
Comprehensive Characterization of Human Lung Large Cell Carcinoma Identifies Transcriptomic Signatures with Potential Implications in Response to Immunotherapy

 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Lung Large Cell Carcinoma Samples and Patient Cohort
2.2. DNA Extraction, Quantification and Quality Measurement
2.3. RNA Extraction, Quantification and Quality Measurement
2.4. Tumor Genetic Sequencing
2.5. Gene Expression Analysis with the Oncomine Immune Response Research Assay
2.6. Immunohistochemistry
2.7. Bioinformatic Analyses
2.8. Statistical Analyses
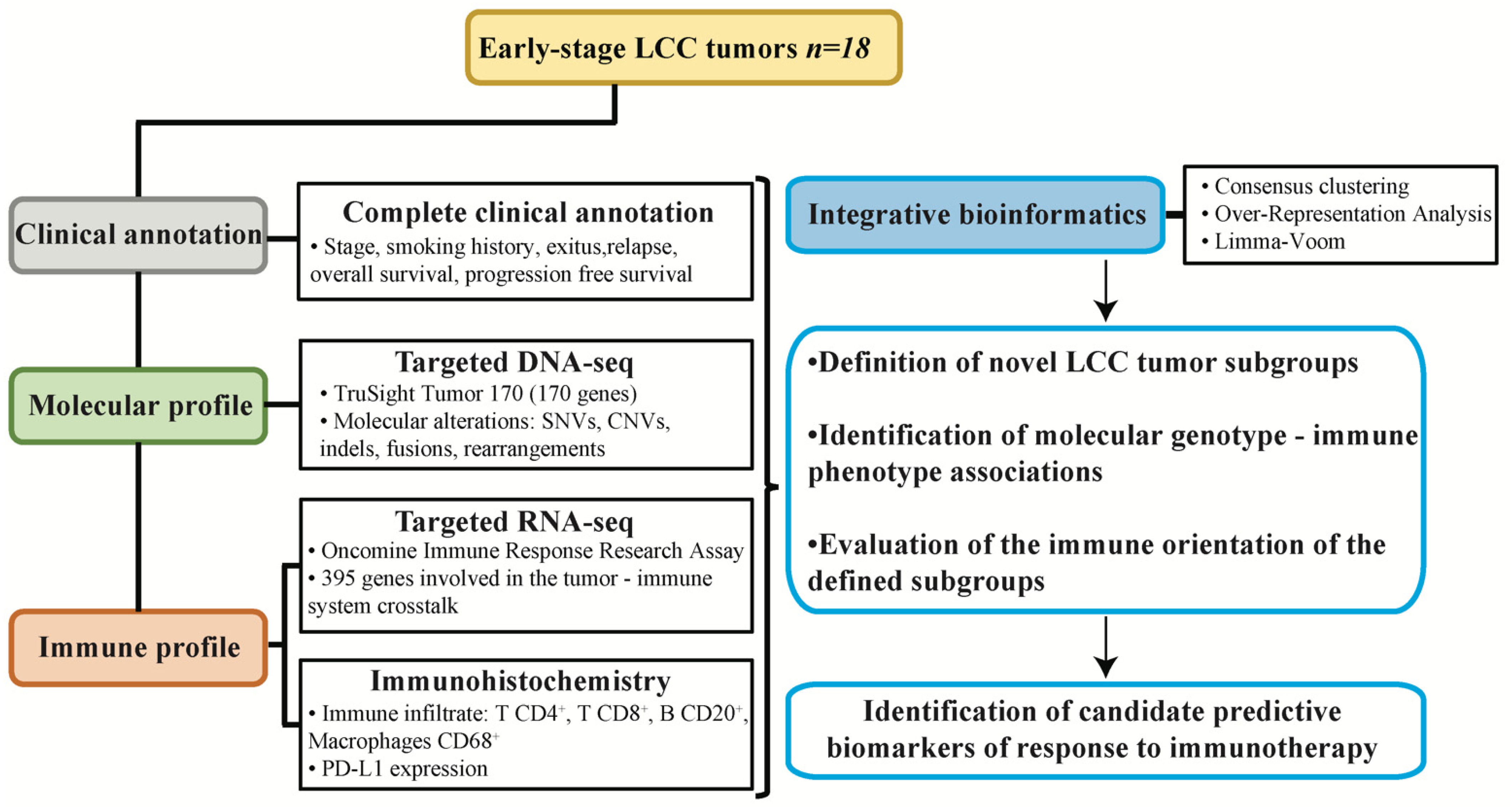
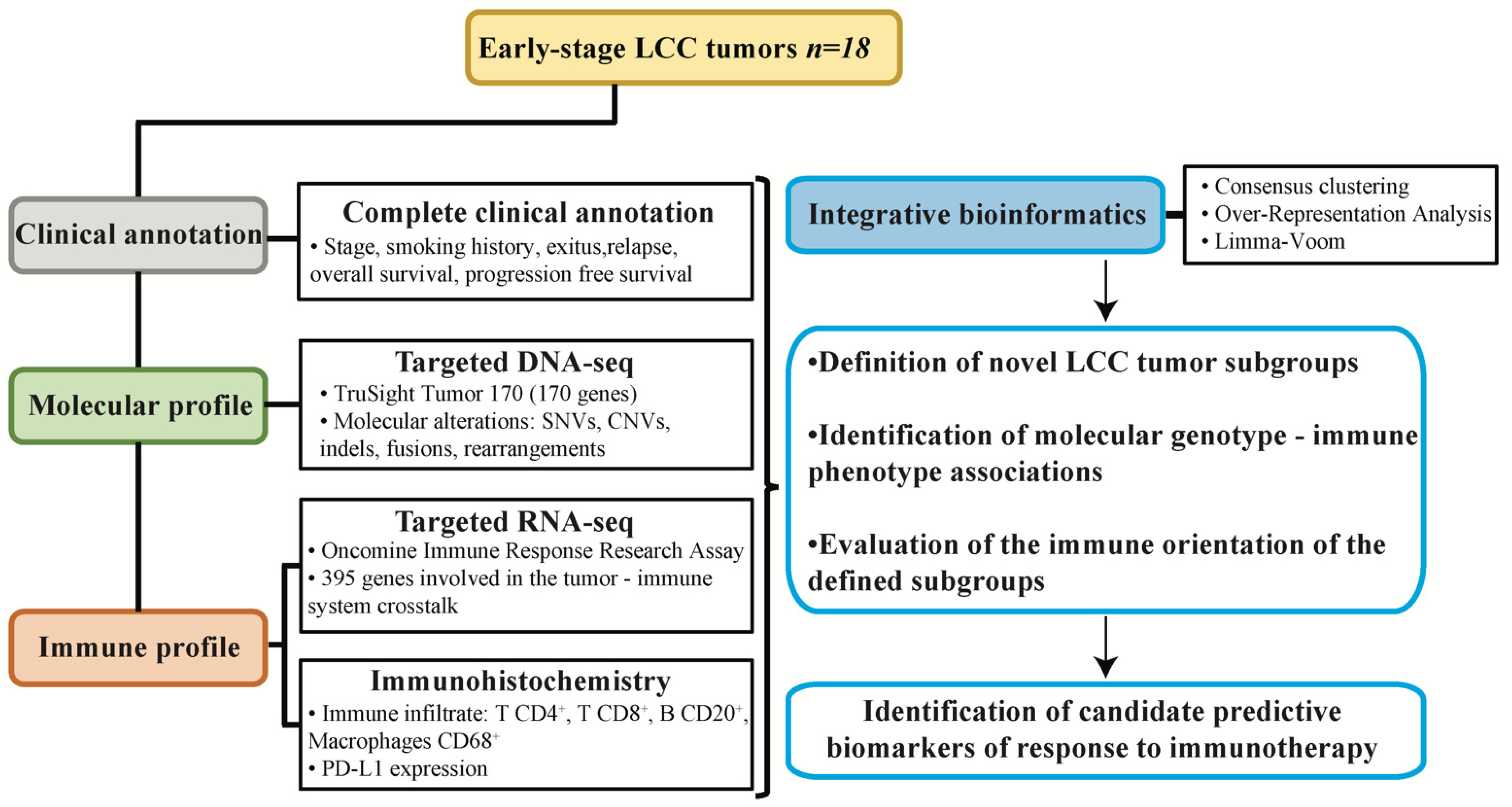
2.9. Experimental Design
3. Results
3.1. Description of the LCC Cohort
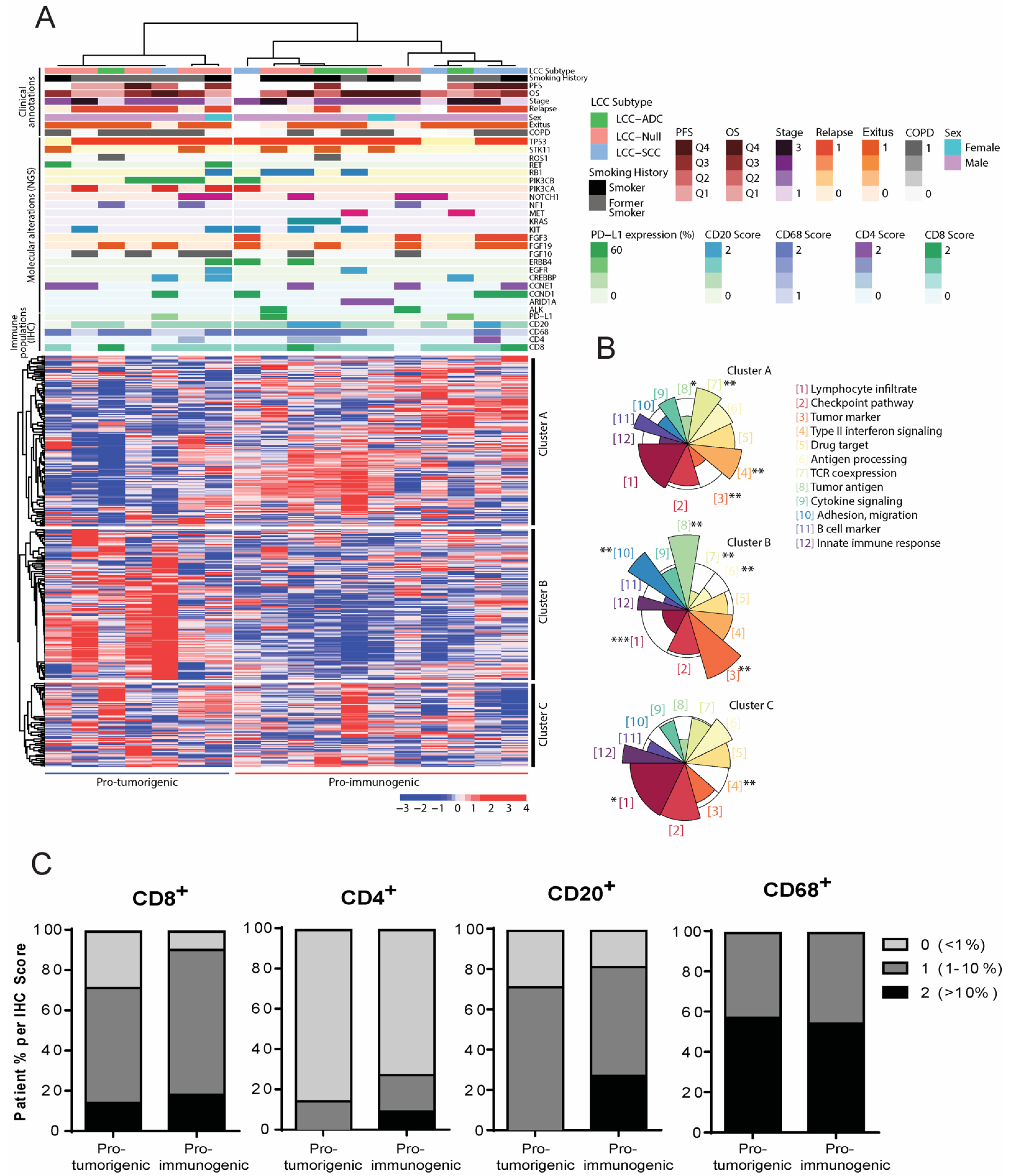
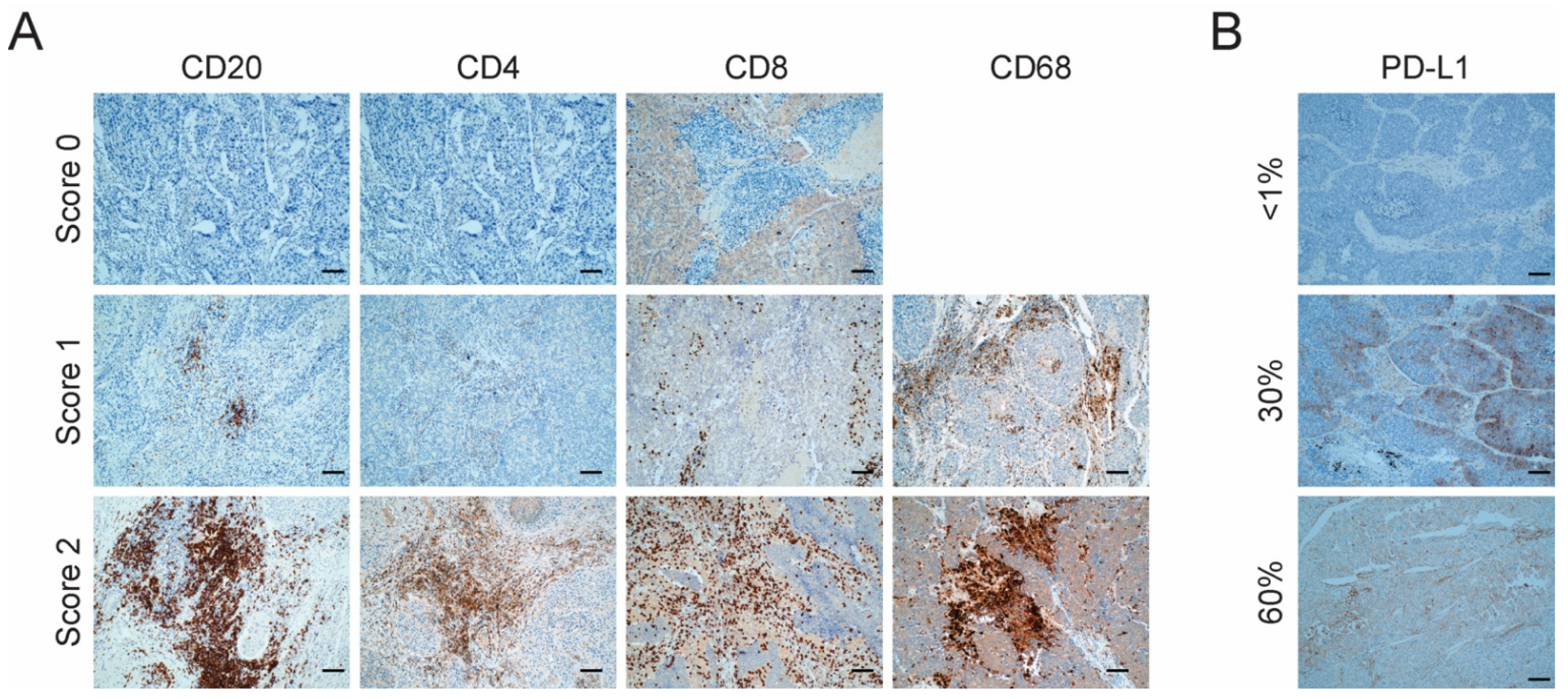
3.2. Evaluation of the Level of Infiltration of Immune Cell Populations and the Expression of PD-L1 Detected by Immunohistochemistry
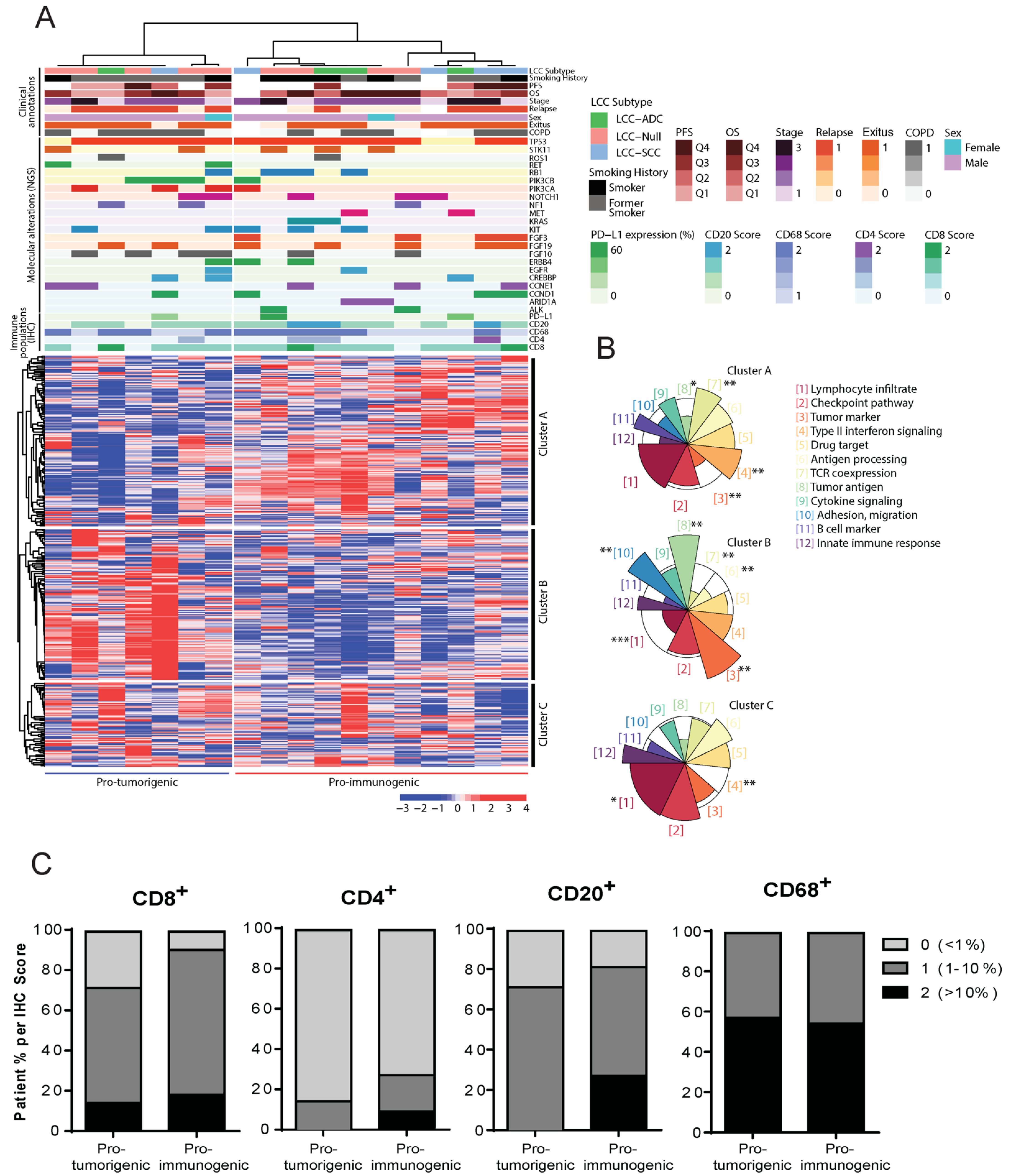
3.3. Definition of Novel LCC Tumor Subgroups by Consensus Clustering with Specific Molecular, Immune and Clinical Features
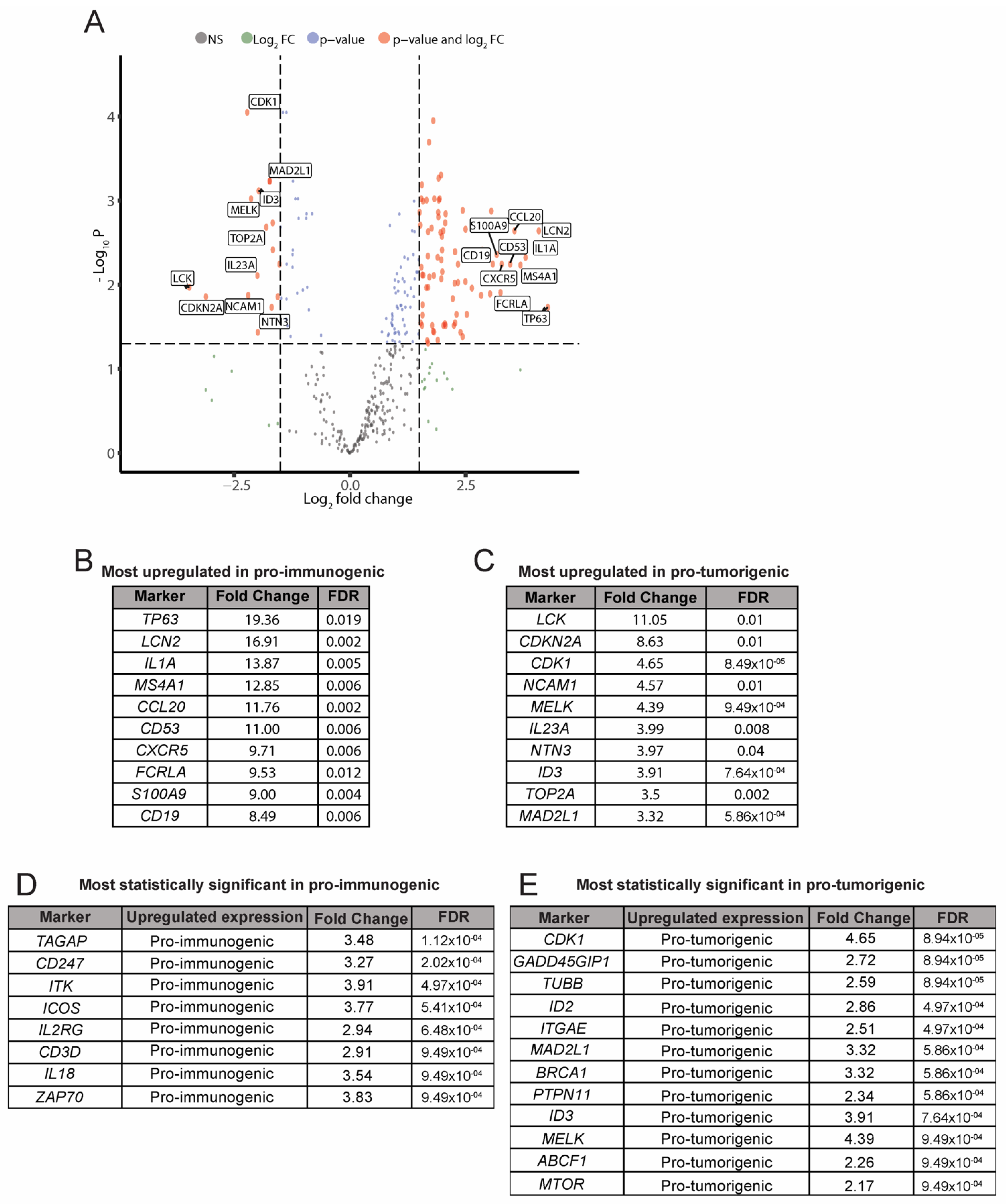
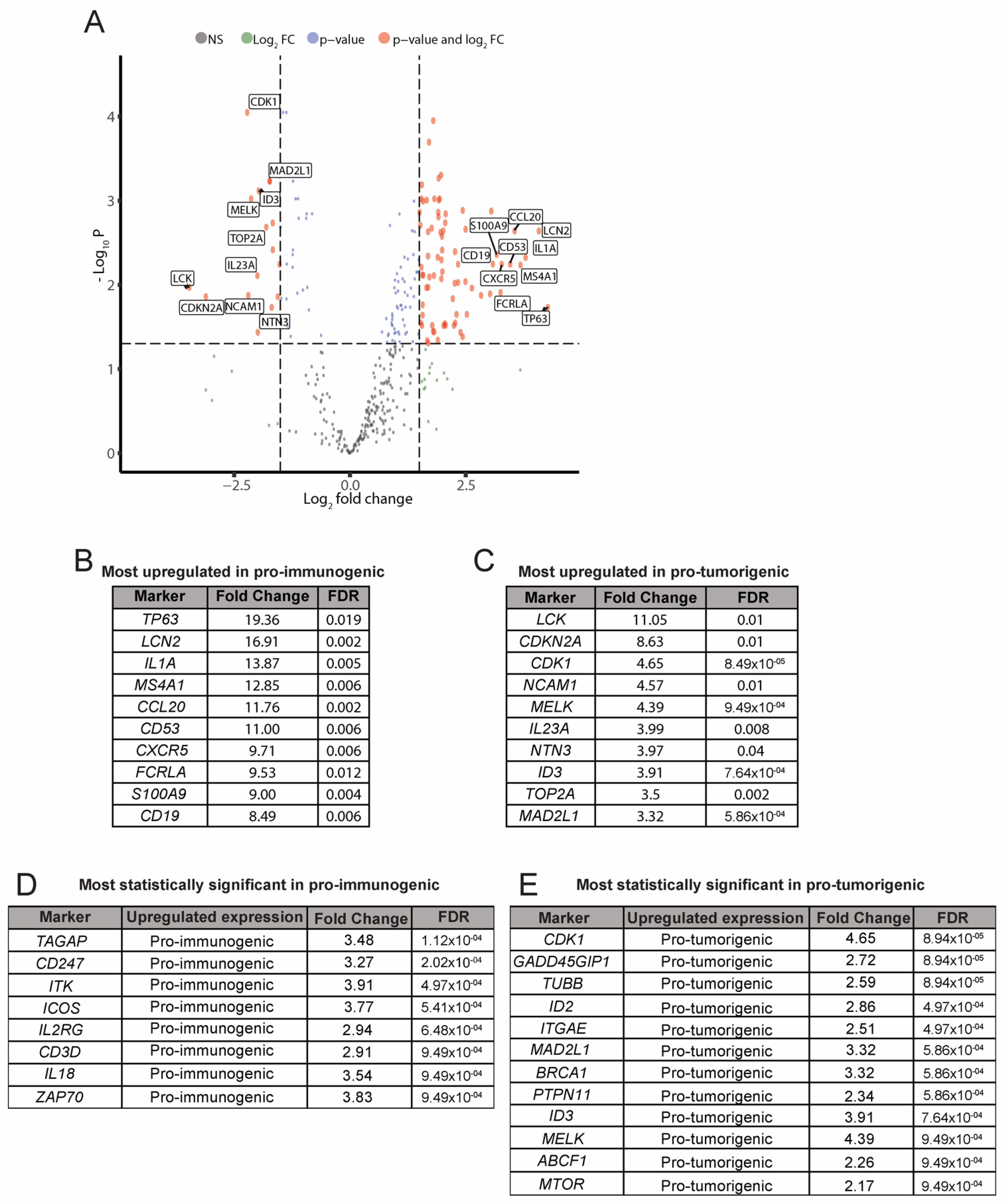
3.4. Analysis of Differentially Expressed Genes between the Pro-Immunogenic and Pro-Tumorigenic LCC Subgroups
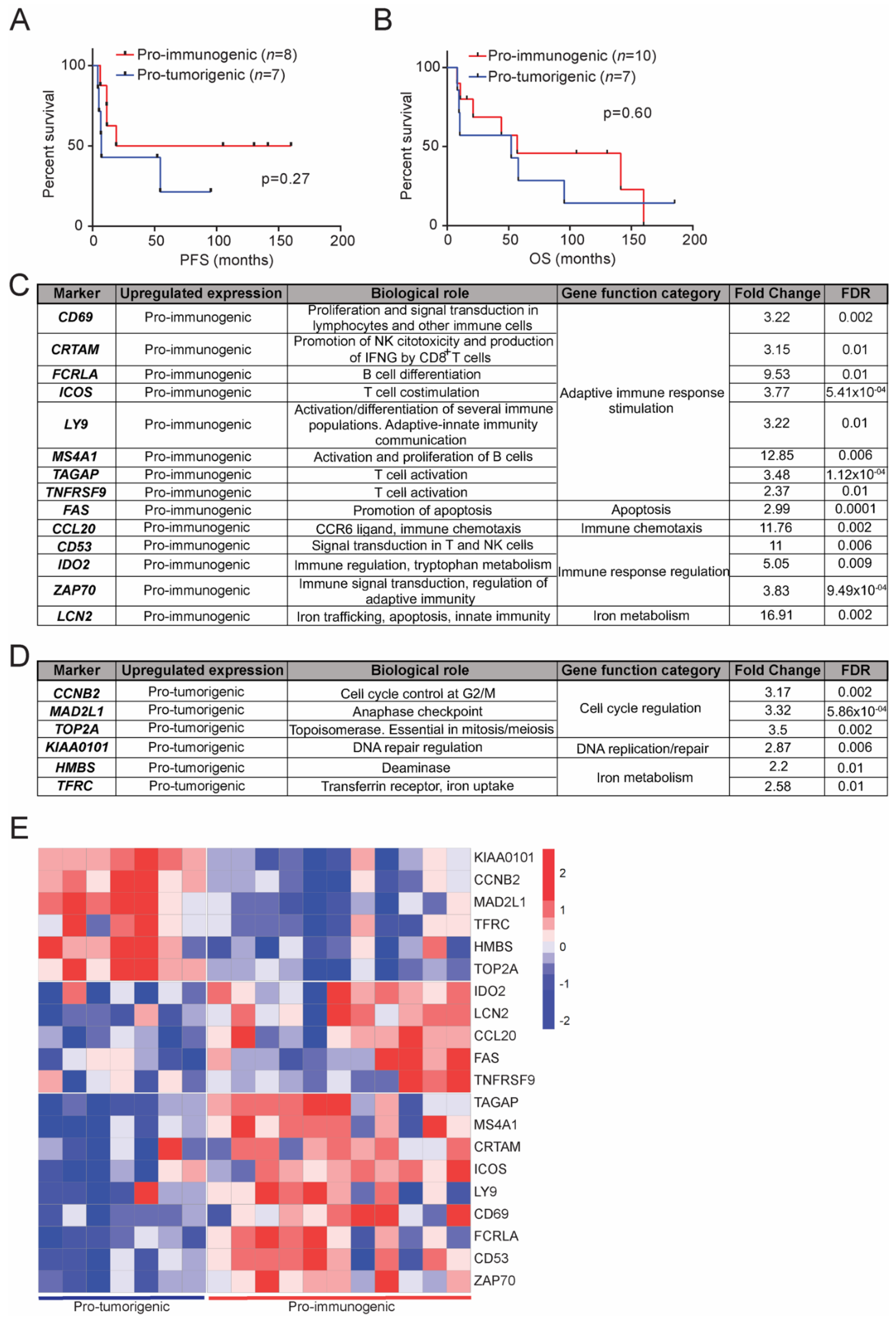
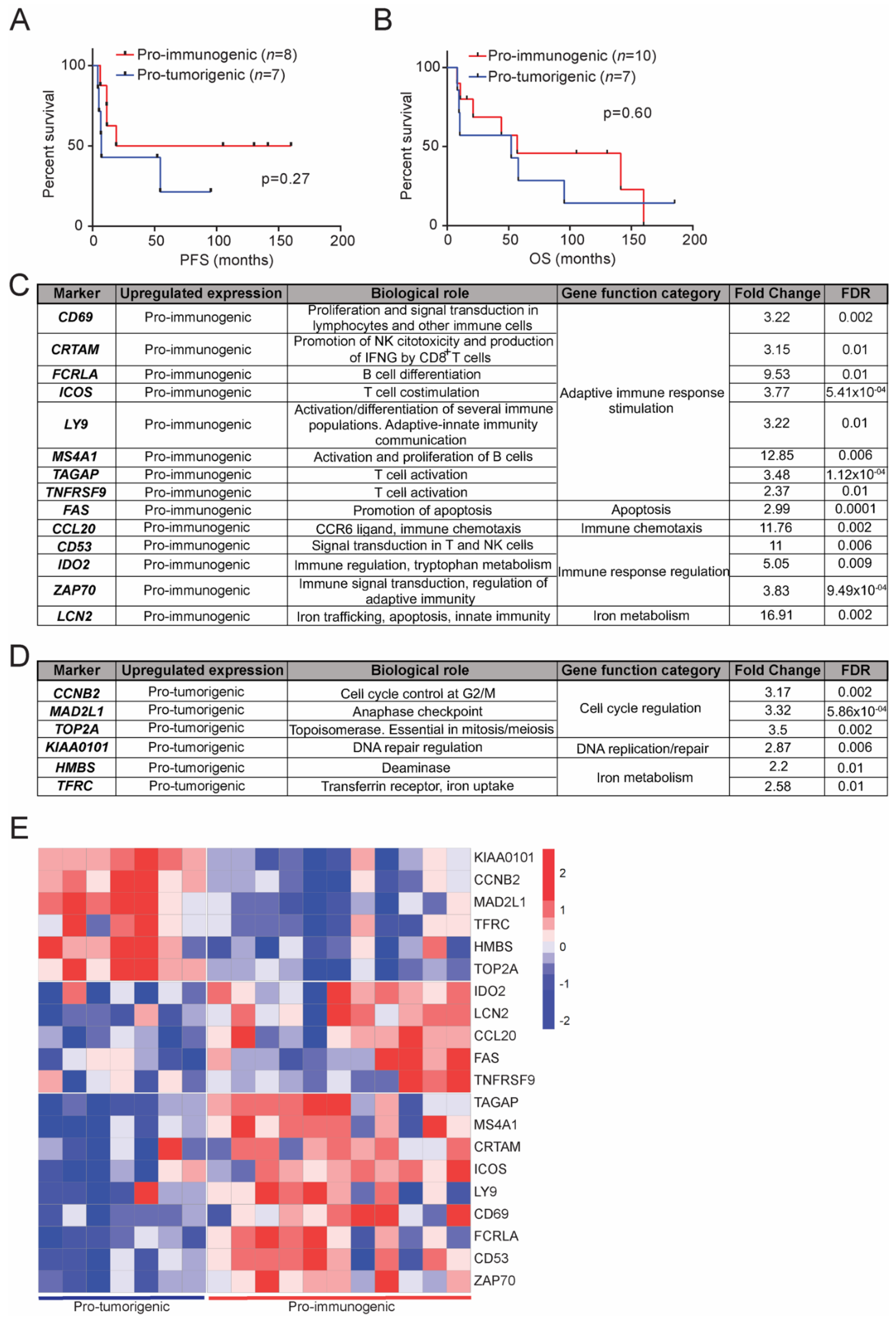
3.5. Clinical Relevance of the Defined LCC Subgroups
3.6. Transcriptional Signatures of the Defined LCC Subgroups and Potential Implications in Response to Immunotherapy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.K. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.M.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelosi, G.; Barbareschi, M.; Cavazza, A.; Graziano, P.; Rossi, G.; Papotti, M. Large cell carcinoma of the lung: A tumor in search of an author. A clinically oriented critical reappraisal. Lung Cancer 2015, 87, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Weissferdt, A. Large cell carcinoma of lung: On the verge of extinction? Semin. Diagn. Pathol. 