
Tumor Budding: The Name is EMT. Partial EMT.

 , ,
, ,
Abstract
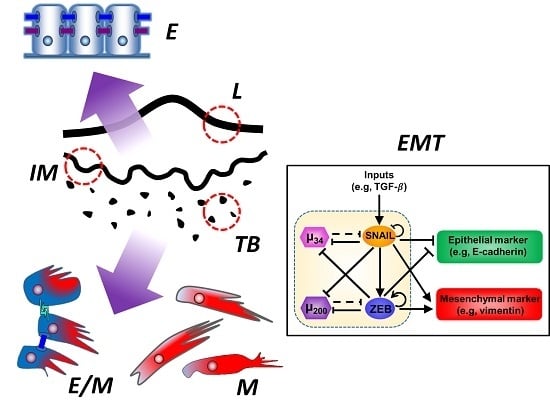
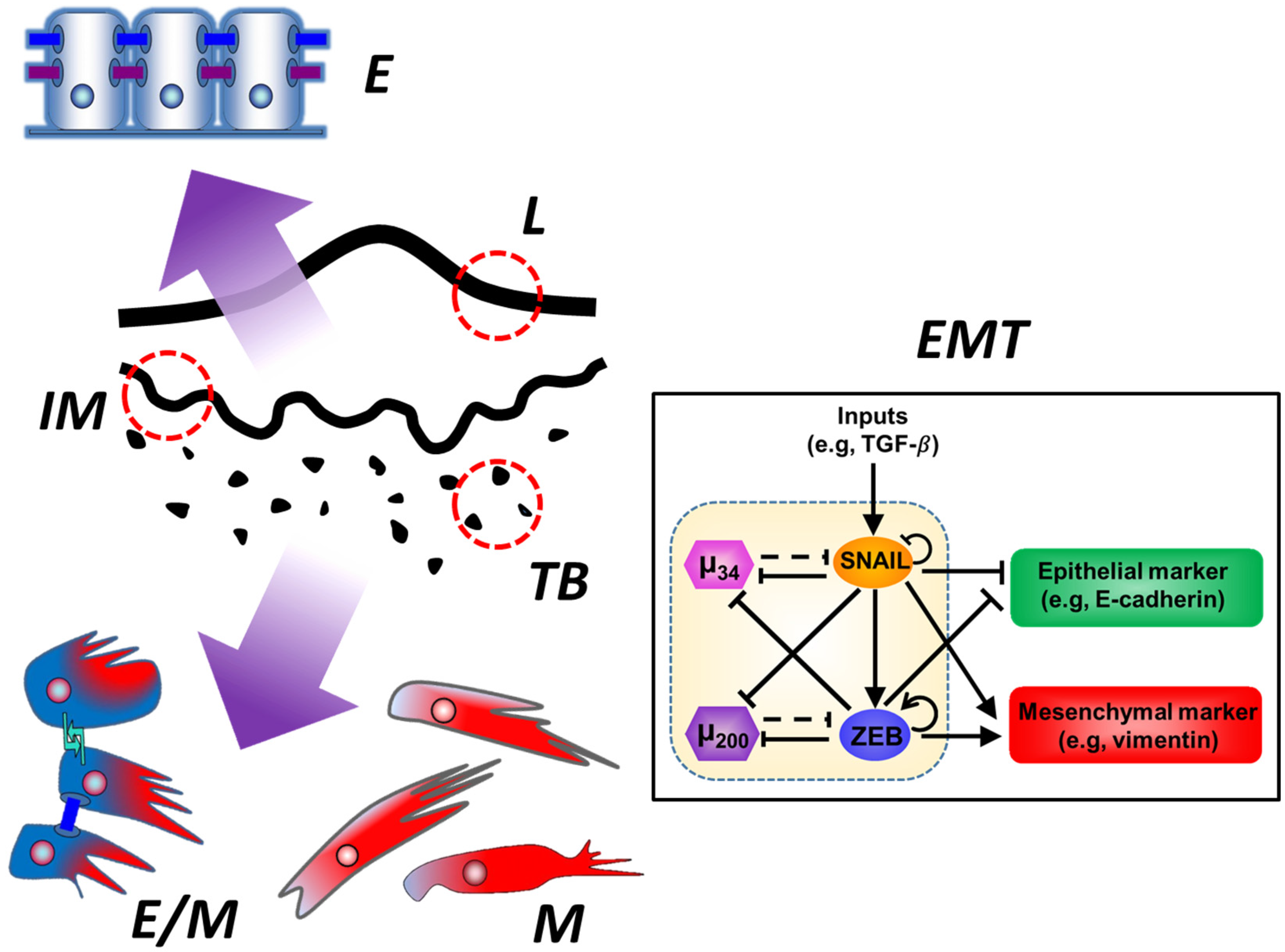
:
{kind=link}
{kind=link}
1. Tumor Budding: Introducing the Concept
2. Tumor Budding in Colorectal Cancer: Concepts and Methodologies
3. Tumor Budding in Colorectal Cancer: Clinical Significance
3.1. Tumor Budding Association with Recurrence and Survival
3.2. Tumor Budding Association with Other Factors Traditionally Linked to Poor Prognosis
4. Tumor Budding in Colorectal Cancer: Implementation in Clinical Practice
5. Tumor Budding in Other Cancer Types
6. Intra-Tumoral Budding
7. Epithelial-Mesenchymal Transition (EMT) and Partial EMT
8. Tumor Budding and Partial EMT
9. Proliferation/Quiescence and Cancer Stem Cells in Tumor Buds: Further Connection with EMT
10. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| CRC | colorectal cancer |
| CSS | cancer-specific survival |
| DFS | disease-free survival |
| E/M | epithelial/mesenchymal |
| EMT | epithelial-mesenchymal transition |
| H&E | hematoxylin and eosin |
| PDAC | pancreatic ductal adenocarcinoma |
| RFS | recurrence-free survival |
| OS | overall survival |
| RC | rectal cancer |
References
- De Smedt, L.; Palmans, S.; Sagaert, X. Tumour budding in colorectal cancer: What do we know and what can we do? Virchows Arch. 2016, 468, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Van Wyk, H.C.; Park, J.; Roxburgh, C.; Horgan, P.; Foulis, A.; McMillan, D.C. The role of tumour budding in predicting survival in patients with primary operable colorectal cancer: A systematic review. Cancer Treat. Rev. 2015, 41, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Koelzer, V.H.; Langer, R.; Zlobec, I.; Lugli, A. Tumor budding in colorectal cancer—Ready for diagnostic practice? Hum. Pathol. 2016, 47, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Imai, T. Growth patterns in human carcinoma. Their classification and relation to prognosis. Obstet. Gynecol. 1960, 16, 296–308. [Google Scholar] [PubMed]
- Hase, K.; Shatney, C.; Johnson, D.; Trollope, M.; Vierra, M. Prognostic value of tumor “budding” in patients with colorectal cancer. Dis. Colon Rectum 1993, 36, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Morodomi, T.; Isomoto, H.; Shirouzu, K.; Kakegawa, K.; Irie, K.; Morimatsu, M. An index for estimating the probability of lymph node metastasis in rectal cancers. Lymph node metastasis and the histopathology of actively invasive regions of cancer. Cancer 1989, 63, 539–543. [Google Scholar] [CrossRef]
- Gabbert, H.; Wagner, R.; Moll, R.; Gerharz, C.D. Tumor dedifferentiation: An important step in tumor invasion. Clin. Exp. Metastasis 1985, 3, 257–279. [Google Scholar] [CrossRef] [PubMed]
- Carr, I.; Levy, M.; Watson, P. The invasive edge: Invasion in colorectal cancer. Clin. Exp. Metastasis 1986, 4, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Amato, A.; Scucchi, L.; Pescatori, M. Tumor budding and recurrence of colorectal cancer. Dis. Colon Rectum 1994, 37, 514–515. [Google Scholar] [CrossRef] [PubMed]
- Bronsert, P.; Enderle-Ammour, K.; Bader, M.; Timme, S.; Kuehs, M.; Csanadi, A.; Kayser, G.; Kohler, I.; Bausch, D.; Hoeppner, J.; et al. Cancer cell invasion and EMT marker expression—A three-dimensional study of the human cancer-host interface. J. Pathol. 2014, 234, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Compton, C.C. Pathology and prognostic determinants of colorectal cancer. In UpToDate; Savarese, D.M., Ed.; UpToDate: Waltham, MA, USA, 2016. [Google Scholar]
- Schmoll, H.J.; Van Cutsem, E.; Stein, A.; Valentini, V.; Gliemelius, B.; Haustermans, K.; Nordlinger, B.; van de Velde, C.J.; Balmana, J.; Regula, J.; et al. ESMO Consensus Guidelines for management of patients with colon and rectal cancer. A personalized approach to clinical decision making. Ann. Oncol. 2012, 23, 2479–2516. [Google Scholar] [CrossRef] [PubMed]
- Puppa, G.; Senore, C.; Sheahan, K.; Vieth, M.; Lugli, A.; Zlobec, I.; Pecori, S.; Wang, L.M.; Langner, C.; Mitomi, H.; et al. Diagnostic reproducibility of tumour budding in colorectal cancer: A multicentre, multinational study using virtual microscopy. Histopathology 2012, 61, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, T.; Oya, M.; Ishikawa, H. Budding as a useful prognostic marker in pT3 well- or moderately-differentiated rectal adenocarcinoma. J. Surg. Oncol. 2003, 83, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-S.; Liang, W.-Y.; Lin, T.-C.; Chen, W.-S.; Jiang, J.-K.; Yang, S.-H.; Chang, S.-C.; Lin, J.-K. Curative resection of T1 colorectal carcinoma: Risk of lymph node metastasis and long-term prognosis. Dis. Colon Rectum 2005, 48, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Ueno, H.; Murphy, J.; Jass, J.R.; Mochizuki, H.; Talbot, I.C. Tumour “budding” as an index to estimate the potential of aggressiveness in rectal cancer. Histopathology 2002, 40, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.-S.; Choi, H.-J.; Park, K.-J.; Kim, J.-M.; Kim, S.-H.; Roh, Y.-H.; Kwon, H.-C.; Roh, M.-S. Intensity of tumor budding as an index for the malignant potential in invasive rectal carcinoma. Cancer Res. Treat. 2005, 37, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Park, K.J.; Choi, H.J.; Roh, M.S.; Kwon, H.C.; Kim, C. Intensity of tumor budding and its prognostic implications in invasive colon carcinoma. Dis. Colon Rectum 2005, 48, 1597–1602. [Google Scholar] [CrossRef] [PubMed]
