Microbiota Dysbiosis in Fungal Rhinosinusitis

 ,
,  ,
, 
Abstract
1. Introduction
2. Material and Methods
2.1. Study Design and Population
2.2. Sample Collection
2.3. DNA Extraction
2.4. 16S Metagenomics Sequencing
2.5. Microbial Community Analysis and Statistical Analysis
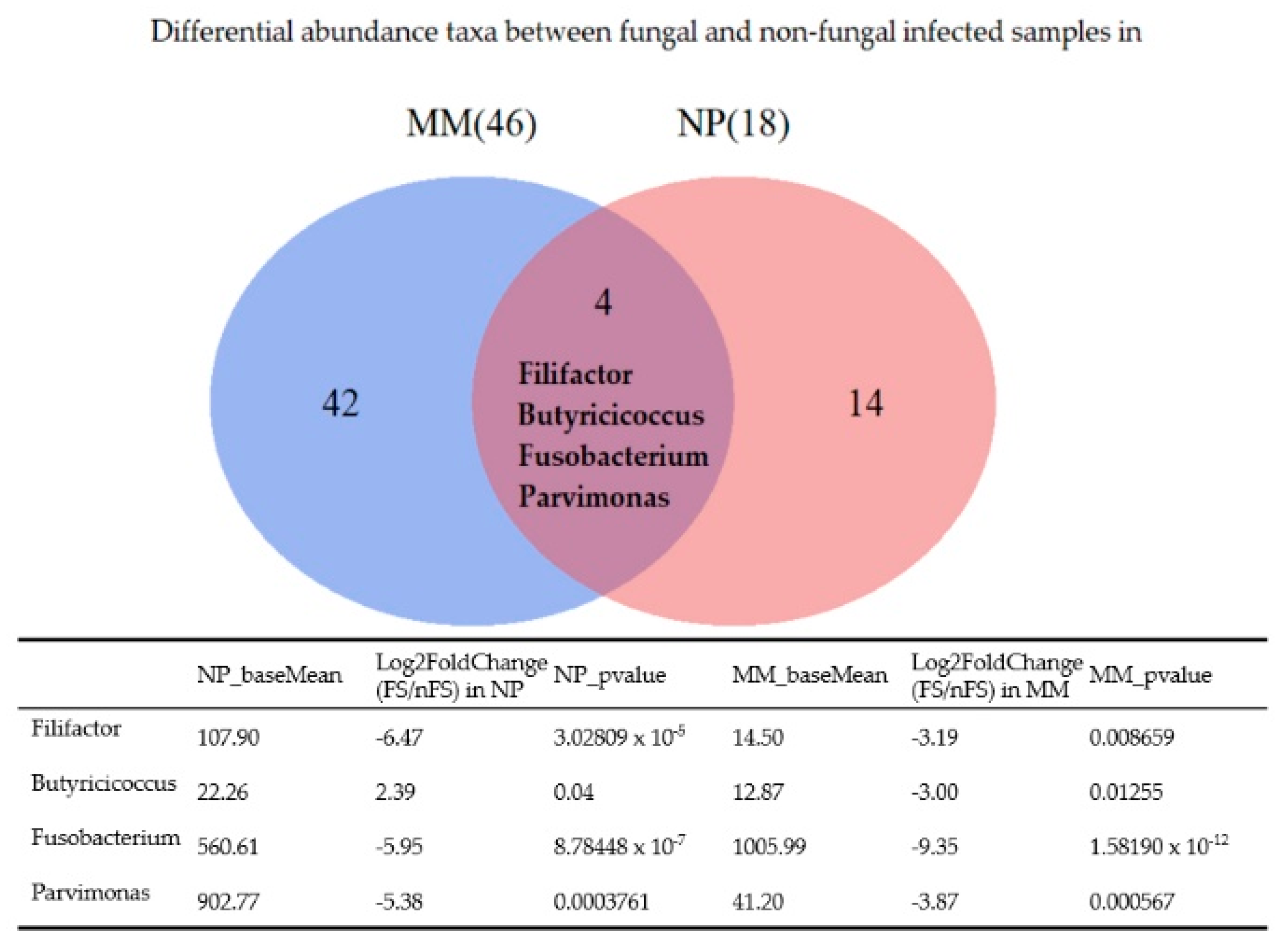
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lam, K.; Schleimer, R.; Kern, R.C. The Etiology and pathogenesis of chronic rhinosinusitis: A review of current hypotheses. Curr. Allergy Asthma Rep. 2015, 15, 41. [Google Scholar] [CrossRef] [PubMed]
- Hauser, L.J.; Feazel, L.M.; Ir, D.; Fang, R.; Wagner, B.D.; Robertson, C.E.; Frank, D.N.; Ramakrishnan, V.R. Sinus culture poorly predicts resident microbiota. Int. Forum Allergy Rhinol. 2015, 5, 3–9. [Google Scholar] [CrossRef]
- Hoggard, M.; Biswas, K.; Zoing, M.; Wagner Mackenzie, B.; Taylor, M.W.; Douglas, R.G. Evidence of microbiota dysbiosis in chronic rhinosinusitis. Int. Forum Allergy Rhinol. 2017, 7, 230–239. [Google Scholar] [CrossRef]
- Zhao, Y.C.; Bassiouni, A.; Tanjararak, K.; Vreugde, S.; Wormald, P.J.; Psaltis, A.J. Role of fungi in chronic rhinosinusitis through ITS sequencing. Laryngoscope 2018, 128, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, R.R.; Kingdom, T.T.; Hwang, P.H.; Smith, T.L.; Alt, J.A.; Baroody, F.M.; Batra, P.S.; Bernal-Sprekelsen, M.; Bhattacharyya, N.; Chandra, R.K.; et al. International Consensus Statement on Allergy and Rhinology: Rhinosinusitis. Int. Forum Allergy Rhinol. 2016, 6, 22–209. [Google Scholar] [CrossRef]
- Ramakrishnan, V.R.; Gitomer, S.; Kofonow, J.M.; Robertson, C.E.; Frank, D.N. Investigation of sinonasal microbiome spatial organization in chronic rhinosinusitis. Int. Forum Allergy Rhinol. 2017, 7, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Montone, K.T. Pathology of Fungal Rhinosinusitis: A Review. Head Neck Pathol. 2016, 10, 40–46. [Google Scholar] [CrossRef]
- Nicolai, P.; Lombardi, D.; Tomenzoli, D.; Villaret, A.B.; Piccioni, M.; Mensi, M.; Maroldi, R. Fungus ball of the paranasal sinuses: Experience in 160 patients treated with endoscopic surgery. Laryngoscope 2009, 119, 2275–2279. [Google Scholar] [CrossRef] [PubMed]
- Frey-Klett, P.; Burlinson, P.; Deveau, A.; Barret, M.; Tarkka, M.; Sarniguet, A. Bacterial-fungal interactions: Hyphens between agricultural, clinical, environmental, and food microbiologists. Microbiol. Mol. Biol. Rev. 2011, 75, 583–609. [Google Scholar] [CrossRef]
- Wang, Z.K.; Yang, Y.S.; Stefka, A.T.; Sun, G.; Peng, L.H. Review article: Fungal microbiota and digestive diseases. Aliment Pharm. Ther. 2014, 39, 751–766. [Google Scholar] [CrossRef]