2014, 31, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Sholl, L.M. Large-cell carcinoma of the lung: A diagnostic category redefined by immunohistochemistry and genomics. Curr. Opin. Pulm. Med. 2014, 20, 324–331. [Google Scholar] [CrossRef]
- Osmani, L.; Askin, F.; Gabrielson, E.; Li, Q.K. Current WHO guidelines and the critical role of immunohistochemical markers in the subclassification of non-small cell lung carcinoma (NSCLC): Moving from targeted therapy to immunotherapy. Semin. Cancer Biol. 2018, 52 Pt 1, 103–109. [Google Scholar] [CrossRef]
- Chan, A.W.; Chau, S.L.; Tong, J.H.; Chow, C.; Kwan, J.S.H.; Chung, L.Y.; Lung, R.W.; Tong, C.Y.; Tin, E.K.; Law, P.P.; et al. The Landscape of Actionable Molecular Alterations in Immunomarker-Defined Large-Cell Carcinoma of the Lung. J. Thorac. Oncol. 2019, 14, 1213–1222. [Google Scholar] [CrossRef]
- Rekhtman, N.; Travis, W.D. Large No More: The Journey of Pulmonary Large Cell Carcinoma from Common to Rare Entity. J. Thorac. Oncol. 2019, 14, 1125–1127. [Google Scholar] [CrossRef]
- Wang, F.; Lu, J.B.; Wu, X.Y.; Feng, Y.F.; Shao, Q.; An, X.; Wang, H.-Y. Clinical genetic features and related survival implications in patients with surgically resected large-cell lung cancer. Cancer Manag. Res. 2019, 11, 5489–5499. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Cui, X.; Wang, D.; Xia, G.; Xing, M.; Cheng, L.; Sheng, L.; Du, X. Molecular Characterization and Prognostication of Large Cell Neuroendocrine Carcinoma and Large Cell Carcinoma. Front. Oncol. 2021, 11, 664397. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, G.; Fabbri, A.; Papotti, M.; Rossi, G.; Cavazza, A.; Righi, L.; Tamborini, E.; Perrone, F.; Settani, G.; Busico, A.; et al. Dissecting Pulmonary Large-Cell Carcinoma by Targeted Next Generation Sequencing of Several Cancer Genes Pushes Genotypic-Phenotypic Correlations to Emerge. J. Thorac. Oncol. 