- Shinto, E.; Jass, J.R.; Tsuda, H.; Sato, T.; Ueno, H.; Hase, K.; Mochizuki, H.; Matsubara, O. Differential prognostic significance of morphologic invasive markers in colorectal cancer: Tumor budding and cytoplasmic podia. Dis. Colon Rectum 2006, 49, 1422–1430. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Park, K.J.; Shin, J.S.; Roh, M.S.; Kwon, H.C.; Lee, H.S. Tumor budding as a prognostic marker in stage-III rectal carcinoma. Int. J. Colorectal Dis. 2007, 22, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, K.; Koyama, F.; Tamura, T.; Enomoto, Y.; Fujii, H.; Mukogawa, T.; Nakagawa, T.; Uchimoto, K.; Nakamura, S.; Nonomura, A.; et al. Prognostic value of immunohistochemical analysis of tumor budding in colorectal carcinoma. Anticancer Res. 2008, 28, 1831–1836. [Google Scholar] [PubMed]
- Wang, L.M.; Kevans, D.; Mulcahy, H.; O’Sullivan, J.; Fennelly, D.; Hyland, J.; O’Donoghue, D.; Sheahan, K. Tumor budding is a strong and reproducible prognostic marker in T3N0 colorectal cancer. Am. J. Surg. Pathol. 2009, 33, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Sy, J.; Fung, C.L.-S.; Dent, O.F.; Chapuis, P.H.; Bokey, L.; Chan, C. Tumor budding and survival after potentially curative resection of node-positive colon cancer. Dis. Colon Rectum 2010, 53, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Zlobec, I.; Molinari, F.; Martin, V.; Mazzucchelli, L.; Saletti, P.; Trezzi, R.; De Dosso, S.; Vlajnic, T.; Frattini, M.; Lugli, A. Tumor budding predicts response to anti-EGFR therapies in metastatic colorectal cancer patients. World J. Gastroenterol. 2010, 16, 4823–4831. [Google Scholar] [CrossRef] [PubMed]
- Betge, J.; Kornprat, P.; Pollheimer, M.J.; Lindtner, R.A.; Schlemmer, A.; Rehak, P.; Vith, M.; Langner, C. Tumor budding is an independent predictor of outcome in AJCC/UICC stage II colorectal cancer. Ann. Surg. Oncol. 2012, 19, 3706–3712. [Google Scholar] [CrossRef] [PubMed]
- Giger, O.T.; Comtesse, S.C.M.; Lugli, A.; Zlobec, I.; Kurrer, M.O. Intra-tumoral budding in preoperative biopsy specimens predicts lymph node and distant metastasis in patients with colorectal cancer. Mod. Pathol. 2012, 25, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.-H.; Wu, L.-C.; Li, P.-S.; Wu, W.-H.; Yang, S.-B.; Xia, P.; He, X.-X.; Xiao, L.-B. Tumour budding is a reproducible index for risk stratification of patients with Stage II colon cancer. Colorectal Dis. 2014, 16, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.P.; Vierkant, R.A.; Tillmans, L.S.; Wang, A.H.; Laird, P.W.; Weisenberger, D.J.; Lynch, C.F.; French, A.J.; Slager, S.L.; Raissian, Y.; et al. Tumor budding in colorectal carcinoma: Confirmation of prognostic significance and histologic cutoff in a population-based cohort. Am. J. Surg. Pathol. 2015, 39, 1340–1346. [Google Scholar] [CrossRef] [PubMed]
- Prall, F.; Nizze, H.; Barten, M. Tumour budding as prognostic factor in stage I/II colorectal carcinoma. Histopathology 2005, 47, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Zlobec, I.; Hädrich, M.; Dawson, H.; Koelzer, V.H.; Borner, M.; Mallaev, M.; Schnüriger, B.; Inderbitzin, D.; Lugli, A. Intratumoural budding (ITB) in preoperative biopsies predicts the presence of lymph node and distant metastases in colon and rectal cancer patients. Br. J. Cancer 2014, 110, 1008–1013. [Google Scholar] [CrossRef] [PubMed]
- Koelzer, V.H.; Zlobec, I.; Berger, M.D.; Cathomas, G.; Dawson, G.; Dirschmid, K.; Hädrich, M.; Inderbitzin, D.; Offner, F.; Puppa, G.; et al. Tumor budding in colorectal cancer revisited: Results of a multicenter interobserver study. Virchows Arch. 2015, 466, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Max, N.; Harbaum, L.; Pollheimer, M.J.; Lindtner, R.A.; Kornprat, P.; Langer, C. Tumour budding with and without admixed inflammation: Two different sides of the same coin? Br. J. Cancer 2016, 114, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Mitrovic, B.; Schaeffer, D.F.; Riddell, R.H.; Kirsch, R. Tumor budding in colorectal carcinoma: Time to take notice. Mod. Pathol. 2012, 25, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Horcic, M.; Koelzer, V.H.; Karamitopoulou, E.; Terracciano, L.; Puppa, G.; Zlobec, I.; Lugli, A. Tumor budding score based on 10 high-power fields is a promising basis for a standardized prognostic scoring system in stage II colorectal cancer. Hum. Pathol. 2013, 44, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.D.; Maguire, A.; Conlon, N.; Gibbons, D.; Wang, L.M.; Sheahan, K. Reproducibility of the rapid bud count method for assessment of tumor budding in stage II colorectal cancer. Am. J. Surg. Pathol. 2010, 34, 746–748. [Google Scholar] [CrossRef] [PubMed]