- Zhang, J.; He, S.; Li, Y.; Lv, M.; Wei, H.; Qu, B.; Zheng, Y.; Hu, C. Distinguishing the dominant species of pathogen in ethmoidal sinusitis by sequencing DNA dataset analysis. Exp. Med. 2016, 16, 4207–4212. [Google Scholar] [CrossRef] [PubMed]
- Papathanasiou, A.; Abdel-Fahim, R.; Evangelou, N. Dual infectious brainstem encephalitis with Aspergillus flavus and Haemophilus influenza in an immunocompetent patient. Neurol. Sci. 2018, 39, 1795–1796. [Google Scholar] [CrossRef] [PubMed]
- Dall’Igna, C.; Palombini, B.C.; Anselmi, F.; Araújo, E.; Dall’Igna, D.P. Fungal rhinosinusitis in patients with chronic sinusal disease. Braz. J. Otorhinolaryngol. 2005, 71, 712–720. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Y.; Lu, X.; Wang, X.; Zang, H.; Wang, T.; Zhou, B.; Zhang, L. Analysis of fungal ball rhinosinusitis by culturing fungal clumps under endoscopic surgery. Int. J. Clin. Exp. Med. 2015, 8, 5925–5930. [Google Scholar] [PubMed]
- Sass, G.; Ansari, S.R.; Dietl, A.-M.; Déziel, E.; Haas, H.; Stevens, D.A. Intermicrobial interaction: Aspergillus fumigatus siderophores protect against competition by Pseudomonas aeruginosa. PLoS ONE 2019, 14, e0216085. [Google Scholar] [CrossRef] [PubMed]
- Sass, G.; Nazik, H.; Penner, J.; Shah, H.; Ansari, S.R.; Clemons, K.V.; Groleau, M.-C.; Dietl, A.-M.; Visca, P.; Haas, H. Aspergillus-Pseudomonas interaction, relevant to competition in airways. Med. Mycol. 2019, 57, S228–S232. [Google Scholar] [CrossRef]
- Brook, I. Microbiology of chronic rhinosinusitis. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 1059–1068. [Google Scholar] [CrossRef]
- Bor, B.; Cen, L.; Agnello, M.; Shi, W.; He, X. Morphological and physiological changes induced by contact-dependent interaction between Candida albicans and Fusobacterium nucleatum. Sci. Rep. 2016, 6, 27956. [Google Scholar] [CrossRef]
- Yang, C.Y.; Yeh, Y.M.; Yu, H.Y.; Chin, C.Y.; Hsu, C.W.; Liu, H.; Huang, P.J.; Hu, S.N.; Liao, C.T.; Chang, K.P.; et al. Oral Microbiota Community Dynamics Associated with Oral Squamous Cell Carcinoma Staging. Front. Microbiol. 2018, 9, 862. [Google Scholar] [CrossRef]
- Schlafer, S.; Riep, B.; Griffen, A.L.; Petrich, A.; Hübner, J.; Berning, M.; Friedmann, A.; Göbel, U.B.; Moter, A. Filifactor alocis—involvement in periodontal biofilms. BMC Microbiol. 2010, 10, 66. [Google Scholar] [CrossRef]