2015, 10, 1560–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Zhu, L.; Niu, X.; Shen, S.; Zhao, Y.; Zhang, J.; Ye, J.; Zhang, H.-H.; Liu, J.; Liu, C.; et al. Comparison of genomic landscapes of large cell neuroendocrine carcinoma, small cell lung carcinoma, and large cell carcinoma. Thorac. Cancer 2019, 10, 839–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charkiewicz, R.; Niklinski, J.; Claesen, J.; Sulewska, A.; Kozlowski, M.; Michalska-Falkowska, A.; Reszec, J.; Moniuszko, M.; Naumnik, W.; Niklinska, W. Gene Expression Signature Differentiates Histology But Not Progression Status of Early-Stage NSCLC. Transl. Oncol. 2017, 10, 450–458. [Google Scholar] [CrossRef]
- Sheng, M.; Xie, X.; Wang, J.; Gu, W. A Pathway-Based Strategy to Identify Biomarkers for Lung Cancer Diagnosis and Prognosis. Evol. Bioinform. 2019, 15, 1176934319838494. [Google Scholar] [CrossRef]
- Li, X.; Shi, G.; Chu, Q.; Jiang, W.; Liu, Y.; Zhang, S.; Zhang, Z.; Wei, Z.; He, F.; Guo, Z.; et al. A qualitative transcriptional signature for the histological reclassification of lung squamous cell carcinomas and adenocarcinomas. BMC Genom. 2019, 20, 881. [Google Scholar] [CrossRef] [Green Version]
- Niemira, M.; Collin, F.; Szalkowska, A.; Bielska, A.; Chwialkowska, K.; Reszec, J.; Niklinski, J.; Kwasniewski, M.; Kretowski, A. Molecular Signature of Subtypes of Non-Small-Cell Lung Cancer by Large-Scale Transcriptional Profiling: Identification of Key Modules and Genes by Weighted Gene Co-Expression Network Analysis (WGCNA). Cancers 2019, 12, 37. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.; Abbas, H.A.; Negrao, M.V.; Ramineni, M.; Hu, X.; Hubert, S.M.; Fujimoto, J.; Reuben, A.; Varghese, S.; Zhang, J.; et al. The histologic phenotype of lung cancers is associated with transcriptomic features rather than genomic characteristics. Nat. Commun. 2021, 12, 7081. [Google Scholar] [CrossRef]