- Karamitopoulou, E.; Zlobec, I.; Kölzer, V.; Kondi-Pafiti, A.; Patsouris, E.S.; Gennatas, K.; Lugli, A. Proposal for a 10-high-power-fields scoring methods for the assessment of tumor budding in colorectal cancer. Mod. Pathol. 2013, 26, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, T.; Oya, M.; Yamaguchi, M. Budding (sprouting) as a useful prognostic marker in colorectal mucinous carcinoma. Jpn. J. Clin. Oncol. 2002, 32, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Hashiguchi, Y.; Ueno, H.; Hase, K.; Mochizuki, H. Tumor budding at the invasive margin can predict patients at high risk of recurrence after curative surgery for stage II, T3 colon cancer. Dis. Colon Rectum 2003, 46, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Mitomi, H.; Kanazawa, H.; Ohkura, Y.; Watanabe, M. Tumor budding as an index to identify high-risk patients with stage II colon cancer. Dis. Colon Rectum 2008, 51, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, H.; Mitomi, H.; Nishiyama, Y.; Kishimoto, I.; Fukui, N.; Nakamura, T.; Watanabe, M. Tumour budding at invasive margins and outcome in colorectal cancer. Colorectal Dis. 2008, 10, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Ueno, H.; Mochizuki, H.; Hatsuse, K.; Hase, K.; Yamamoto, T. Indicators for treatment strategies of colorectal liver metastases. Ann. Surg. 2000, 231, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, F.; Pezzica, E.; Cabiddu, M.; Coinu, A.; Borgonovo, K.; Ghilardi, M.; Lonati, V.; Corti, D.; Barni, S. Tumour budding and survival in stage II colorectal cancer: A systematic review and pooled analysis. J. Gastrointest. Cancer 2015, 46, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Benson, A.B., 3rd; Schrag, D.; Somerfield, M.R.; Cohen, A.M.; Figueredo, A.T.; Flynn, P.J.; Kryzanowska, M.K.; Maroun, J.; McAllister, P.; Van Cutsem, E.; et al. American Society of Clinical Oncology recommendations on adjuvant chemotherapy for stage II colon cancer. J. Clin. Oncol. 2004, 22, 3408–3419. [Google Scholar] [CrossRef] [PubMed]
- Kai, K.; Aishima, S.; Aoki, S.; Takase, Y.; Uchihashi, K.; Masuda, M.; Nishijima-Matsunobu, A.; Yamamoto, M.; Ide, K.; Nakayama, A.; et al. Cytokeratin immunohistochemistry improves interobserver variability between unskilled pathologists in the evaluation of tumor budding in T1 colorectal cancer. Pathol. Int. 2016, 66, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Roh, M.S.; Lee, J.I.; Choi, P.J. Tumor budding as a useful prognostic marker in esophageal squamous cell carcinoma. Dis. Esophagus 2004, 17, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Koike, M.; Kodera, Y.; Itoh, Y.; Nakayama, G.; Fujiwara, M.; Hamajima, N.; Nakao, A. Multivariate analysis of the pathologic features of esophageal squamous cell cancer: Tumor budding is a significant independent prognostic factor. Ann. Surg. Oncol. 2008, 15, 1977–1982. [Google Scholar] [CrossRef] [PubMed]
- Miyata, H.; Yoshioka, A.; Yamasaki, M.; Nushijima, Y.; Takiguchi, S.; Fujiwara, Y.; Nishida, T.; Mano, M.; Mori, M.; Doki, Y. Tumor budding in tumor invasive front predicts prognosis and survival of patients with esophageal squamous cell carcinomas receiving neoadjuvant chemotherapy. Cancer 2009, 115, 3324–3334. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; Sillah, K.; Griffiths, E.A.; Swindell, R.; West, C.M.; Page, R.D.; Welch, I.M.; Pritchard, S.A. Tumour budding and a low host inflammatory response are associated with a poor prognosis in oesophageal and gastro-oesophageal junction cancers. Histopathology 2010, 56, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, Y.; Ohara, M.; Doumen, H.; Kimura, N.; Ishidate, T.; Kondo, S. Correlation between tumor budding and post-resection prognosis in patients with invasive squamous cell carcinoma of the thoracic esophagus. World J. Surg. 2011, 35, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Teramoto, H.; Koike, M.; Tanaka, C.; Yamada, S.; Nakayama, G.; Fujii, T.; Sugimoto, H.; Fujiwara, M.; Suzuki, Y.; Kodera, Y. Tumor budding as a useful prognostic marker in T1-stage squamous cell carcinoma of the esophagus. J. Surg. Oncol. 