|
| HP | Multiprep | HP | Multiprep | |
| NM | 104.1 ± 8.4 | 95.5 ± 10.9 | 97.0 ± 2.9 | 97.8 ± 4.0 |
| Liver | 96.2 ± 2.2 | 88.1 ± 3.6 | 91.4 ± 2.2 | 86.8 ± 3.0 |
| Spleen | 86.7 ± 4.5 | 87.3 ± 1.7 | 85.0 ± 3.7 | 87.8 ± 3.1 |
| Sample | Control | NM-200 | NM-203 | |||
|---|---|---|---|---|---|---|
| Min | Max | Min | Max | Min | Max | |
| Liver | ≤0.3 | 0.6 (0.1) | 0.9 (0.1) | 116.9 (3.5) | 0.4 (0.1) | 114.1 (3.4) |
| Spleen | ≤0.4 | ≤0.4 | ≤0.4 | 55.8 (1.7) | ≤0.4 | 270.6 (8.1) |
| Lungs | ≤0.5 | 1.1 (0.1) | ≤0.5 | 54.3 (1.6) | ≤0.5 | 25.0 (1.1) |
| Heart | ≤0.4 | 0.8 (0.1) | ≤0.4 | 1.9 (0.1) | ≤0.4 | 2.6 (0.2) |
| Brain | ≤0.2 | 0.7 (0.1) | ≤0.2 | 0.5 (0.1) | ≤0.2 | 1.5 (0.1) |
| Kidneys | ≤0.3 | 0.6 (0.1) | ≤0.3 | 1.5 (0.1) | ≤0.3 | 1.5 (0.1) |
| Testis | 1.0 (0.1) | 1.6 (0.2) | 0.8 (0.1) | 1.3 (0.1) | 0.8 (0.1) | 1.4 (0.1) |
| Ovaries | ≤1.8 | ≤1.8 | ≤1.8 | ≤1.8 | ≤1.8 | ≤1.8 |
| Sample | Control | NM-200 | NM-203 | |||
|---|---|---|---|---|---|---|
| Min | Max | Min | Max | Min | Max | |
| Liver | ≤0.3 | 0.5 (0.1) | 12.2 (0.9) | 419.8 (12.6) | 7.8 (0.5) | 283.0 (8.5) |
| Spleen | ≤0.4 | ≤0.4 | 12.2 (0.9) | 227.1 (6.8) | 17.9 (1.3) | 424.0 (12.7) |
| Lungs | ≤0.5 | 1.2 (0.1) | 2.9 (0.2) | 101.5 (3.0) | 1.4 (0.1) | 82.5 (3.7) |
| Heart | ≤0.4 | 0.8 (0.1) | ≤0.4 | 4.0 (0.3) | ≤0.4 | 9.0 (0.6) |
| Brain | ≤0.2 | 0.7 (0.1) | 0.3 (0.1) | 0.7 (0.1) | 0.3 | 0.9 (0.1) |
| Kidneys | ≤0.3 | 0.6 (0.1) | 0.4 (0.1) | 3.2 (0.2) | ≤0.3 | 7.6 (0.5) |
| Testis | 1.0 (0.1) | 1.6 (0.1) | 0.7 (0.1) | 1.4 (0.1) | 0.6 (0.1) | 2.3 (0.2) |
| Ovaries | ≤1.8 | ≤1.8 | ≤1.8 | ≤1.8 | ≤1.8 | ≤1.8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aureli, F.; Ciprotti, M.; D’Amato, M.; do Nascimento da Silva, E.; Nisi, S.; Passeri, D.; Sorbo, A.; Raggi, A.; Rossi, M.; Cubadda, F. Determination of Total Silicon and SiO2 Particles Using an ICP-MS Based Analytical Platform for Toxicokinetic Studies of Synthetic Amorphous Silica. Nanomaterials 2020, 10, 888. https://doi.org/10.3390/nano10050888
Aureli F, Ciprotti M, D’Amato M, do Nascimento da Silva E, Nisi S, Passeri D, Sorbo A, Raggi A, Rossi M, Cubadda F. Determination of Total Silicon and SiO2 Particles Using an ICP-MS Based Analytical Platform for Toxicokinetic Studies of Synthetic Amorphous Silica. Nanomaterials. 2020; 10(5):888. https://doi.org/10.3390/nano10050888
Chicago/Turabian StyleAureli, Federica, Maria Ciprotti, Marilena D’Amato, Emanueli do Nascimento da Silva, Stefano Nisi, Daniele Passeri, Angela Sorbo, Andrea Raggi, Marco Rossi, and Francesco Cubadda. 2020. "Determination of Total Silicon and SiO2 Particles Using an ICP-MS Based Analytical Platform for Toxicokinetic Studies of Synthetic Amorphous Silica" Nanomaterials 10, no. 5: 888. https://doi.org/10.3390/nano10050888
APA StyleAureli, F., Ciprotti, M., D’Amato, M., do Nascimento da Silva, E., Nisi, S., Passeri, D., Sorbo, A., Raggi, A., Rossi, M., & Cubadda, F. (2020). Determination of Total Silicon and SiO2 Particles Using an ICP-MS Based Analytical Platform for Toxicokinetic Studies of Synthetic Amorphous Silica. Nanomaterials, 10(5), 888. https://doi.org/10.3390/nano10050888