- Pastuszak-Lewandoska, D.; Domańska-Senderowska, D.; Antczak, A.; Kordiak, J.; Górski, P.; Czarnecka, K.H.; Migdalska-Sęk, M.; Nawrot, E.; Kiszałkiewicz, J.M.; Brzeziańska-Lasota, E. The Expression Levels of IL-4/IL-13/STAT6 Signaling Pathway Genes and SOCS3 Could Help to Differentiate the Histopathological Subtypes of Non-Small Cell Lung Carcinoma. Mol. Diagn. Ther. 2018, 22, 621–629. [Google Scholar] [CrossRef]
- Karlsson, A.; Brunnström, H.; Micke, P.; Veerla, S.; Mattsson, J.; La Fleur, L.; Botling, J.; Jönsson, M.; Reuterswärd, C.; Planck, M.; et al. Gene Expression Profiling of Large Cell Lung Cancer Links Transcriptional Phenotypes to the New Histological WHO 2015 Classification. J. Thorac. Oncol. 2017, 12, 1257–1267. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer. 2016, 16, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Şenler, F.Ç.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Wehler, T.; Orlandi, F.; Nogami, N.; Barone, C.; Moro-Sibilot, D.; Shtivelband, M.; Larriba, J.L.G.; Rothenstein, J.; Früh, M.; et al. Safety and Patient-Reported Outcomes of Atezolizumab Plus Chemotherapy With or Without Bevacizumab Versus Bevacizumab Plus Chemotherapy in Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2020, 38, 2530. [Google Scholar] [CrossRef]
- Gadgeel, S.; Rodríguez-Abreu, D.; Speranza, G.; Esteban, E.; Felip, E.; Dómine, M.; Hui, R.; Hochmair, M.J.; Clingan, P.; Powell, S.F.; et al. Updated Analysis from KEYNOTE-189: Pembrolizumab or Placebo Plus Pemetrexed and Platinum for Previously Untreated Metastatic Nonsquamous Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2020, 38, 1505–1517. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Updated Analysis of KEYNOTE-024: Pembrolizumab Versus Platinum-Based Chemotherapy for Advanced Non-Small-Cell Lung Cancer With PD-L1 Tumor Proportion Score of 50% or Greater. J. Clin. Oncol. 2019, 37, 537–546. [Google Scholar] [CrossRef]