- Aruni, A.W.; Zhang, K.; Dou, Y.; Fletcher, H. Proteome analysis of coinfection of epithelial cells with Filifactor alocis and Porphyromonas gingivalis shows modulation of pathogen and host regulatory pathways. Infect. Immun. 2014, 82, 3261–3274. [Google Scholar] [CrossRef] [PubMed]
- Aruni, A.W.; Mishra, A.; Dou, Y.; Chioma, O.; Hamilton, B.N.; Fletcher, H.M. Filifactor alocis—A new emerging periodontal pathogen. Microbes Infect. 2015, 17, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Al-Hebshi, N.N.; Shuga-Aldin, H.M.; Al-Sharabi, A.K.; Ghandour, I. Subgingival periodontal pathogens associated with chronic periodontitis in Yemenis. BMC Oral Health 2014, 14, 13. [Google Scholar] [CrossRef] [PubMed]
- Signat, B.; Roques, C.; Poulet PDuffaut, D. Role of Fusobacterium nucleatum in Periodontal Health and Disease. Curr. Issues Mol. Biol. 2011, 13, 25–36. [Google Scholar] [PubMed]
- Jain, R.; Hoggard, M.; Biswas, K.; Zoing, M.; Jiang, Y.; Douglas, R. Changes in the bacterial microbiome of patients with chronic rhinosinusitis after endoscopic sinus surgery. Int. Forum Allergy Rhinol. 2017, 7, 7–15. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fungal Rhinosinusitis (n = 7) | Non-Fungal Rhinosinusitis (n = 18) | p-Value | |
|---|---|---|---|
| Gender (M:F) | 4:3 | 11:7 | 0.856 |
| Asthma | 0 | 0 | NA |
| DM 1 | 0 | 2 | 1.00 |
| Smoking | 0 | 4 | 0.294 |
| Previous FESS 2 | 1 | 2 | 0.826 |
| Endoscope features | |||
| Purulence | 7 | 13 | 0.294 |
| Polyp | 2 | 11 | 0.202 |
| Unilateral/Bilateral CT 3 features Discrete calcification Sample Origin: Nasopharynx Middle meatus | 7/0 7 6 7 | 9/9 0 18 17 | 0.057 0.000 |
| Taxonomy | Mean Proportion-Fungal | Log2foldchange | p-Value |
|---|---|---|---|
| Pseudomonas | 0.157621 | 7.041475 | 1.34 × 10−16 |
| Fusobacterium | 9.39 × 10−5 | −10.4598 | 1.58 × 10−12 |
| Enterobacteriaceae | 0.112324 | 6.760324 | 2.39 × 10−11 |
| Haemophilus | 0.39881 | 3.39837 | 1.93 × 10−9 |
| Corynebacterium 1 | 9.39 × 10−5 | −8.62602 | 5.8 × 10−7 |
| Faecalibacterium | 0.001241 | −4.01124 | 3.06 × 10−6 |
| Lawsonella | 9.39 × 10−5 | −6.46435 | 1.04 × 10−5 |
| Roseburia | 9.39 × 10−5 | −6.3614 | 1.09 × 10−5 |
| Prevotella 2 | 9.39 × 10−5 | −8.5811 | 1.85 × 10−5 |
| Dialister | 0.003497 | −2.87925 | 2.44 × 10−5 |
| Neisseria | 0.077839 | 3.064623 | 2.71 × 10−5 |
| Phascolarctobacterium | 9.39 × 10−5 | −5.8985 | 5.28 × 10−5 |
| Bifidobacterium | 9.39 × 10−5 | −5.55629 | 8.48 × 10−5 |
| Sphingomonas | 0.002662 | −3.92634 | 0.000283 |
| Parasutterella | 0.000142 | -6.25574 | 0.000274 |
| Parvimonas | 0.000105 | −7.56253 | 0.000576 |
| Cutibacterium | 0.004112 | −2.95335 | 0.001021 |
| Prevotellaceae UCG-001 | 9.39 × 10−5 | −5.67206 | 0.000922 |