2013, 108, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Niwa, Y.; Yamada, S.; Koike, M.; Kanda, M.; Fujii, T.; Nakayama, G.; Sugimoto, H.; Nomoto, S.; Fujiwara, M.; Kodera, Y. Epithelial to mesenchymal transition correlates with tumor budding and predicts prognosis in esophageal squamous cell carcinoma. J. Surg. Oncol. 2014, 110, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Almangush, A.; Karhunen, M.; Hautaniemi, S.; Salo, T.; Leivo, I. Prognostic value of tumour budding in oesophageal cancer: A meta-analysis. Histopathology 2016, 68, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Masugi, Y.; Yamazaki, K.; Hibi, T.; Aiura, K.; Kitagawa, Y.; Sakamoto, M. Solitary cell infiltration is a novel indicator of poor prognosis and epithelial-mesenchymal transition in pancreatic cancer. Hum. Pathol. 2010, 41, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Kohler, I.; Bronsert, P.; Timme, S.; Werner, M.; Brabletz, T.; Hopt, U.T.; Schilling, O.; Bausch, D.; Keck, T.; Wellner, U.F. Detailed analysis of epithelial-mesenchymal transition and tumor budding identifies predictors of long-term survival in pancreatic ductal adenocarcinoma. J. Gastroenterol. Hepatol. 2015, 30 (Suppl. 1), 78–84. [Google Scholar] [PubMed]
- O’Connor, K.; Li-Chang, H.H.; Kalloger, S.E.; Peixoto, R.D.; Webber, D.L.; Owen, D.A.; Driman, D.K.; Kirsch, R.; Serra, S.; Scudamore, C.H.; et al. Tumor budding is an independent adverse prognostic factor in pancreatic ductal adenocarcinoma. Am. J. Surg. Pathol. 2015, 39, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Karamitopoulou, E.; Zlobec, I.; Born, D.; Kondi-Pafiti, A.; Lykoudis, P.; Mellou, A.; Gennatas, K.; Gloor, B.; Lugli, A. Tumour budding is a strong and independent prognostic factor in pancreatic cancer. Eur. J. Cancer 2013, 49, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Sarioglu, S.; Acara, C.; Akman, F.C.; Dag, N.; Ecevit, C.; Ikiz, A.O.; Cetinayak, O.H.; Ada, E. Tumor budding as a prognostic marker in laryngeal carcinoma. Pathol. Res. Pract. 2010, 206, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Huang, H.; Huang, Z.; Wang, A.; Chen, X.; Huang, L.; Zhou, X.; Liu, X. Tumor budding correlates with poor prognosis and epithelial-mesenchymal transition in tongue squamous cell carcinoma. J. Oral Pathol. Med. 2011, 40, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Almangush, A.; Salo, T.; Hagström, J.; Leivo, I. Tumour budding in head and neck squamous cell carcinoma—A systematic review. Histopathology 2014, 65, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Ishii, G.; Kojima, M.; Yoh, K.; Otsuka, H.; Otaki, Y.; Aokage, K.; Yanagi, S.; Nagai, K.; Nishiwaki, Y.; et al. Histopathologic features of the tumor budding in adenocarcinoma of the lung: Tumor budding as an index to predict the potential aggressiveness. J. Thorac. Oncol. 2010, 5, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Masuda, R.; Kijima, H.; Imamura, N.; Aruga, N.; Nakamura, Y.; Masuda, D.; Takeichi, H.; Kato, N.; Nakagawa, T.; Tanaka, M.; et al. Tumor budding is a significant indicator of a poor prognosis in lung squamous cell carcinoma patients. Mol. Med. Rep. 2012, 6, 937–943. [Google Scholar] [PubMed]
- Taira, T.; Ishii, G.; Nagai, K.; Yoh, K.; Takahashi, Y.; Matsumura, Y.; Kojima, M.; Ohmatsu, H.; Goto, K.; Niho, S.; et al. Characterization of the immunophenotype of the tumor budding and its prognostic implications in squamous cell carcinoma of the lung. Lung Cancer 2012, 76, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Cao, W.; Wang, Y.; Li, L.; Zhang, G.; Wang, Z. The prognostic value of tumor budding in invasive breast cancer. Pathol. Res. Pract. 2013, 209, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Gujam, F.J.A.; McMillan, D.C.; Mohammed, Z.M.A.; Edwards, J.; Going, J.J. The relationship between tumour budding, the tumour microenvironment and survival in patients with invasive ductal breast cancer. Br. J. Cancer 2015, 113, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Kai, K.; Kohya, N.; Kitahara, K.; Masuda, M.; Miyoshi, A.; Ide, T.; Tokunaga, O.; Miyazaki, K.; Noshiro, H. Tumor budding and dedifferentiation in gallbladder carcinoma: Potential for the prognostic factors in T2 lesions. Virchows Arch. 2011, 459, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Koyuncuoglu, M.; Okyay, E.; Saatli, B.; Olgan, S.; Akin, M.; Saygili, U. Tumor budding and E-Cadherin expression in endometrial carcinoma: Are they prognostic factors in endometrial cancer? Gynecol. Oncol. 2012, 125, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Attramadal, C.G.; Kumar, S.; Boysen, M.E.; Dhakal, H.P.; Nesland, J.M.; Bryne, M. Tumor budding, EMT and cancer stem cells in T1–2/N0 oral squamous cell carcinomas. Anticancer Res. 2015, 35, 6111–6120. [Google Scholar] [PubMed]