- Hanna, N.H.; Temin, S.; Masters, G. Therapy for Stage IV Non-Small-Cell Lung Cancer Without Driver Alterations: ASCO and OH (CCO) Joint Guideline Update Summary. JCO Oncol. Pract. 2020, 16, e844–e848. [Google Scholar] [CrossRef]
- Arbour, K.C.; Riely, G.J. Systemic Therapy for Locally Advanced and Metastatic Non-Small Cell Lung Cancer: A Review. JAMA 2019, 322, 764–774. [Google Scholar] [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, F.R.; McElhinny, A.; Stanforth, D.; Ranger-Moore, J.; Jansson, M.; Kulangara, K.; Richardson, W.; Towne, P.; Hanks, D.; Vennapusa, B.; et al. PD-L1 Immunohistochemistry Assays for Lung Cancer: Results from Phase 1 of the Blueprint PD-L1 IHC Assay Comparison Project. J. Thorac. Oncol. 2017, 12, 208–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsao, M.S.; Kerr, K.M.; Kockx, M.; Beasley, M.B.; Borczuk, A.C.; Botling, J.; Bubendorf, L.; Chirieac, L.; Chen, G.; Chou, J.-H.; et al. PD-L1 Immunohistochemistry Comparability Study in Real-Life Clinical Samples: Results of Blueprint Phase 2 Project. J. Thorac. Oncol. 2018, 13, 1302–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; Creelan, B.C.; Sanchez-Vega, F.; Ahuja, A.; Ni, A.; Novik, J.B.; Mangarin, L.M.B.; Abu-Akeel, M.; et al. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell 2018, 33, 843–852.e4. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Carbone, D.P.; Gandara, D.R.; Antonia, S.J.; Zielinski, C.; Paz-Ares, L. Non-Small-Cell Lung Cancer: Role of the Immune System and Potential for Immunotherapy. J. Thorac. Oncol. 2015, 10, 974–984. [Google Scholar] [CrossRef] [Green Version]
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2018, 359, 582–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Roach, C.; Zhang, N.; Corigliano, E.; Jansson, M.; Toland, G.; Ponto, G.; Dolled-Filhart, M.; Emancipator, K.; Stanforth, D.; Kulangara, K. Development of a Companion Diagnostic PD-L1 Immunohistochemistry Assay for Pembrolizumab Therapy in Non-Small-cell Lung Cancer. Appl. Immunohistochem. Mol. Morphol. 2016, 24, 392–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [Green Version]
