Loss of Prefrontal Cortical Higher Cognition with Uncontrollable Stress: Molecular Mechanisms, Changes with Age, and Relevance to Treatment


Abstract
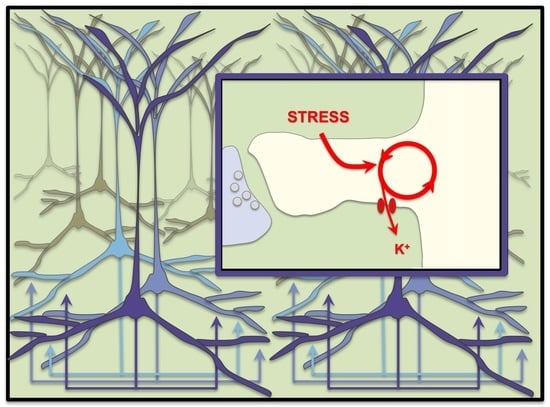
{kind=link}
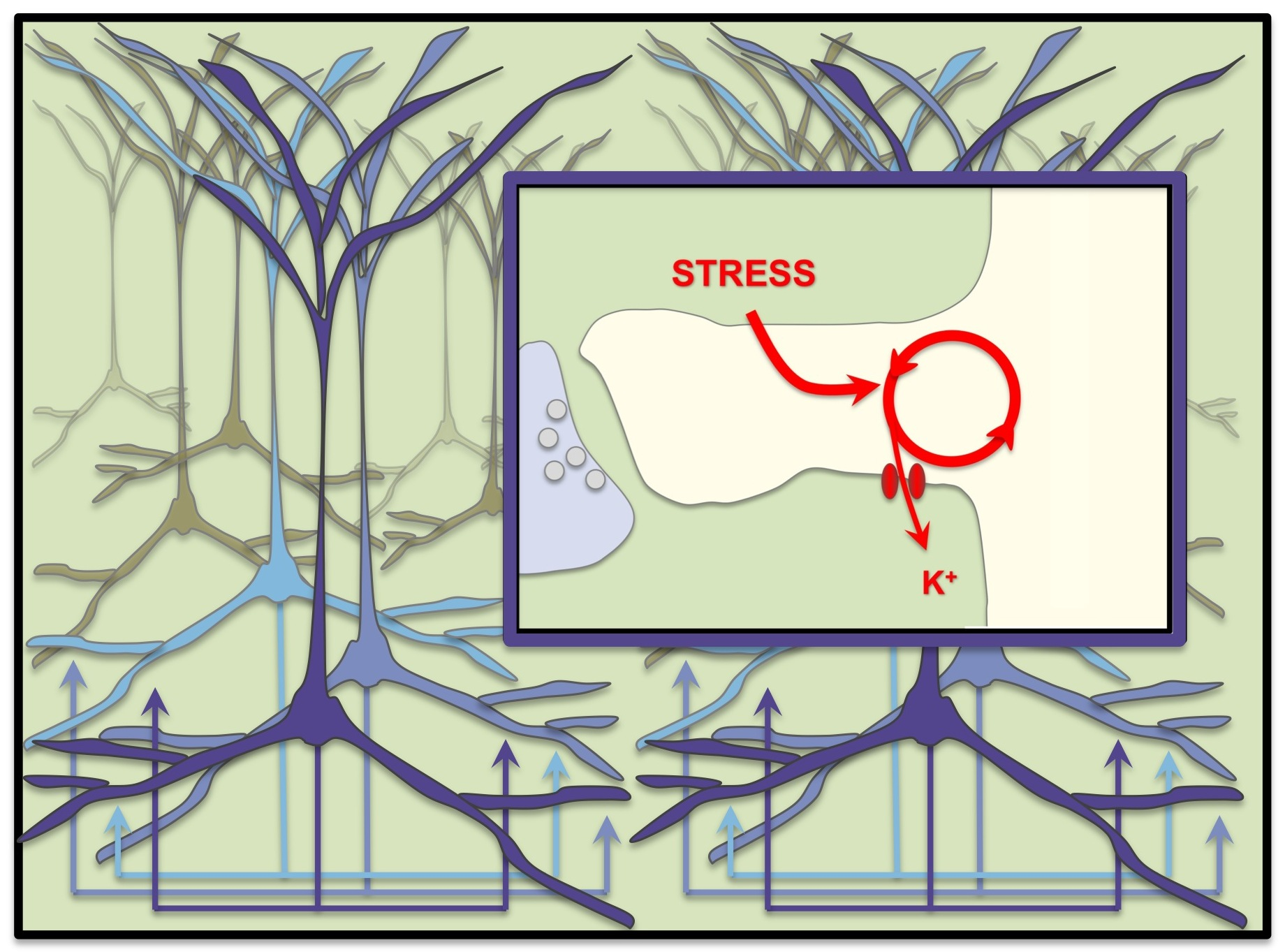
{kind=link}
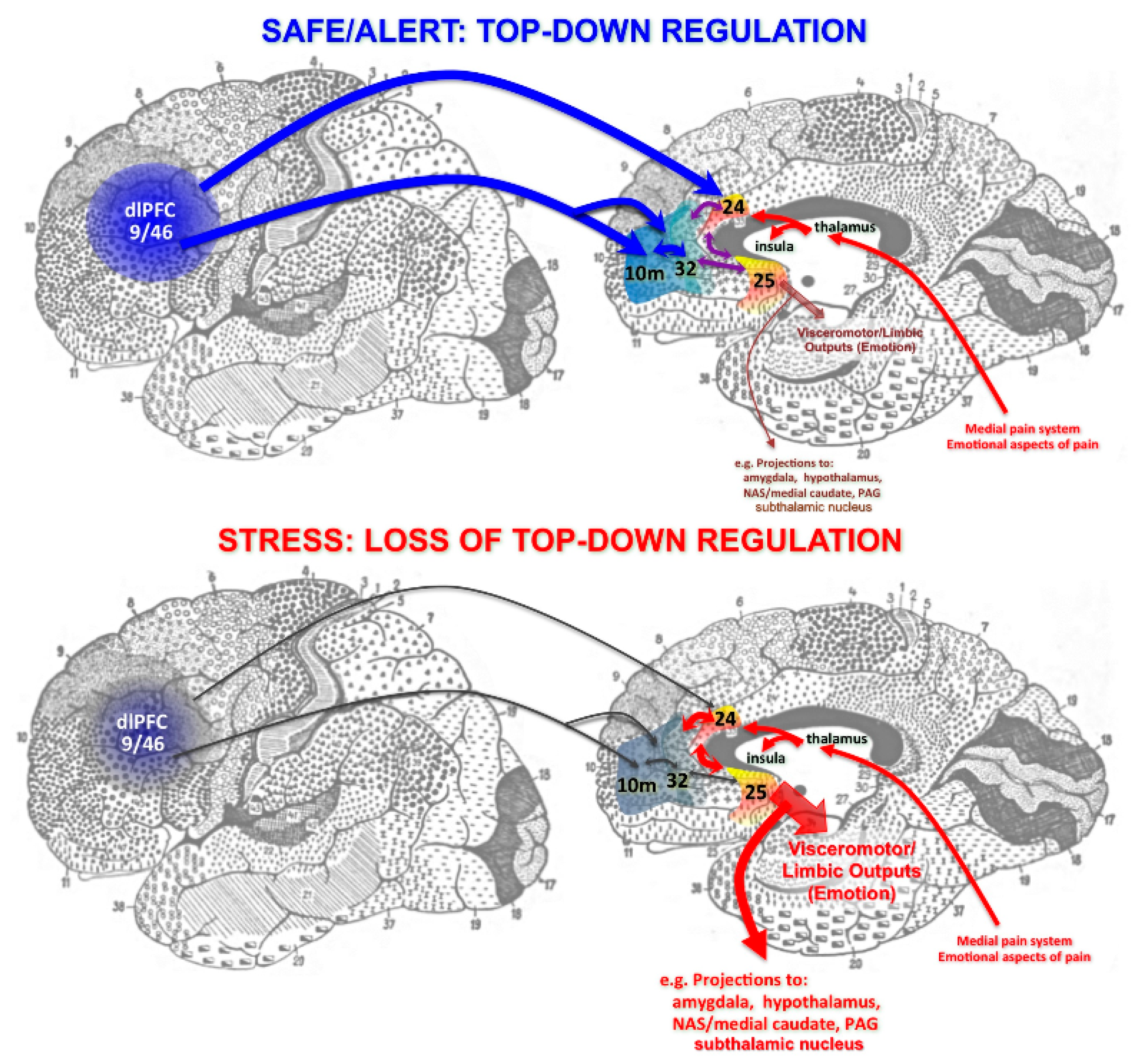
{kind=link}
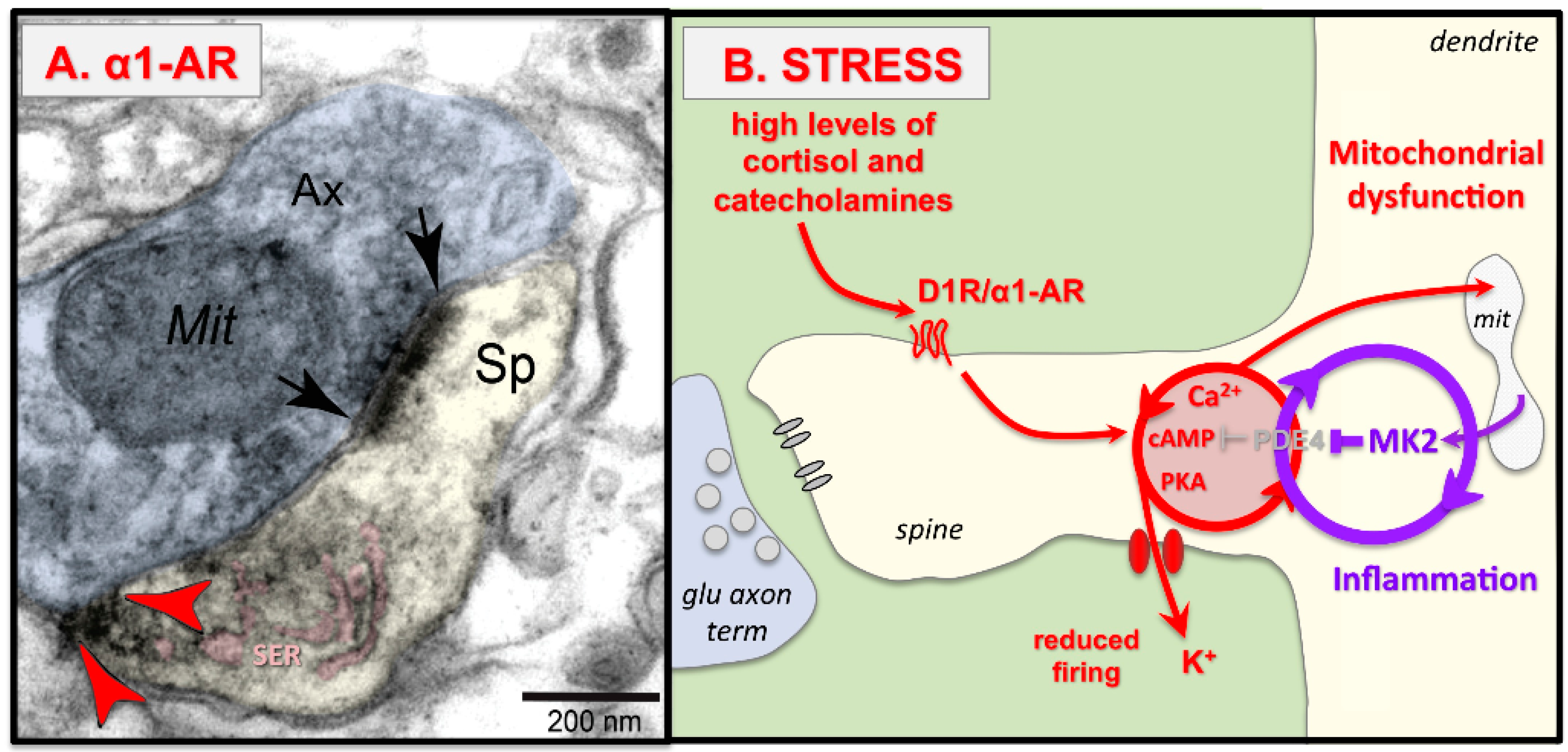{kind=link}
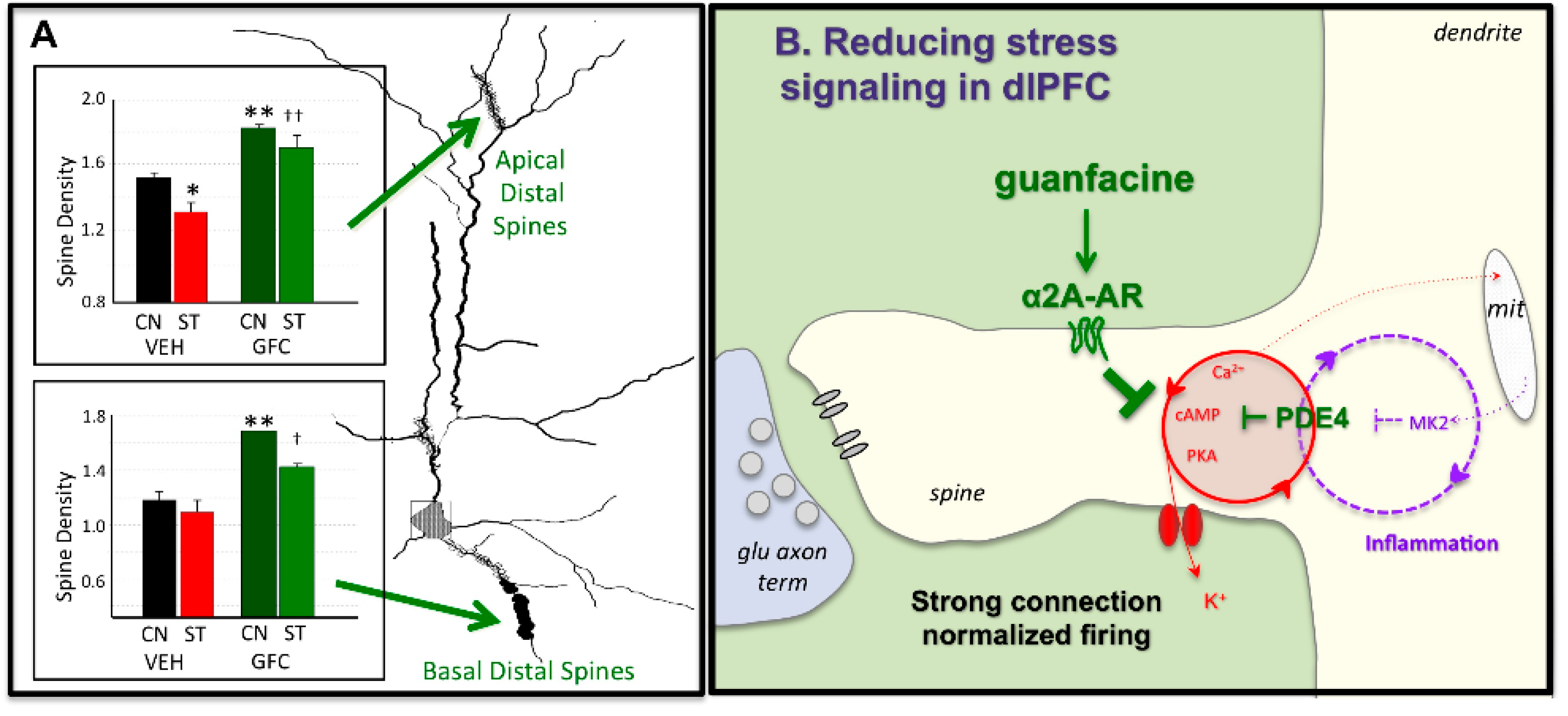{kind=link}
1. Introduction
2. The PFC Circuitry in Primates Serving Top-Down Control
3. The Microcircuitry for Generating Top-Down Goals for Regulating Thought, Action, and Emotion
4. The Unique Neurotransmission and Neuromodulation of dlPFC Synapses
5. Stress Rapidly Takes PFC Circuits “Offline”
6. Architectural Changes with Chronic Stress
7. Females Have a Greater Stress Response Than Males
8. Increased Susceptibility to Stress-Induced PFC Dysfunction During Adolescence
9. Increased Vulnerability During Aging Loss of Brakes on Stress Signaling Pathways
10. Successful Translation to Clinical Treatments
11. Outstanding Questions and Future Directions
Funding
Conflicts of Interest
References
- Goldman-Rakic, P.S. The prefrontal landscape: Implications of functional architecture for understanding human mentation and the central executive. Phil. Trans. R. Soc. London 1996, 351, 1445–1453. [Google Scholar]
- Goldman-Rakic, P.S. Circuitry of the primate prefrontal cortex and the regulation of behavior by representational memory. In Handbook of Physiology, The Nervous System, Higher Functions of the Brain; Plum, F., Ed.; American Physiological Society: Bethesda, MD, USA, 1987; pp. 373–417. [Google Scholar]
- Ongür, D.; Price, J.L. The organization of networks within the orbital and medial prefrontal cortex of rats, monkeys and humans. Cereb. Cortex 2000, 10, 206–219. [Google Scholar] [CrossRef]
- Ghashghaei, H.T.; Barbas, H. Pathways for emotion: Interactions of prefrontal and anterior temporal pathways in the amygdala of the rhesus monkey. Neuroscience 2002, 115, 1261–1279. [Google Scholar] [CrossRef]
- Barbas, H.; Medalla, M.; Alade, O.; Suski, J.; Zikopoulos, B.; Lera, P. Relationship of prefrontal connections to inhibitory systems in superior temporal areas in the rhesus monkey. Cereb. Cortex 2005, 15, 1356–1370. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Wang, M.; Paspalas, C.D. Neuromodulation of thought: Flexibilities and vulnerabilities in prefrontal cortical network synapses. Neuron 2012, 76, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Barbas, H.; Saha, S.; Rempel-Clower, N.; Ghashghaei, T. Serial pathways from primate prefrontal cortex to autonomic areas may influence emotional expression. BMC Neurosci. 2003, 4, 25. [Google Scholar] [CrossRef] [PubMed]
- Amodio, D.M.; Frith, C.D. Meeting of minds: The medial frontal cortex and social cognition. Nat. Rev. Neurosci. 2006, 7, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Fuster, J.M.; Alexander, G.E. Neuron activity related to short-term memory. Science 1971, 173, 652–654. [Google Scholar] [CrossRef]
- Funahashi, S.; Bruce, C.J.; Goldman-Rakic, P.S. Mnemonic coding of visual space in the monkey’s dorsolateral prefrontal cortex. J. Neurophysiol. 1989, 61, 331–349. [Google Scholar] [CrossRef]
- Funahashi, S.; Chafee, M.V.; Goldman-Rakic, P.S. Prefrontal neuronal activity in rhesus monkeys performing a delayed anti-saccade task. Nature 1993, 365, 753–756. [Google Scholar] [CrossRef]
- Suzuki, M.; Gottlieb, J. Distinct neural mechanisms of distractor suppression in the frontal and parietal lobe. Nat. Neurosci. 2013, 16, 98–104. [Google Scholar] [CrossRef]
- Wallis, J.D.; Anderson, K.C.; Miller, E.K. Single neurons in prefrontal cortex encode abstract rules. Nature 2001, 411, 953–956. [Google Scholar] [CrossRef]
- Kim, S.; Hwang, J.; Lee, D. Prefrontal coding of temporally discounted values during intertemporal choice. Neuron 2008, 59, 161–172. [Google Scholar] [CrossRef]
- Miller, E.K. The prefrontal cortex and cognitive control. Nat. Rev. Neurosci. 2000, 1, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Buschman, T.J.; Miller, E.K. Top-down versus bottom-up control of attention in the prefrontal and posterior parietal cortices. Science 2007, 315, 1860–1862. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, Y.; Galvin, V.C.; Yang, S.; Arnsten, A.F.; Wang, M. Nicotinic α4β2 cholinergic receptor influences on dorsolateral prefrontal cortical neuronal firing during a working memory task. J. Neurosci. 2017, 37, 5366–5377. [Google Scholar] [CrossRef]