- Okado, Y.; Aoki, M.; Hamasaki, M.; Koga, K.; Sueta, T.; Shiratsuchi, H.; Oda, Y.; Nakagawa, T.; Nabeshima, K. Tumor budding and laminin5-γ2 in squamous cell carcinoma of the external auditory canal are associated with shorter survival. Springerplus 2015, 4, 814. [Google Scholar] [CrossRef] [PubMed]
- Koelzer, V.H.; Langer, R.; Zlobec, I.; Lugli, A. Tumor budding in upper gastrointestinal carcinomas. Front. Oncol. 2014, 4, 216. [Google Scholar] [CrossRef] [PubMed]
- Thies, S.; Guldener, L.; Slotta-Huspenina, J.; Zlobec, I.; Koelzer, V.H.; Lugli, A.; Kroll, D.; Seiler, C.A.; Feith, M.; Langer, R. Impact of peritumoral and intratumoral budding in esophageal adenocarcinoma. Hum. Pathol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Nowak, J.A.; Agoston, A.; Zheng, Y.; Bueno, R.; Odze, R.D.; Srivastava, A. Tumor budding is a predictor of nodal metastasis and tumor recurrence in T1 esophageal adenocarcinoma. Mod. Pathol. 2013, 26, 169A. [Google Scholar]
- Gabbert, H.E.; Meier, S.; Gerharz, C.D.; Hommel, G. Tumor-cell dissociation at the invasion front: A new prognostic parameter in gastric cancer patients. Int. J. Cancer 1992, 50, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Ohike, N.; Coban, I.; Kim, G.E.; Basturk, O.; Tajiri, T.; Krasinskas, A.; Bandyopadhyay, S.; Morohoshi, T.; Shimada, Y.; Kooby, D.A.; et al. Tumor budding as a strong prognostic indicator in invasive ampullary adenocarcinomas. Am. J. Surg. Pathol. 2010, 34, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.R.; Gao, F.; Li, S.Y.; Yao, K.T. Tumour budding and the expression of cancer stem cell marker aldehyde dehydrogenase 1 in nasopharyngeal carcinoma. Histopathology 2012, 61, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Salhia, B.; Trippel, M.; Pfaltz, K.; Cihoric, N.; Grogg, A.; Lädrach, C.; Zlobec, I.; Tapia, C. High tumor budding stratifies breast cancer with metastatic properties. Breast Cancer Res. Treat. 2015, 150, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Mason, J.; Jewett, A.; Qian, J.; Ding, Y.; Cho, W.C.S.; Zhang, X.; Man, Y.G. Cell budding from normal appearing epithelia: A predictor of colorectal cancer metastasis? Int. J. Biol. Sci. 2013, 9, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Lugli, A.; Vlajnic, T.; Giger, O.; Karamitopoulou, E.; Patsouris, E.S.; Peros, G.; Terracciano, L.M.; Zlobec, I. Intratumoral budding as a potential parameter of tumor progression in mismatch repair-proficient and mismatch repair-deficient colorectal cancer patients. Hum. Pathol. 2011, 42, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Rogers, A.C.; Gibbons, D.; Hanly, A.M.; Hyland, J.M.P.; O’Connell, P.R.; Winter, D.C.; Sheahan, K. Prognostic significance of tumor budding in rectal cancer biopsies before neoadjuvant therapy. Mod. Pathol. 2014, 27, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial-mesenchymal transition in cancer: Parallels between normal development and tumor progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342. [Google Scholar] [CrossRef] [PubMed]
- Schmalhofer, O.; Brabletz, S.; Brabletz, T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009, 28, 151–166. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Jordan, N.V.; Johnson, G.L.; Abell, A.N. Tracking the intermediate stages of epithelial-mesenchymal transition in epithelial stem cells and cancer. Cell Cycle 2011, 10, 2865–2873. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.; Watanabe, K.; Ta, C.H.; Villarreal-Ponce, A.; Nie, Q.; Dai, X. An Ovol2-Zeb1 mutual inhibitory circuit governs bidirectional and multi-step transition between epithelial and mesenchymal states. PLoS Comput. Biol. 2015, 11, e1004569. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.-J.; Wong, M.K.; Tan, T.Z.; Kuay, K.T.; Ng, A.H.C.; Chung, V.Y.; Chu, Y.-S.; Matsumura, N.; Lai, H.-C.; Lee, Y.F.; et al. An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e-cadherin restoration by a src-kinase inhibitor, saracatinib (AZD0530). Cell Death Dis. 2013, 4, e915. [Google Scholar] [CrossRef] [PubMed]
- Morbini, P.; Inghilleri, S.; Campo, I.; Oggionni, T.; Zorzetto, M.; Luisetti, M. Incomplete expression of epithelial-mesenchymal transition markers in idiopathic pulmonary fibrosis. Pathol. Res. Pract. 2011, 207, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Umbreit, C.; Flanjak, J.; Weiss, C.; Erben, P.; Aderhold, C.; Faber, A.; Stern-straeter, J.; Hoermann, K.; Schultz, J.D. Incomplete epithelial-mesenchymal transition in p16-positive squamous cell carcinoma cells correlates with β-catenin expression. Anticancer Res. 2014, 34, 7061–7070. [Google Scholar] [PubMed]