- Feitelson, D.G. Comparing Partitions with Spie Charts; Technical Report 2003, 87; School of Computer Science and Engineering, The Hebrew University of Jerusalem: Jerusalem, Israel, 2003. [Google Scholar]
- Hofmann, M.H.; Gerlach, D.; Misale, S.; Petronczki, M.; Kraut, N. Expanding the Reach of Precision Oncology by Drugging All KRAS Mutants. Cancer Discov. 2022; Online ahead of print. [Google Scholar] [CrossRef]
- Lizotte, P.H.; Ivanova, E.V.; Awad, M.M.; Jones, R.E.; Keogh, L.; Liu, H.; Dries, R.; Almonte, C.; Herter-Sprie, G.S.; Santos, A.; et al. Multiparametric profiling of non-small-cell lung cancers reveals distinct immunophenotypes. JCI Insight 2016, 1, e89014. [Google Scholar] [CrossRef]
- Hwang, S.; Kwon, A.Y.; Jeong, J.Y.; Kim, S.; Kang, H.; Park, J.; Kim, J.-H.; Han, O.J.; Lim, S.M.; An, H.J. Immune gene signatures for predicting durable clinical benefit of anti-PD-1 immunotherapy in patients with non-small cell lung cancer. Sci. Rep. 2020, 10, 643. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Shang, X.; Li, J.; Zhang, S. The Prognosis and Immune Checkpoint Blockade Efficacy Prediction of Tumor-Infiltrating Immune Cells in Lung Cancer. Front. Cell Dev. Biol. 2021, 9, 707143. [Google Scholar] [CrossRef]
- Dong, Z.Y.; Zhong, W.Z.; Zhang, X.C.; Su, J.; Xie, Z.; Liu, S.Y.; Tu, H.-Y.; Chen, H.-J.; Sun, Y.-L.; Zhou, Q.; et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2017, 23, 3012–3024. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pabla, S.; Conroy, J.M.; Nesline, M.K.; Glenn, S.T.; Papanicolau-Sengos, A.; Burgher, B.; Hagen, J.; Giamo, V.; Andreas, J.; Lenzo, F.L.; et al. Proliferative potential and resistance to immune checkpoint blockade in lung cancer patients. J. Immunother. Cancer 2019, 7, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subrahmanyam, P.B.; Dong, Z.; Gusenleitner, D.; Giobbie-Hurder, A.; Severgnini, M.; Zhou, J.; Manos, M.; Eastman, L.M.; Maecker, H.T.; Hodi, F.S. Distinct