- Lee, D.; Seo, H. Neural Basis of Strategic Decision Making. Trends Neurosci. 2016, 39, 40–48. [Google Scholar] [CrossRef]
- Tsujimoto, S.; Genovesio, A.; Wise, S.P. Evaluating self-generated decisions in frontal pole cortex of monkeys. Nat. Neurosci. 2010, 13, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, S.; Genovesio, A.; Wise, S.P. Frontal pole cortex: Encoding ends at the end of the endbrain. Trends Cogn. Sci. 2011, 15, 169–176. [Google Scholar] [CrossRef]
- Szczepanski, S.M.; Knight, R.T. Insights into human behavior from lesions to the prefrontal cortex. Neuron 2014, 83, 1002–1018. [Google Scholar] [CrossRef]
- Rolls, E.T. The orbitofrontal cortex and reward. Cereb. Cortex 2000, 10, 284–294. [Google Scholar] [CrossRef]
- Wallis, J.D.; Miller, E.K. Neuronal activity in primate dorsolateral and orbital prefrontal cortex during performance of a reward preference task. Eur. J. Neurosci. 2003, 18, 2069–2081. [Google Scholar] [CrossRef] [PubMed]
- Bouret, S.; Richmond, B.J. Ventromedial and orbital prefrontal neurons differentially encode internally and externally driven motivational values in monkeys. J. Neurosci. 2010, 30, 8591–8601. [Google Scholar] [CrossRef]
- Rudebeck, P.H.; Saunders, R.C.; Lundgren, D.A.; Murray, E.A. Specialized Representations of Value in the Orbital and Ventrolateral Prefrontal Cortex: Desirability versus Availability of Outcomes. Neuron 2017, 95, 1208–1220. [Google Scholar] [CrossRef]
- Ongür, D.; Ferry, A.; Price, J.L. Architectonic subdivision of the human orbital and medial prefrontal cortex. J. Comp. Neurol. 2003, 460, 425–449. [Google Scholar] [CrossRef] [PubMed]
- Vogt, B.A.; Sikes, R.W. The medial pain system, cingulate cortex, and parallel processing of nociceptive information. Prog. Brain Res. 2000, 22, 223–235. [Google Scholar]
- Bushnell, M.C.; Ceko, M.; Low, L.A. Cognitive and emotional control of pain and its disruption in chronic pain. Nat. Rev. Neurosci. 2013, 14, 502–511. [Google Scholar] [CrossRef]
- Jaggi, A.S.; Singh, N. Role of different brain areas in peripheral nerve injury-induced neuropathic pain. Brain Res. 2011, 1381, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, A.; Harsh, V.; Pereira, E.A.; Aziz, T.Z. Cingulotomy for medically refractory cancer pain. Neurosurg. Focus. 2013, 35, E1. [Google Scholar] [CrossRef] [PubMed]
- Van Veen, V.; Carter, C.S. The anterior cingulate as a conflict monitor: fMRI and ERP studies. Physiol. Behav. 2002, 77, 477–482. [Google Scholar] [CrossRef]
- Mayberg, H.S.; Lozano, A.M.; Voon, V.; McNeely, H.E.; Seminowicz, D.; Hamani, C.; Schwalb, J.M.; Kennedy, S.H. Deep brain stimulation for treatment-resistant depression. Neuron 2005, 45, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Neafsey, E.J. Prefrontal control of the autonomic nervous system: Anatomical and physiological observations. Prog. Brain Res. 1990, 85, 147–165. [Google Scholar]
- An, X.; Bandler, R.; Ongür, D.; Price, J.L. Prefrontal cortical projections to longitudinal columns in the midbrain periaqueductal gray in macaque monkeys. J. Comp. Neurol. 1998, 401, 455–479. [Google Scholar] [CrossRef]
- Sinha, R.; Lacadie, C.M.; Constable, R.T.; Seo, D. Dynamic neural activity during stress signals resilient coping. Proc. Natl. Acad. Sci. USA 2016, 113, 8837–8842. [Google Scholar] [CrossRef] [PubMed]
- Haynes, W.I.; Haber, S.N. The organization of prefrontal-subthalamic inputs in primates provides an anatomical substrate for both functional specificity and integration: Implications for Basal Ganglia models and deep brain stimulation. J. Neurosci. 2013, 33, 4804–4814. [Google Scholar] [CrossRef]
- Birchall, E.L.; Walker, H.C.; Cutter, G.; Guthrie, S.; Joop, A.; Memon, R.A.; Watts, R.L.; Standaert, D.G.; Amara, A.W. The effect of unilateral subthalamic nucleus deep brain stimulation on depression in Parkinson’s disease. Brain Stimul. 2017, 10, 651–656. [Google Scholar] [CrossRef]
- Barbas, H.; Pandya, D.N. Architecture and intrinsic connections of the prefrontal cortex in the rhesus monkey. J. Comp. Neurol. 1989, 286, 353–375. [Google Scholar] [CrossRef]