| Barnesiella | 9.39 × 10−5 | −5.36435 | 0.001773 |
| Prevotella | 0.009641 | −3.5138 | 0.002127 |
| Campylobacter | 0.002497 | −2.78179 | 0.003202 |
| Dubosiella | 0.001719 | −1.34162 | 0.003869 |
| uncultured bacterium | 9.39 × 10−5 | −4.31437 | 0.004209 |
| Herbaspirillum | 0.001283 | −0.91395 | 0.004913 |
| Burkholderia-Caballeronia-Paraburkholderia | 0.001427 | −1.42577 | 0.005533 |
| Murdochiella | 0.000407 | −3.49905 | 0.00606 |
| Thermus | 0.006022 | 0.12931 | 0.007197 |
| Filifactor | 9.39 × 10−5 | −6.65602 | 0.008659 |
| Caulobacter | 9.39 × 10−5 | −5.71 | 0.009229 |
| Granulicatella | 9.39 × 10−5 | −5.01554 | 0.011858 |
| Butyricicoccus | 9.39 × 10−5 | −5.86795 | 0.01255 |
| Ruminococcaceae UCG-013 | 0.000228 | −3.92258 | 0.012505 |
| Parabacteroides | 0.002514 | −2.20143 | 0.01374 |
| Oceanobacillus | 0.000385 | −3.95811 | 0.015384 |
| Lactobacillus | 0.008292 | −1.74028 | 0.015927 |
| Ezakiella | 0.002612 | −2.88786 | 0.017742 |
| Lachnospiraceae | 0.008553 | −1.38552 | 0.02134 |
| Bacteroides | 0.009134 | −2.6368 | 0.026386 |
| uncultured Bacteroidales bacterium | 0.000326 | −5.18799 | 0.034265 |
| Staphylococcus | 0.151025 | 0.366581 | 0.036067 |
| Lachnospiraceae NK4A136 group | 0.004441 | −1.25274 | 0.033673 |
| Veillonella | 0.006022 | −2.41288 | 0.035876 |
| Enhydrobacter | 0.000119 | −5.33978 | 0.035423 |
| uncultured bacterium | 0.010302 | −1.14696 | 0.038908 |
| Ruminiclostridium 5 | 0.00215 | −1.34321 | 0.039208 |
| Ruminococcaceae UCG-014 | 0.011234 | −1.72203 | 0.04779 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Y.-T.; Wang, S.-H.; Liou, M.-L.; Shen, T.-A.; Lu, Y.-C.; Hsin, C.-H.; Yang, S.-F.; Chen, Y.-Y.; Chang, T.-H. Microbiota Dysbiosis in Fungal Rhinosinusitis. J. Clin. Med. 2019, 8, 1973. https://doi.org/10.3390/jcm8111973
Lu Y-T, Wang S-H, Liou M-L, Shen T-A, Lu Y-C, Hsin C-H, Yang S-F, Chen Y-Y, Chang T-H. Microbiota Dysbiosis in Fungal Rhinosinusitis. Journal of Clinical Medicine. 2019; 8(11):1973. https://doi.org/10.3390/jcm8111973
Chicago/Turabian StyleLu, Yen-Ting, Shao-Hung Wang, Ming-Li Liou, Ting-An Shen, Ying-Chou Lu, Chung-Han Hsin, Shun-Fa Yang, Yih-Yuan Chen, and Tzu-Hao Chang. 2019. "Microbiota Dysbiosis in Fungal Rhinosinusitis" Journal of Clinical Medicine 8, no. 11: 1973. https://doi.org/10.3390/jcm8111973
APA StyleLu, Y.-T., Wang, S.-H., Liou, M.-L., Shen, T.-A., Lu, Y.-C., Hsin, C.-H., Yang, S.-F., Chen, Y.-Y., & Chang, T.-H. (2019). Microbiota Dysbiosis in Fungal Rhinosinusitis. Journal of Clinical Medicine, 8(11), 1973. https://doi.org/10.3390/jcm8111973