- Lundgren, K.; Nordenskjöld, B.; Landberg, G. Hypoxia, Snail and incomplete epithelial-mesenchymal transition in breast cancer. Br. J. Cancer 2009, 101, 1769–1781. [Google Scholar] [CrossRef] [PubMed]
- Bednarz-Knoll, N.; Alix-Panabières, C.; Pantel, K. Plasticity of disseminating cancer cells in patients with epithelial malignancies. Cancer Metastasis Rev. 2012, 31, 673–687. [Google Scholar] [CrossRef] [PubMed]
- Sampson, V.B.; David, J.M.; Puig, I.; Patil, P.U.; de Herreros, A.G.; Thomas, G.V.; Rajasekaran, A.K. Wilms’ tumor protein induces an epithelial-mesenchymal hybrid differentiation state in clear cell renal cell carcinoma. PLoS ONE 2014, 9, e102041. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Jolly, M.K.; Levine, H.; Onuchic, J.N.; Ben-Jacob, E. MicroRNA-based regulation of epithelial-hybrid-mesenchymal fate determination. Proc. Natl. Acad. Sci. USA 2013, 110, 18174–18179. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Jolly, M.K.; Boareto, M.; Parsana, P.; Mooney, S.M.; Pienta, K.J.; Levine, H.; Ben-Jacob, E. OVOL guides the epithelial-hybrid-mesenchymal transition. Oncotarget 2015, 6, 15436–15448. [Google Scholar] [CrossRef] [PubMed]
- Schliekelman, M.J.; Taguchi, A.; Zhu, J.; Dai, X.; Rodriguez, J.; Celiktas, M.; Zhang, Q.; Chin, A.; Wong, C.; Wang, H.; et al. Molecular portraits of epithelial, mesenchymal and hybrid states in lung adenocarcinoma and their relevance to survival. Cancer Res. 2015, 75, 1789–1800. [Google Scholar] [CrossRef] [PubMed]
- Steinway, S.N.; Zañudo, J.G.T.; Michel, P.J.; Feith, D.J.; Loughran, T.P.; Albert, R. Combinatorial interventions inhibit TGFβ-driven epithelial-to-mesenchymal transition and support hybrid cellular phenotypes. NPJ Syst. Biol. Appl. 2015, 1, 15014. [Google Scholar] [CrossRef]
- Bastos, L.G.D.R.; de Marcondes, P.G.; de-Freitas-Junior, J.C.M.; Leve, F.; Mencalha, A.L.; de Souza, W.F.; de Araujo, W.M.; Tanaka, M.N.; Abdelhay, E.S.F.W.; Morgado-Díaz, J.A. Progeny from irradiated colorectal cancer cells acquire an EMT-like phenotype and activate Wnt/β-catenin pathway. J. Cell. Biochem. 2014, 115, 2175–2187. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.S.; Liu, X.W.; Chirco, R.; Warner, R.B.; Fridman, R.; Kim, H.R.C. TIMP-1 induces an EMT-like phenotypic conversion in MDCK cells independent of its MMP-inhibitory domain. PLoS ONE 2012, 7, e38773. [Google Scholar] [CrossRef] [PubMed]
- Eades, G.; Yao, Y.; Yang, M.; Zhang, Y.; Chumsri, S.; Zhou, Q. miR-200a regulates SIRT1 expression and epithelial to mesenchymal transition (EMT)-like transformation in mammary epithelial cells. J. Biol. Chem. 2011, 286, 25992–26002. [Google Scholar] [CrossRef] [PubMed]
- Savagner, P. The epithelial-mesenchymal transition (EMT) phenomenon. Ann. Oncol. 2010, 21 (Suppl. 7), vii89–vii92. [Google Scholar] [CrossRef] [PubMed]
- Futterman, M.A.; García, A.J.; Zamir, E.A. Evidence for partial epithelial-to-mesenchymal transition (pEMT) and recruitment of motile blastoderm edge cells during avian epiboly. Dev. Dyn. 2011, 240, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Leroy, P.; Mostov, K.E. Slug is required for cell survival during partial epithelial-mesenchymal transition of HGF-induced tubulogenesis. J. Cell Sci. 2007, 18, 1943–1952. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.-J.; Zhang, H.; Xing, J. Coupled reversible and irreversible bistable switches underlying TGFβ-induced epithelial to mesenchymal transition. Biophys. J. 2013, 105, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tian, X.-J.; Zhang, H.; Teng, Y.; Li, R.; Bai, F.; Elankumaran, S.; Xing, J. TGF-β–induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Sci. Signal. 2014, 7. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.S.; Paredes, J. P-cadherin linking breast cancer stem cells and invasion: A promising marker to identify an “intermediate/metastable” EMT state. Front. Oncol. 2015, 4, 371. [Google Scholar] [CrossRef] [PubMed]
- Savagner, P. Epithelial-mesenchymal transitions: From cell plasticity to concept elasticity. Curr. Top. Dev. Biol. 2015, 112, 273–300. [Google Scholar] [PubMed]
- Grosse-Wilde, A.; Fouquier d’Hérouël, A.; McIntosh, E.; Ertaylan, G.; Skupin, A.; Kuestner, R.E.; del Sol, A.; Walters, K.-A.; Huang, S. Stemness of the hybrid epithelial/mesenchymal state in breast cancer and its association with poor survival. PLoS ONE 2015, 10, e0126522. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Tripathi, S.C.; Jia, D.; Mooney, S.M.; Celiktas, M.; Hanash, S.M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Stability of the hybrid epithelial/mesenchymal phentoype. Oncotarget 2016. [Google Scholar] [CrossRef]
- Arnoux, V.; Côme, C.; Kusewitt, D.F.; Hudson, L.G.; Savagner, P. Cutaneous wound reepithelialization. In Rise and Fall of Epithelial Phenotype; Springer US: New York, NY, USA, 2005; pp. 111–134. [Google Scholar]