- Riva-Posse, P.; Choi, K.S.; Holtzheimer, P.E.; McIntyre, C.C.; Gross, R.E.; Chaturvedi, A.; Crowell, A.L.; Garlow, S.J.; Rajendra, J.K.; Mayberg, H.S. Defining Critical White Matter Pathways Mediating Successful Subcallosal Cingulate Deep Brain Stimulation for Treatment-Resistant Depression. Biol. Psychiatry 2014, 76, 963–969. [Google Scholar] [CrossRef]
- Fox, M.D.; Buckner, R.L.; White, M.P.; Greicius, M.D.; Pascual-Leone, A. Efficacy of transcranial magnetic stimulation targets for depression is related to intrinsic functional connectivity with the subgenual cingulate. Biol. Psychiatry 2012, 72, 595–603. [Google Scholar] [CrossRef]
- Goldman-Rakic, P.S. Cellular basis of working memory. Neuron 1995, 14, 477–485. [Google Scholar] [CrossRef]
- González-Burgos, G.; Barrionuevo, G.; Lewis, D.A. Horizontal synaptic connections in monkey prefrontal cortex: An in vitro electrophysiological study. Cereb. Cortex 2000, 10, 82–92. [Google Scholar] [CrossRef] [PubMed]
- González-Burgos, G.; Krimer, L.S.; Povysheva, N.V.; Barrionuevo, G.; Lewis, D.A. Functional properties of fast spiking interneurons and their synaptic connections with pyramidal cells in primate dorsolateral prefrontal cortex. J. Neurophysiol. 2005, 93, 942–953. [Google Scholar] [CrossRef]
- Elston, G.N.; Benavides-Piccione, R.; Elston, A.; Zietsch, B.; Defelipe, J.; Manger, P.; Casagrande, V.; Kaas, J.H. Specializations of the granular prefrontal cortex of primates: Implications for cognitive processing. Anat Rec. A Discov. Mol. Cell Evol. Biol. 2006, 288, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Glausier, J.R.; Lewis, D.A. Mapping pathologic circuitry in schizophrenia. Handb. Clin. Neurol. 2018, 150, 389–417. [Google Scholar]
- Bussière, T.; Giannakopoulos, P.; Bouras, C.; Perl, D.P.; Morrison, J.H.; Hof, P.R. Progressive degeneration of nonphosphorylated neurofilament protein-enriched pyramidal neurons predicts cognitive impairment in Alzheimer’s disease: Stereologic analysis of prefrontal cortex area 9. J. Comp. Neurol. 2003, 463, 281–302. [Google Scholar] [CrossRef]
- Yang, S.T.; Wang, M.; Paspalas, C.P.; Crimins, J.L.; Altman, M.T.; Mazer, J.A.; Arnsten, A.F. Core differences in synaptic signaling between primary visual and dorsolateral prefrontal cortex. Cereb. Cortex 2018, 28, 1458–1471. [Google Scholar] [CrossRef]
- Liu, X.B.; Murray, K.D.; Jones, E.G. Switching of NMDA receptor 2A and 2B subunits at thalamic and cortical synapses during early postnatal development. J. Neurosci. 2004, 24, 8885–8895. [Google Scholar] [CrossRef]
- Wang, M.; Yang, Y.; Wang, C.J.; Gamo, N.J.; Jin, L.E.; Mazer, J.A.; Morrison, J.H.; Wang, X.-J.; Arnsten, A.F. NMDA receptors subserve working memory persistent neuronal firing In dorsolateral prefrontal cortex. Neuron 2013, 77, 736–749. [Google Scholar] [CrossRef]
- Rotaru, D.C.; Yoshino, H.; Lewis, D.A.; Ermentrout, G.B.; Gonzalez-Burgos, G. Glutamate receptor subtypes mediating synaptic activation of prefrontal cortex neurons: Relevance for schizophrenia. J. Neurosci. 2011, 31, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Datta, D.; Arion, D.; Lewis, D.A. Developmental Expression Patterns of GABAA Receptor Subunits in Layer 3 and 5 Pyramidal Cells of Monkey Prefrontal Cortex. Cereb. Cortex 2014, 25, 2295–2305. [Google Scholar] [CrossRef]
- Yang, Y.; Paspalas, C.D.; Jin, L.E.; Picciotto, M.R.; Arnsten, A.F.T.; Wang, M. Nicotinic α7 receptors enhance NMDA cognitive circuits in dorsolateral prefrontal cortex. Proc. Nat. Acad. Sci. USA 2013, 110, 12078–12083. [Google Scholar] [CrossRef] [PubMed]
- Brennan, A.R.; Dolinsky, B.; Vu, M.A.; Stanley, M.; Yeckel, M.F.; Arnsten, A.F. Blockade of IP3-mediated SK channel signaling in the rat medial prefrontal cortex improves spatial working memory. Learn. Mem. 2008, 15, 93–96. [Google Scholar] [CrossRef]
- Wang, M.; Ramos, B.; Paspalas, C.; Shu, Y.; Simen, A.; Duque, A.; Vijayraghavan, S.; Brennan, A.; Dudley, A.G.; Nou, E.; et al. Alpha2A-adrenoceptor stimulation strengthens working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell 2007, 129, 397–410. [Google Scholar] [CrossRef]
- Arnsten, A.F. Stress weakens prefrontal networks: Molecular insults to higher cognition. Nat. Neurosci. 2015, 18, 1376–1385. [Google Scholar] [CrossRef]