predictive biomarker candidates for response to anti-CTLA-4 and anti-PD-1 immunotherapy in melanoma patients. J. Immunother. Cancer 2018, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Hodkinson, B.P.; Schaffer, M.; Brody, J.D.; Jurczak, W.; Carpio, C.; Ben-Yehuda, D.; Avivi, I.; Forslund, A.; Özcan, M.; Alvarez, J.; et al. Biomarkers of response to ibrutinib plus nivolumab in relapsed diffuse large B-cell lymphoma, follicular lymphoma, or Richter's transformation. Transl. Oncol. 2021, 14, 100977. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | N | (%) | Characteristic | N | (%) |
|---|---|---|---|---|---|
| Total LCC cases | 18 | 100.00 | Total LCC cases | 18 | 100.00 |
| Sex | Stage | ||||
| Male | 16 | 88.89 | I | 5 | 27.78 |
| Female | 2 | 11.11 | II III | 9 4 | 50.00 22.22 |
| Average age at diagnosis | 65.7 | NA | Neoadjuvant therapy | ||
| Smoking History | Yes | 1 | 5.56 | ||
| Current smoker | 7 | 38.89 | No ND | 15 2 | 83.33 11.11 |
| Former smoker | 9 | 50.00 | Adjuvant therapy | ||
| Never smoker | 0 | 0.00 | Yes | 9 | 50.00 |
| ND | 2 | 11.11 | No ND | 7 2 | 38.89 11.11 |
| Pack-year group | Exitus | ||||
| <10 | 0 | 0.00 | Yes | 13 | 72.22 |
| 10 to 20 | 1 | 5.56 | No | 4 | 22.22 |
| 20 to 40 | 1 | 5.56 | ND | 1 | 5.56 |
| >40 | 5 | 27.78 | Average OS (months) | 65.4 | NA |
| NA | 0 | 0.00 | Relapse | ||
| ND | 11 | 61.11 | Yes | 9 | 50.00 |
| COPD | No ND | 6 3 | 33.33 16.67 | ||
| Yes | 10 | 55.56 | Average PFS (months) | 54 | NA |
| No | 6 | 33.33 | |||
| ND | 2 | 11.11 |
| Marker | Pro-Immunogenic Tumors N = 11 | Pro-Tumorigenic Tumors N = 7 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Score | 0 | 1 | 2 | Positive (%) | Negative (%) | Avg. Score | 0 | 1 | 2 | Positive (%) | Negative (%) | Avg. Score |
| CD8 | 1 | 8 | 2 | 10 (90.91) | 1 (9.09) | 1.09 | 2 | 4 | 1 | 5 (71.43) | 2 (28.57) | 0.85 |
| CD4 | 8 | 2 | 1 | 3 (27.27) | 8 (72.73) | 0.36 | 6 | 1 | 0 | 1 (14.29) | 6 (85.71) | 0.14 |