- Watanabe, K.; Villarreal-Ponce, A.; Sun, P.; Salmans, M.L.; Fallahi, M.; Andersen, B.; Dai, X. Mammary morphogenesis and regeneration require the inhibition of EMT at terminal end buds by Ovol2 transcriptional repressor. Dev. Cell 2014, 29, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Sarioglu, A.F.; Aceto, N.; Kojic, N.; Donaldson, M.C.; Zeinali, M.; Hamza, B.; Engstrom, A.; Zhu, H.; Sundaresan, T.K.; Miyamoto, D.T.; et al. A microfluidic device for label-free, physical capture of circulating tumor cell clusters. Nat. Methods 2015, 12, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Andriani, F.; Bertolini, G.; Facchinetti, F.; Baldoli, E.; Moro, M.; Casalini, P.; Caserini, R.; Milione, M.; Leone, G.; Pelosi, G.; et al. Conversion to stem-cell state in response to microenvironmental cues is regulated by balance between epithelial and mesenchymal features in lung cancer cells. Mol. Oncol. 2015, 10, 253–271. [Google Scholar] [CrossRef] [PubMed]
- Jeevan, D.S.; Cooper, J.B.; Braun, A.; Murali, R.A.J.; Jhanwar-uniyal, M. Molecular pathways mediating metastases to the brain via epithelial-to-mesenchymal transition: Genes, proteins, and functional analysis. Anticancer Res. 2016, 36, 523–532. [Google Scholar] [PubMed]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Lugli, A.; Karamitopoulou, E.; Panayiotides, I.; Karakitsos, P.; Rallis, G.; Peros, G.; Iezzi, G.; Spagnoli, G.; Bihl, M.; Terracciano, L.; et al. CD8+ lymphocytes/tumour-budding index: An independent prognostic factor representing a “pro-/anti-tumour” approach to tumour host interaction in colorectal cancer. Br. J. Cancer 2009, 101, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Paterson, E.L.; Kazenwadel, J.; Bert, A.G.; Khew-Goodall, Y.; Ruszkiewicz, A.; Goodall, G.J. Down-regulation of the miRNA-200 family at the invasive front of colorectal cancers with degraded basement membrane indicates EMT is involved in cancer progression. Neoplasia 2013, 15, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Jensen, D.H.; Dabelsteen, E.; Specht, L.; Fiehn, A.M.; Therkildsen, M.H.; Jønson, L.; Vikesaa, J.; Nielsen, F.C.; von Buchwald, C. Molecular profiling of tumour budding implicates TGF-β-mediated epithelial-mesenchymal transition as a therapeutic target in oral squamous cell carcinoma. J. Pathol. 2015, 236, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bracken, C.P.; Smith, E.; Bert, A.G.; Wright, J.A.; Roslan, S.; Morris, M.; Wyatt, L.; Farshid, G.; Lim, Y.-Y.; et al. An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol. Biol. Cell 2011, 22, 1686–1698. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Jolly, M.K.; Lu, M.; Tsarfaty, I.; Onuchic, J.N.; Ben-Jacob, E. Modeling the transitions between collective and solitary migration phenotypes in cancer metastasis. Sci. Rep. 2015, 5, 17379. [Google Scholar] [CrossRef] [PubMed]
- Kainulainen, T.; Hakkinen, L.; Hamidi, S.; Larjava, K.; Kallioinen, M.; Peltonen, J.; Salo, T.; Larjava, H.; Oikarinen, A. Laminin-5 expression is independent of the injury and the microenvironment during reepithelialization of wounds. J. Histochem. Cytochem. 1998, 46, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, S.; Brabletz, T. The ZEB/miR-200 feedback loop—A motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the hybrid epithelial/mesenchymal phenotype in metastasis. Front. Oncol. 2015, 5, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Galván, J.A.; Zlobec, I.; Wartenberg, M.; Lugli, A.; Gloor, B.; Perren, A.; Karamitopoulou, E. Expression of E-cadherin repressors SNAIL, ZEB1 and ZEB2 by tumour and stromal cells influences tumour-budding phenotype and suggests heterogeneity of stromal cells in pancreatic cancer. Br. J. Cancer 2015, 112, 1944–1950. [Google Scholar] [CrossRef] [PubMed]
- Dawson, H.; Lugli, A. Molecular and pathogenetic aspects of tumor budding in colorectal cancer. Front. Med. 2015, 2, 11. [Google Scholar] [CrossRef] [PubMed]
- Ocaña, O.H.; Córcoles, R.; Fabra, A.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, R.; Rutegard, J.N.; Bozoky, B.; Landberg, G.; Stenling, R. Human colorectal cancers with an intact p16/cyclin D1/pRb pathway have up-regulated p16 expression and decreased proliferation in small invasive tumor clusters. Am. J. Pathol. 2000, 157, 1947–1953. [Google Scholar] [CrossRef]
- Jung, A.; Schrauder, M.; Oswald, U.; Knoll, C.; Sellberg, P.; Palmqvist, R.; Niedobitek, G.; Brabletz, T.; Kirchner, T. The invasion front of human colorectal adenocarcinomas shows co-localization of nuclear beta-catenin, cyclin D1, and p16INK4A and is a region of low proliferation. Am. J. Pathol. 2001, 159, 1613–1617. [Google Scholar] [PubMed]