- Paspalas, C.D.; Min Wang, M.; Arnsten, A.F.T. Constellation of HCN Channels and cAMP regulating proteins in dendritic spines of the primate prefrontal cortex–Potential substrate for working memory deficits in schizophrenia. Cereb. Cortex 2013, 23, 1643–1654. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Wang, M.; Paspalas, C.D. Dopamine’s actions in primate prefrontal cortex: Challenges for treating cognitive disorders. Pharmacological Rev. 2015, 67, 681–696. [Google Scholar] [CrossRef]
- Carlyle, B.C.; Nairn, A.C.; Wang, M.; Yang, Y.; Jin, L.E.; Simen, A.A.; Ramos, B.P.; Bordner, K.A.; Craft, G.E.; Davies, P.; et al. cAMP-PKA phosphorylation of tau confers risk for degeneration in aging association cortex. Proc. Natl. Acad. Sci. USA 2014, 111, 5036–5041. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.E.; Wang, M.; Galvin, V.C.; Lightbourne, T.C.; Conn, P.J.; Arnsten, A.F.T.; Paspalas, C.D. mGluR2 vs. mGluR3 in Primate Prefrontal Cortex: Postsynaptic mGluR3 Strengthen Cognitive Networks. Cereb. Cortex 2018, 28, 974–987. [Google Scholar] [CrossRef]
- Minor, T.R.; Jackson, R.L.; Maier, S.F. Effects of task-irrelevant cues and reinforcement delay on choice-escape learning following inescapable shock: Evidence for a deficit in selective attention. J. Exp. Psychol. Anim. Behav. Process. 1984, 10, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Arnsten, A.F.; Berridge, C.W.; Segal, D.S. Stress produces opioid-like effects on investigatory behavior. Pharmacol Biochem Behav. 1985, 22, 803–809. [Google Scholar] [CrossRef]
- Bland, S.T.; Hargrave, D.; Pepin, J.L.; Amat, J.; Watkins, L.R.; Maier, S.F. Stressor controllability modulates stress-induced dopamine and serotonin efflux and morphine-induced serotonin efflux in the medial prefrontal cortex. Neuropsychopharmacology 2003, 28, 1589–1596. [Google Scholar] [CrossRef] [PubMed]
- Deutch, A.Y.; Roth, R.H. The determinants of stress-induced activation of the prefrontal cortical dopamine system. Prog. Brain Res. 1990, 85, 367–403. [Google Scholar]
- Finlay, J.M.; Zigmond, M.J.; Abercrombie, E.D. Increased dopamine and norepinephrine release in medial prefrontal cortex induced by acute and chronic stress: Effects of diazepam. Neuroscience 1995, 64, 619–628. [Google Scholar] [CrossRef]
- Goldstein, L.E.; Rasmusson, A.M.; Bunney, S.B.; Roth, R.H. Role of the amygdala in the coordination of behavioral, neuroendocrine and prefrontal cortical monoamine responses to psychological stress in the rat. J. Neurosci. 1996, 16, 4787–4798. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.L.; Arnsten, A.F.T.; Goldman-Rakic, P.S.; Roth, R.H. Increased dopamine turnover in the prefrontal cortex impairs spatial working memory performance in rats and monkeys. Proc. Nat. Acad. Sci. U.S.A. 1996, 93, 1325–1329. [Google Scholar] [CrossRef]
- Birnbaum, S.G.; Gobeske, K.T.; Auerbach, J.; Taylor, J.R.; Arnsten, A.F.T. A role for norepinephrine in stress-induced cognitive deficits: Alpha-1-adrenoceptor mediation in prefrontal cortex. Biol. Psychiatry 1999, 46, 1266–1274. [Google Scholar] [CrossRef]
- Birnbaum, S.B.; Yuan, P.; Wang, M.; Vijayraghavan, S.; Bloom, A.; Davis, D.; Gobeske, K.; Sweatt, D.; Manji, H.K.; Arnsten, A.F.T. Protein kinase C overactivity impairs prefrontal cortical regulation of working memory. Science 2004, 306, 882–884. [Google Scholar] [CrossRef]
- Gamo, N.J.; Lur, G.; Higley, M.J.; Wang, M.; Paspalas, C.D.; Vijayraghavan, S.; Yang, Y.; Ramos, B.P.; Peng, K.; Kata, A.; et al. Stress impairs prefrontal cortical function via D1 dopamine receptor interactions with HCN channels. Biol. Psychiatry 2015, 78, 860–870. [Google Scholar] [CrossRef]
- Datta, D.; Yang, S.T.; Galvin, V.C.; Solder, J.; Luo, F.; Morozov, Y.M.; Arellano, J.; Duque, A.; Rakic, P.; Arnsten, A.F.T.; et al. Noradrenergic α1-Adrenoceptor Actions in the Primate Dorsolateral Prefrontal Cortex. J. Neurosci. 2019, 39, 2722–2734. [Google Scholar] [CrossRef] [PubMed]
- Smiley, J.F.; Levey, A.I.; Ciliax, B.J.; Goldman-Rakic, P.S. D1 dopamine receptor immunoreactivity in human and monkey cerebral cortex: Predominant and extrasynaptic localization in dendritic spines. Proc. Natl. Acad. Sci. USA 1994, 91, 5720–5724. [Google Scholar] [CrossRef]
- Vijayraghavan, S.; Wang, M.; Birnbaum, S.G.; Bruce, C.J.; Williams, G.V.; Arnsten, A.F.T. Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat. Neurosci. 2007, 10, 376–384. [Google Scholar] [CrossRef]