| CD68 | 0 | 5 | 6 | 11 (100) | 0 (0) | 1.55 | 0 | 3 | 4 | 7 (100) | 0 (0) | 1.57 |
| CD20 | 2 | 6 | 3 | 9 (81.82) | 2 (18.18) | 1.09 | 2 | 5 | 0 | 5 (71.43) | 2 (28.57) | 0.71 |
| PD-L1 | NA | NA | NA | 2 (18.18) | 9 (81.82) | NA | NA | NA | NA | 1 (14.29) | 6 (85.71) | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramos-Paradas, J.; Gómez-Sánchez, D.; Rosado, A.; Ucero, A.C.; Ferrer, I.; García-Luján, R.; Zugazagoitia, J.; Carrizo, N.; Enguita, A.B.; Conde, E.; et al. Comprehensive Characterization of Human Lung Large Cell Carcinoma Identifies Transcriptomic Signatures with Potential Implications in Response to Immunotherapy. J. Clin. Med. 2022, 11, 1500. https://doi.org/10.3390/jcm11061500
Ramos-Paradas J, Gómez-Sánchez D, Rosado A, Ucero AC, Ferrer I, García-Luján R, Zugazagoitia J, Carrizo N, Enguita AB, Conde E, et al. Comprehensive Characterization of Human Lung Large Cell Carcinoma Identifies Transcriptomic Signatures with Potential Implications in Response to Immunotherapy. Journal of Clinical Medicine. 2022; 11(6):1500. https://doi.org/10.3390/jcm11061500
Chicago/Turabian StyleRamos-Paradas, Javier, David Gómez-Sánchez, Aranzazu Rosado, Alvaro C. Ucero, Irene Ferrer, Ricardo García-Luján, Jon Zugazagoitia, Nuria Carrizo, Ana B. Enguita, Esther Conde, and et al. 2022. "Comprehensive Characterization of Human Lung Large Cell Carcinoma Identifies Transcriptomic Signatures with Potential Implications in Response to Immunotherapy" Journal of Clinical Medicine 11, no. 6: 1500. https://doi.org/10.3390/jcm11061500
APA StyleRamos-Paradas, J., Gómez-Sánchez, D., Rosado, A., Ucero, A. C., Ferrer, I., García-Luján, R., Zugazagoitia, J., Carrizo, N., Enguita, A. B., Conde, E., Garrido-Martin, E. M., & Paz-Ares, L. (2022). Comprehensive Characterization of Human Lung Large Cell Carcinoma Identifies Transcriptomic Signatures with Potential Implications in Response to Immunotherapy. Journal of Clinical Medicine, 11(6), 1500. https://doi.org/10.3390/jcm11061500