- Jass, J.R.; Barker, M.; Fraser, L.; Walsh, M.D.; Whitehall, V.L.J.; Gabrielli, B.; Young, J.; Leggett, B.A. APC mutation and tumour budding in colorectal cancer. J. Clin. Pathol. 2003, 56, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Rubio, C.A. Arrest of cell proliferation in budding tumor cells ahead of the invading edge of colonic carcinomas. A preliminary report. Anticancer Res. 2008, 28, 2417–2420. [Google Scholar] [PubMed]
- Dawson, H.; Koelzer, V.H.; Karamitopoulou, E.; Economou, M.; Hammer, C.; Muller, D.E.; Lugli, A.; Zlobec, I. The apoptotic and proliferation rate of tumour budding cells in colorectal cancer outlines a heterogeneous population of cells with various impacts on clinical outcome. Histopathology 2014, 64, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Jung, A.; Reu, S.; Porzner, M.; Hlubek, F.; Kunz-Schughart, L.A.; Knuechel, R.; Kirchner, T. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc. Natl. Acad. Sci. USA 2001, 98, 10356–10361. [Google Scholar] [CrossRef] [PubMed]
- Hatzikirou, H.; Basanta, D.; Simon, M.; Schaller, K.; Deutsch, A. “Go or grow”: The key to the emergence of invasion in tumour progression? Math. Med. Biol. 2012, 29, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Matus, D.Q.; Lohmer, L.L.; Kelley, L.C.; Schindler, A.J.; Kohrman, A.Q.; Barkoulas, M.; Zhang, W.; Chi, Q.; Sherwood, D.R. Invasive Cell Fate Requires G1 Cell-Cycle Arrest and Histone Deacetylase-Mediated Changes in Gene Expression. Dev. Cell 2015, 35, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.-C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Robson, E.J.D.; Khaled, W.T.; Abell, K.; Watson, C.J. Epithelial-to-mesenchymal transition confers resistance to apoptosis in three murine mammary epithelial cell lines. Differentiation 2006, 74, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Vega, S.; Morales, A.V.; Ocaña, O.H.; Valdés, F.; Fabregat, I.; Nieto, M.A. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004, 18, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Sanitarias, I.; Valdés, F.; Álvarez, A.M.; Locascio, A.; Vega, S.; Herrera, B.; Alberto, M.A.; Benito, M.; Nieto, M.A.; et al. The epithelial mesenchymal transition confers resistance to the apoptotic effects of transforming growth factor Beta in fetal rat hepatocytes. Mol. Cancer Res. 2002, 1, 68–78. [Google Scholar]
- Radisky, D.C. miR-200c at the nexus of epithelial-mesenchymal transition, resistance to apoptosis, and the breast cancer stem cell phenotype. Breast Cancer Res. 2011, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Lugli, A.; Karamitopoulou, E.; Zlobec, I. Tumour budding: A promising parameter in colorectal cancer. Br. J. Cancer 2012, 106, 1713–1717. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Huang, B.; Lu, M.; Mani, S.A.; Levine, H.; Ben-Jacob, E. Towards elucidating the connection between epithelial-mesenchymal transitions and stemness. J. R. Soc. Interface 2014, 11. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Jia, D.; Boareto, M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Coupling the modules of EMT and stemness: A tunable “stemness window” model. Oncotarget 2015, 6, 25161–25174. [Google Scholar] [CrossRef] [PubMed]
- Goldman, A.; Majumder, B.; Dhawan, A.; Ravi, S.; Goldman, D.; Kohandel, M.; Majumder, P.K.; Sengupta, S. Temporally sequenced anticancer drugs overcome adaptive resistance by targeting a vulnerable chemotherapy-induced phenotypic transition. Nat. Commun. 2015, 6, 6139. [Google Scholar] [CrossRef] [PubMed]
- Swetha, G.; Chandra, V.; Phadnis, S.; Bhonde, R. Glomerular parietal epithelial cells of adult murine kidney undergo EMT to generate cells with traits of renal progenitors. J. Cell. Mol. Med. 2011, 15, 396–413. [Google Scholar] [PubMed]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Migrating cancer stem cells—An integrated concept of malignant tumour progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grigore, A.D.; Jolly, M.K.; Jia, D.; Farach-Carson, M.C.; Levine, H. Tumor Budding: The Name is EMT. Partial EMT. J. Clin. Med. 2016, 5, 51. https://doi.org/10.3390/jcm5050051
Grigore AD, Jolly MK, Jia D, Farach-Carson MC, Levine H. Tumor Budding: The Name is EMT. Partial EMT. Journal of Clinical Medicine. 2016; 5(5):51. https://doi.org/10.3390/jcm5050051
Chicago/Turabian StyleGrigore, Alexandru Dan, Mohit Kumar Jolly, Dongya Jia, Mary C. Farach-Carson, and Herbert Levine. 2016. "Tumor Budding: The Name is EMT. Partial EMT." Journal of Clinical Medicine 5, no. 5: 51. https://doi.org/10.3390/jcm5050051
APA StyleGrigore, A. D., Jolly, M. K., Jia, D., Farach-Carson, M. C., & Levine, H. (2016). Tumor Budding: The Name is EMT. Partial EMT. Journal of Clinical Medicine, 5(5), 51. https://doi.org/10.3390/jcm5050051