- MacKenzie, K.F.; Wallace, D.A.; Hill, E.V.; Anthony, D.F.; Henderson, D.J.; Houslay, D.M.; Arthur, J.S.; Baillie, G.S.; Houslay, M.D. Phosphorylation of cAMP-specific PDE4A5 (phosphodiesterase-4A5) by MK2 (MAPKAPK2) attenuates its activation through protein kinase A phosphorylation. Biochem. J. 2011, 435, 755–769. [Google Scholar] [CrossRef]
- Houslay, K.F.; Christian, F.; MacLeod, R.; Adams, D.R.; Houslay, M.D.; Baillie, G.S. Identification of a multifunctional docking site on the catalytic unit of phosphodiesterase-4 (PDE4) that is utilised by multiple interaction partners. Biochem. J. 2017, 474, 597–609. [Google Scholar] [CrossRef]
- Cahill, L.; McGaugh, J.L. Modulation of memory storage. Curr. Opin. Neurobiol. 1996, 6, 237–242. [Google Scholar] [CrossRef]
- Packard, M.G.; Teather, L.A. Amygdala modulation of multiple memory systems: Hippocampus and caudate-putamen. Neurobiol. Learning Mem. 1998, 69, 163–203. [Google Scholar] [CrossRef]
- Ferry, B.; Roozendaal, B.; McGaugh, J.L. Basolateral amygdala noradrenergic influences on memory storage are mediated by an interaction between beta- and alpha-1-adrenoceptors. J. Neurosci. 1999, 19, 5119–5123. [Google Scholar] [CrossRef]
- Waterhouse, B.D.; Moises, H.C.; Woodward, D.J. Alpha-receptor-mediated facilitation of somatosensory cortical neuronal responses to excitatory synaptic inputs and iontophoretically applied acetylcholine. Neuropharmacology 1981, 20, 907–920. [Google Scholar] [CrossRef]
- Arnsten, A.F.T. Stress signaling pathways that impair prefrontal cortex structure and function. Nature Reviews Neuroscience 2009, 10, 410–422. [Google Scholar] [CrossRef]
- Grundemann, D.; Schechinger, B.; Rappold, G.A.; Schomig, E. Molecular identification of the cortisone-sensitive extraneuronal catecholamine transporter. Nat. Neurosci. 1998, 1, 349–351. [Google Scholar] [CrossRef]
- Barsegyan, A.; Mackenzie, S.M.; Kurose, B.D.; McGaugh, J.L.; Roozendaal, B. Glucocorticoids in the prefrontal cortex enhance memory consolidation and impair working memory by a common neural mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 16655–16660. [Google Scholar] [CrossRef]
- Roozendaal, B.; Quirarte, G.L.; McGaugh, J.L. Glucocorticoids interact with the basolateral amygdala beta-adrenoceptor-cAMP/cAMP/PKA system in influencing memory consolidation. Eur. J. Neurosci. 2002, 15, 553–560. [Google Scholar] [CrossRef]
- Qin, S.; Hermans, E.J.; Van Marle, H.J.F.; Lou, J.; Fernandez, G. Acute psychological stress reduces working memory-related activity in the dorsolateral prefrontal cortex. Biol. Psychiatry 2009, 66, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Cousijn, H.; Rijpkema, M.; Luo, J.; Franke, B.; Hermans, E.J.; Fernández, G. The effect of moderate acute psychological stress on working memory-related neural activity is modulated by a genetic variation in catecholaminergic function in humans. Front. Integr. Neurosci. 2012, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Magariños, A.M.; McEwen, B.S. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: Comparison of stressors. Neuroscience 1995, 69, 83–88. [Google Scholar] [CrossRef]
- Izquierdo, A.; Wellman, C.L.; Holmes, A. Brief uncontrollable stress causes dendritic retraction in infralimbic cortex and resistance to fear extinction in mice. J. Neurosci. 2006, 26, 5733–5738. [Google Scholar] [CrossRef]
- Liston, C.; Miller, M.M.; Goldwater, D.S.; Radley, J.J.; Rocher, A.B.; Hof, P.R.; Morrison, J.H.; McEwen, B.S. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J. Neurosci. 2006, 26, 7870–7874. [Google Scholar] [CrossRef]
- Radley, J.J.; Rocher, A.B.; Miller, M.; Janssen, W.G.; Liston, C.; Hof, P.R.; McEwen, B.S.; Morrison, J.H. Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb. Cortex 2006, 16, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Shansky, R.M.; Hamo, C.; Hof, P.R.; McEwen, B.S.; Morrison, J.H. Stress-induced dendritic remodeling in the prefrontal cortex is circuit specific. Cereb. Cortex 2009, 106, 17957–17962. [Google Scholar] [CrossRef]
- Licznerski, P.; Duman, R.S. Remodeling of axo-spinous synapses in the pathophysiology and treatment of depression. Neuroscience 2013, 251, 33–50. [Google Scholar] [CrossRef]
- Vyas, A.; Mitra, R.; Shankaranarayana Rao, B.S.; Chattarji, S. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J. Neurosci. 2002, 22, 6810–6818. [Google Scholar] [CrossRef]
- Hains, A.B.; Vu, M.A.; Maciejewski, P.K.; Van Dyck, C.H.; Gottron, M.; Arnsten, A.F. Inhibition of protein kinase C signaling protects prefrontal cortex dendritic spines and cognition from the effects of chronic stress. Proc. Natl. Acad. Sci. USA 2009, 106, 17957–17962. [Google Scholar] [CrossRef]
- Hains, A.B.; Yabe, Y.; Arnsten, A.F.T. Chronic stimulation of alpha-2A-adrenoceptors with guanfacine protects rodent prefrontal cortex dendritic spines and cognition from the effects of chronic stress. Neurobiol. Stress 2015, 2, 1–9. [Google Scholar] [CrossRef]
- Bloss, E.B.; Janssen, W.G.; Ohm, D.T.; Yuk, F.J.; Wadsworth, S.; Saardi, K.M.; McEwen, B.S.; Morrison, J.H. Evidence for reduced experience-dependent dendritic spine plasticity in the aging prefrontal cortex. J. Neurosci. 2011, 31, 7831–7839. [Google Scholar] [CrossRef]
- Moda-Sava, R.N.; Murdock, M.H.; Parekh, P.K.; Fetcho, R.N.; Huang, B.S.; Huynh, T.N.; Witztum, J.; Shaver, D.C.; Rosenthal, D.L.; Always, E.J.; et al. Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science 2019, 364. [Google Scholar] [CrossRef]
- Ansell, E.B.; Rando, K.; Tuit, K.; Guarnaccia, J.; Sinha, R. Cumulative adversity and smaller gray matter volume in medial prefrontal, anterior cingulate, and insula regions. Biol. Psychiatry 2012, 72, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Liston, C.; McEwen, B.S.; Casey, B.J. Psychosocial stress reversibly disrupts prefrontal processing and attentional control. Proc. Nat. Acad. Sci. USA 2009, 106, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Hart, H.; Lim, L.; Mehta, M.A.; Chatzieffraimidou, A.; Curtis, C.; Xu, X.; Breen, G.; Simmons, A.; Mirza, K.; Rubia, K. Reduced functional connectivity of fronto-parietal sustained attention networks in severe childhood abuse. PLoS ONE 2017, 12, e0188744. [Google Scholar] [CrossRef]
- Calabrese, B.; Halpain, S. Essential role for the PKC target MARCKS in maintaining dendritic spine morphology. Neuron 2005, 48, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Manji, H.K.; Lenox, R.H. Signaling: Cellular insights into the pathophysiology of bipolar disorder. Biol. Psychiatry 2000, 48, 518–530. [Google Scholar] [CrossRef]
- Moore, G.J.; Bebchuk, J.M.; Wilds, I.B.; Chen, G.; Manji, H.K. Lithium-induced increase in human brain gray matter. The Lancet. 2000, 356, 1241–1242. [Google Scholar] [CrossRef]
- Liu, R.J.; Fuchikami, M.; Dwyer, J.M.; Lepack, A.E.; Duman, R.S.; Aghajanian, G.K. GSK-3 inhibition potentiates the synaptogenic and antidepressant-like effects of subthreshold doses of ketamine. Neuropsychopharmacology. 2013, 38, 2268–2277. [Google Scholar] [CrossRef]
- Bangasser, D.A.; Valentino, R.J. Sex differences in molecular and cellular substrates of stress. Cell Mol. Neurobiol. 2012, 32, 709–723. [Google Scholar] [CrossRef]
- Küppers, E.; Ivanova, T.; Karolczak, M.; Beyer, C. Estrogen: A multifunctional messenger to nigrostriatal dopaminergic neurons. J. Neurocytol. 2000, 29, 375–385. [Google Scholar] [CrossRef]
- Tunbridge, E.M. The catechol-O-methyltransferase gene: Its regulation and polymorphisms. Int. Rev. Neurobiol. 2010, 95, 7–27. [Google Scholar]
- Shansky, R.M.; Glavis-Bloom, C.; Lerman, D.; McRae, P.; Benson, C.; Miller, K.; Cosand, L.; Horvath, T.L.; Arnsten, A.F.T. Estrogen mediates sex differences in stress-induced prefrontal cortex dysfunction. Mol. Psychiatry 2004, 9, 531–538. [Google Scholar] [CrossRef]
- Shansky, R.M.; Rubinow, K.; Brennan, A.; Arnsten, A.F. The effects of sex and hormonal status on restraint-stress-induced working memory impairment. Behav. Brain Funct. 2006, 2, 8. [Google Scholar] [CrossRef]
- Shansky, R.M.; Hamo, C.; Hof, P.R.; Lou, W.; McEwen, B.S.; Morrison, J.H. Estrogen promotes stress sensitivity in a prefrontal cortex-amygdala pathway. Cereb. Cortex 2010, 20, 2560–2567. [Google Scholar] [CrossRef]
- Weiss, E.L.; Longhurst, J.G.; Mazure, C.M. Childhood sexual abuse as a risk factor for depression in women: Psychosocial and neurobiological correlates. Am. J. Psychiatry 1999, 156, 816–828. [Google Scholar] [CrossRef]
- Bebbington, P.; Dunn, G.; Jenkins, R.; Lewis, G.; Brugha, T.; Farrell, M.; Meltzer, H. The influence of age and sex on the prevalence of depressive conditions: Report from the National Survey of Psychiatric Morbidity. Int. Rev. Psychiatry. 2003, 15, 74–83. [Google Scholar] [CrossRef]
- Breslau, N. The epidemiology of trauma, PTSD, and other posttrauma disorders. Trauma Violence Abuse 2009, 10, 198–210. [Google Scholar] [CrossRef]
- Johnson, D.P.; Whisman, M.A. Gender differences in rumination: A meta-analysis. Pers. Individ. Dif. 2013, 55, 367–374. [Google Scholar] [CrossRef]
- Arnsten, A.F.; Shansky, R.M. Adolescence: Vulnerable period for stress-induced prefrontal cortical function? Introduction to part IV. Ann. N.Y. Acad. Sci. 2004, 1021, 143–147. [Google Scholar] [CrossRef]
- Duckworth, A.L.; Kim, B.; Tsukayama, E. Life stress impairs self-control in early adolescence. Front. Psychol. 2012, 3, 608. [Google Scholar] [CrossRef]
- Hanson, J.L.; Chung, M.K.; Avants, B.B.; Rudolph, K.D.; Shirtcliff, E.A.; Gee, J.C.; Davidson, R.J.; Pollak, S.D. Structural variations in prefrontal cortex mediate the relationship between early childhood stress and spatial working memory. J. Neurosci. 2012, 32, 7917–7925. [Google Scholar] [CrossRef]
- Hammond, C.J.; Mayes, L.C.; Potenza, M.N. Neurobiology of Adolescent Substance Use and Addictive Behaviors: Prevention and Treatment Implications. Adolesc. Med. State Art Rev. 2014, 25, 15–32. [Google Scholar]
- Rosenberg, D.R.; Lewis, D.A. Changes in the dopaminergic innervation of monkey prefrontal cortex during late postnatal development: A tyosine hydroxylase immunohistochemical study. Biol. Psychiat. 1994, 36, 272–277. [Google Scholar] [CrossRef]
- Rosenberg, D.R.; Lewis, D.A. Postnatal maturation of the dopaminergic innervation of monkey prefrontal cortices: A tyrosine hydroxylase immunohistochemical analysis. J. Comp. Neurol. 1995, 358, 383–400. [Google Scholar] [CrossRef]
- Brenhouse, H.C.; Sonntag, K.C.; Andersen, S.L. Transient D1 dopamine receptor expression on prefrontal cortex projection neurons: Relationship to enhanced motivational salience of drug cues in adolescence. J. Neurosci. 2008, 28, 2375–2382. [Google Scholar] [CrossRef]
- Morrison, J.H.; Baxter, M.G. The ageing cortical synapse: Hallmarks and implications for cognitive decline. Nat. Rev. Neurosci. 2012, 13, 240–250. [Google Scholar] [CrossRef]
- Otte, C.; Hart, S.; Neylan, T.C.; Marmar, C.R.; Yaffe, K.; Mohr, D.C. A meta-analysis of cortisol response to challenge in human aging: Importance of gender. Psychoneuroendocrinology 2005, 30, 80–91. [Google Scholar] [CrossRef]
- Goldman-Rakic, P.S.; Brown, R.M. Regional changes of monoamines in cerebral cortex and subcortical structures of aging rhesus monkeys. Neuroscience 1981, 6, 177–187. [Google Scholar] [CrossRef]
- Hara, Y.; Yuk, F.; Puri, R.; Janssen, W.G.; Rapp, P.R.; Morrison, J.H. Presynaptic mitochondrial morphology in monkey prefrontal cortex correlates with working memory and is improved with estrogen treatment. Proc. Natl. Acad. Sci. USA 2014, 111, 486–491. [Google Scholar] [CrossRef]
- Morozov, Y.M.; Datta, D.; Paspalas, C.D.; Arnsten, A.F. Ultrastructural evidence for impaired mitochondrial fission in the aged rhesus monkey dorsolateral prefrontal cortex. Neurobiol. Aging 2017, 51, 9–18. [Google Scholar] [CrossRef]
- Paspalas, C.D.; Carlyle, B.; Leslie, S.; Preuss, T.M.; Crimins, J.L.; Huttner, A.J.; Van Dyck, C.H.; Rosene, D.L.; Nairn, A.C.; Arnsten, A.F.T. The aged rhesus macaque manifests Braak-stage III/IV Alzheimer’s-like pathology. Alzheimer’s Dementia 2018, 14, 680–691. [Google Scholar] [CrossRef]
- Johansson, L.; Guo, X.; Hällström, T.; Norton, M.C.; Waern, M.; Ostling, S.; Bengtsson, C.; Skoog, I. Common psychosocial stressors in middle-aged women related to longstanding distress and increased risk of Alzheimer’s disease: A 38-year longitudinal population study. BMJ Open 2013, 3, e003142. [Google Scholar] [CrossRef]
- Johansson, L.; Guo, X.; Duberstein, P.R.; Hällström, T.; Waern, M.; Ostling, S.; Skoog, I. Midlife personality and risk of Alzheimer disease and distress: A 38-year follow-up. Neurology 2014, 83, 1538–1544. [Google Scholar] [CrossRef]
- Flatt, J.D.; Gilsanz, P.; Quesenberry, C.P.J.; Albers, K.B.; Whitmer, R.A. Post-traumatic stress disorder and risk of dementia among members of a health care delivery system. Alzheimers Dement. 2018, 14, 28–34. [Google Scholar] [CrossRef]
- Ennis, G.E.; An, Y.; Resnick, S.M.; Ferrucci, L.; O’Brien, R.J.; Moffat, S.D. Long-term cortisol measures predict Alzheimer disease risk. Neurology 2017, 88, 371–378. [Google Scholar] [CrossRef]
- Altmann, A.; Tian, L.; Henderson, V.W.; Greicius, M.D.; Alzheimer’s Disease Neuroimaging Initiative Investigators. Sex modifies the APOE-related risk of developing Alzheimer disease. Ann. Neurol. 2014, 75, 563–573. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2014 Alzheimer’s disease facts and figures. Alzheimers Dement. 2014, 10, e47–e92. [Google Scholar] [CrossRef]
- Birnbaum, S.G.; Podell, D.M.; Arnsten, A.F.T. Noradrenergic alpha-2 receptor agonists reverse working memory deficits induced by the anxiogenic drug, FG7142, in rats. Pharmacol. Biochem. Behav. 2000, 67, 397–403. [Google Scholar] [CrossRef]
- Gyoneva, S.; Traynelis, S.F. Norepinephrine modulates the motility of resting and activated microglia via different adrenergic receptors. J. Biol. Chem. 2013, 288, 15291–15302. [Google Scholar] [CrossRef]
- Connor, D.F.; Grasso, D.J.; Slivinsky, M.D.; Pearson, G.S.; Banga, A. An open-label study of guanfacine extended release for traumatic stress related symptoms in children and adolescents. J. Child. Adolesc. Psychopharmacol. 2013, 23, 244–251. [Google Scholar] [CrossRef]
- Connor, D.F.; Findling, R.L.; Kollins, S.H.; Sallee, F.; López, F.A.; Lyne, A.; Tremblay, G. Effects of guanfacine extended release on oppositional symptoms in children aged 6–12 years with attention-deficit hyperactivity disorder and oppositional symptoms: A randomized, double-blind, placebo-controlled trial. CNS Drugs 2010, 24, 755–768. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Raskind, M.; Taylor, F.B.; Connor, D.F. The effects of stress exposure on prefrontal cortex: Translating basic research into successful treatments for Post-Traumatic Stress Disorder. Neurobiol. Stress 2015, 1, 89–99. [Google Scholar] [CrossRef]
- Raskind, M.A.; Peterson, K.; Williams, T.; Hoff, D.J.; Hart, K.; Holmes, H.; Homas, D.; Hill, J.; Daniels, C.; Calohan, J.; et al. A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am. J. Psychiatry. 2013, 170, 1003–1010. [Google Scholar] [CrossRef]
- Taylor, F.B.; Lowe, K.; Thompson, C.; McFall, M.M.; Peskind, E.R.; Kanter, E.D.; Allison, N.; Williams, J.A.; Martin, P.; Raskind, M.A. Daytime prazosin reduces psychological distress to trauma specific cues in civilian trauma posttraumatic stress disorder. Biol Psychiatry 2006, 59, 577–581. [Google Scholar] [CrossRef]
- Koola, M.M.; Varghese, S.P.; Fawcett, J.A. High-dose prazosin for the treatment of post-traumatic stress disorder. Ther. Adv. Psychopharmacol. 2014, 4, 43–47. [Google Scholar] [CrossRef]
- Simpson, T.L.; Saxon, A.J.; Meredith, C.W.; Malte, C.A.; McBride, B.; Ferguson, L.C.; Gross, C.A.; Hart, K.L.; Raskind, M. A pilot trial of the alpha-1 adrenergic antagonist, prazosin, for alcohol dependence. Alcohol Clin. Exp. Res. 2009, 33, 255–263. [Google Scholar] [CrossRef]
- Fox, H.C.; Anderson, G.M.; Tuit, K.; Hansen, J.; Kimmerling, A.; Siedlarz, K.M.; Morgan, P.T.; Sinha, R. Prazosin effects on stress- and cue-induced craving and stress response in alcohol-dependent individuals: Preliminary findings. Alcohol Clin. Exp. Res. 2012, 36, 351–360. [Google Scholar] [CrossRef]
- Gabbott, P.L.; Warner, T.A.; Jays, P.R.; Salway, P.; Busby, S.J. Prefrontal cortex in the rat: Projections to subcortical autonomic, motor, and limbic centers. J. Comp. Neurol 2005, 492, 145–177. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Datta, D.; Arnsten, A.F.T. Loss of Prefrontal Cortical Higher Cognition with Uncontrollable Stress: Molecular Mechanisms, Changes with Age, and Relevance to Treatment. Brain Sci. 2019, 9, 113. https://doi.org/10.3390/brainsci9050113
Datta D, Arnsten AFT. Loss of Prefrontal Cortical Higher Cognition with Uncontrollable Stress: Molecular Mechanisms, Changes with Age, and Relevance to Treatment. Brain Sciences. 2019; 9(5):113. https://doi.org/10.3390/brainsci9050113
Chicago/Turabian StyleDatta, Dibyadeep, and Amy F. T. Arnsten. 2019. "Loss of Prefrontal Cortical Higher Cognition with Uncontrollable Stress: Molecular Mechanisms, Changes with Age, and Relevance to Treatment" Brain Sciences 9, no. 5: 113. https://doi.org/10.3390/brainsci9050113
APA StyleDatta, D., & Arnsten, A. F. T. (2019). Loss of Prefrontal Cortical Higher Cognition with Uncontrollable Stress: Molecular Mechanisms, Changes with Age, and Relevance to Treatment. Brain Sciences, 9(5), 113. https://doi.org/10.3390/brainsci9050113