
A Simple Method for Assessing Diversity and Dynamics of Microbial Community: Comparison of Dairy Phages from Industrial and Spontaneous Fermentation

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Featured Application
Abstract
1. Introduction
2. Materials and Methods
2.1. Cheese Samples and DNA Isolation
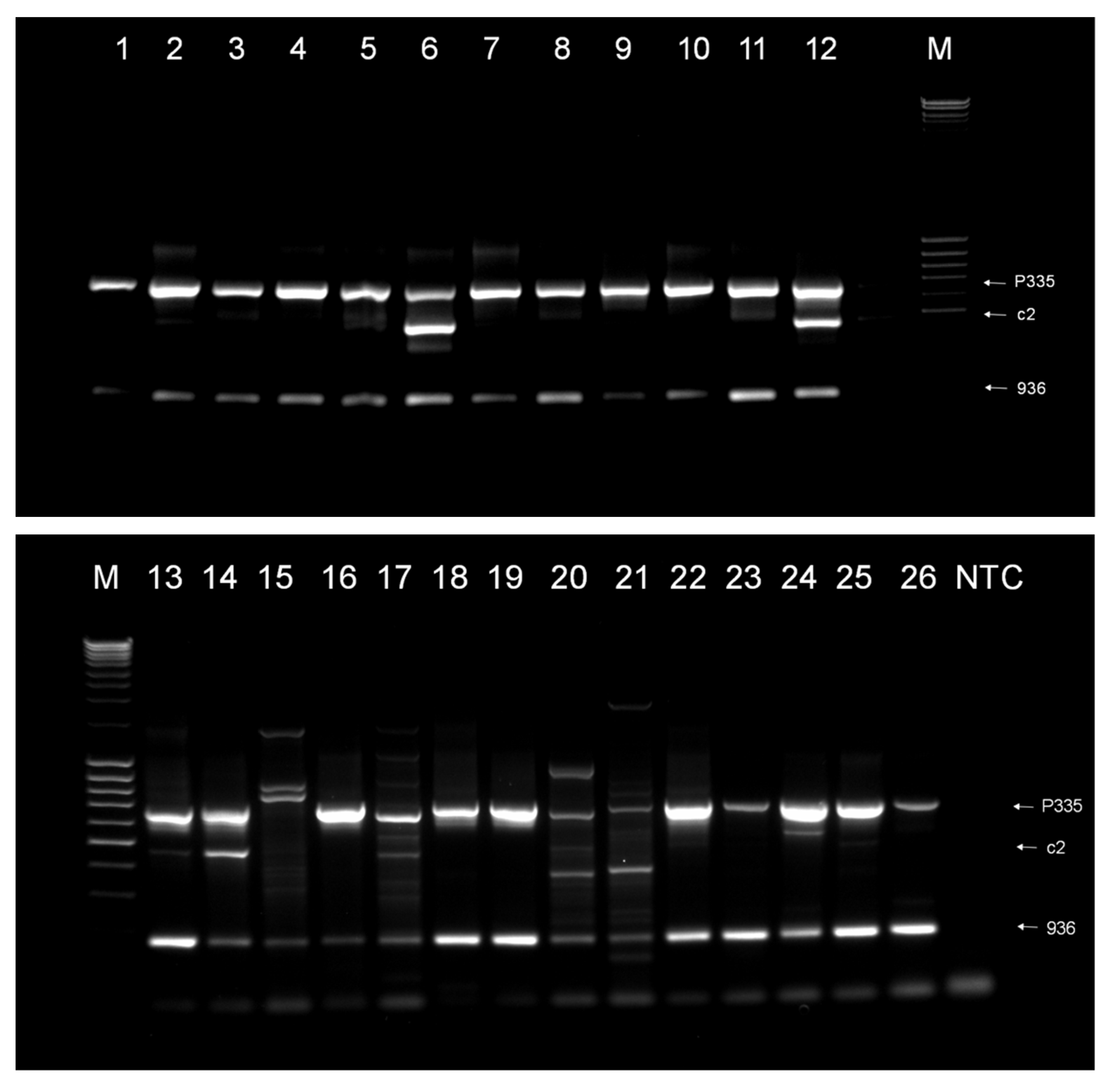
2.2. PCR Amplification and Sequencing
2.3. Sequence Analysis
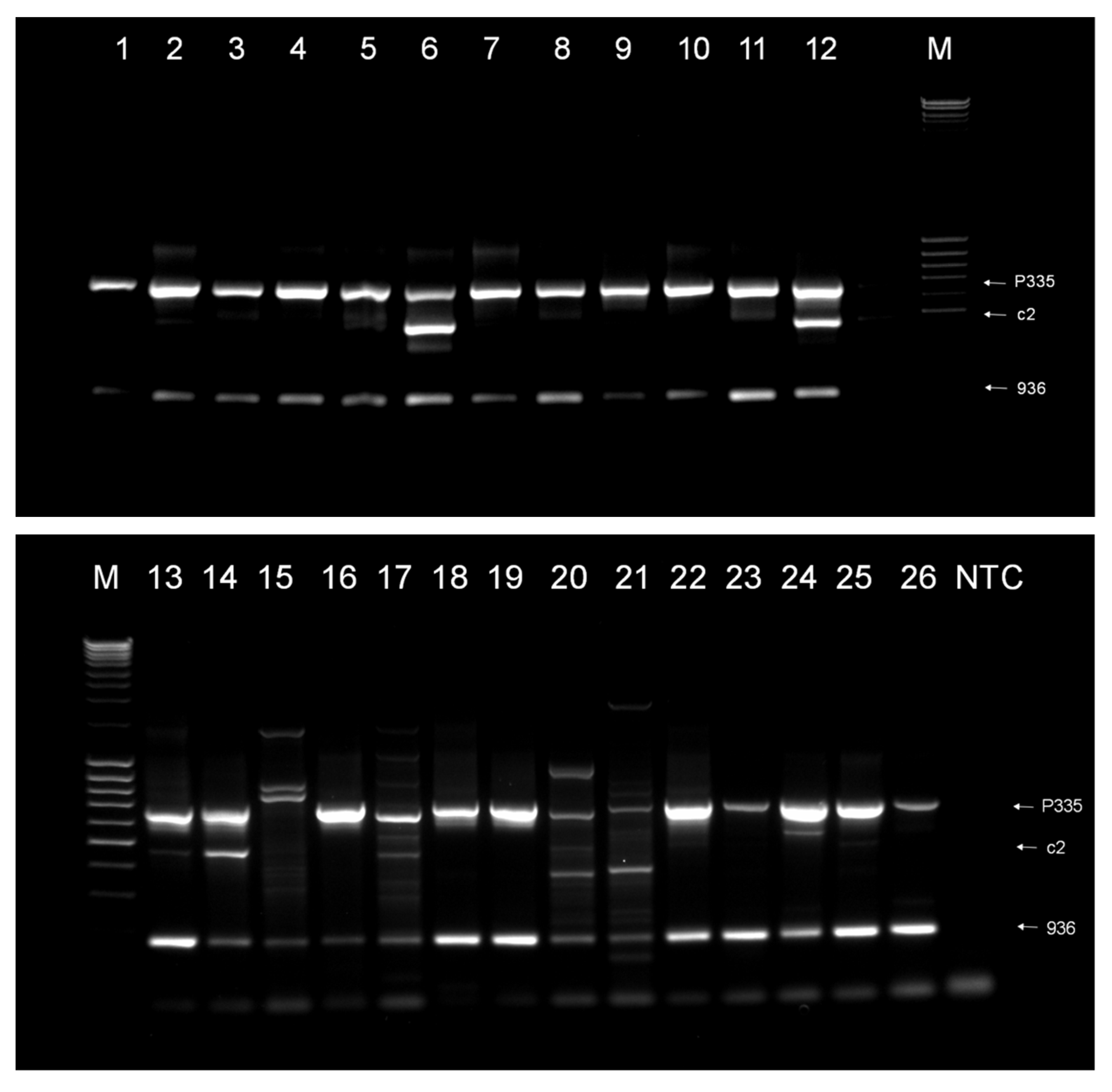
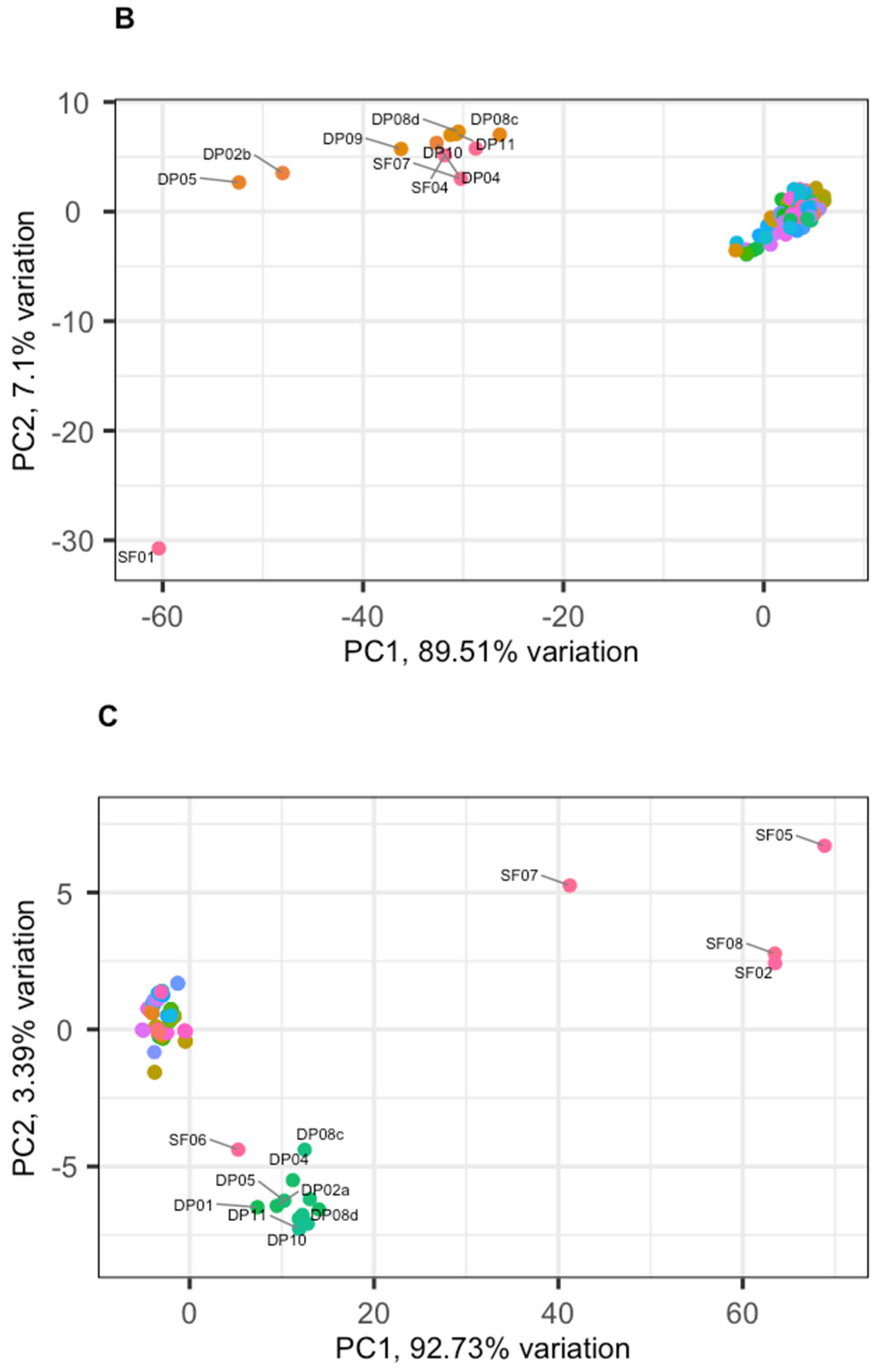
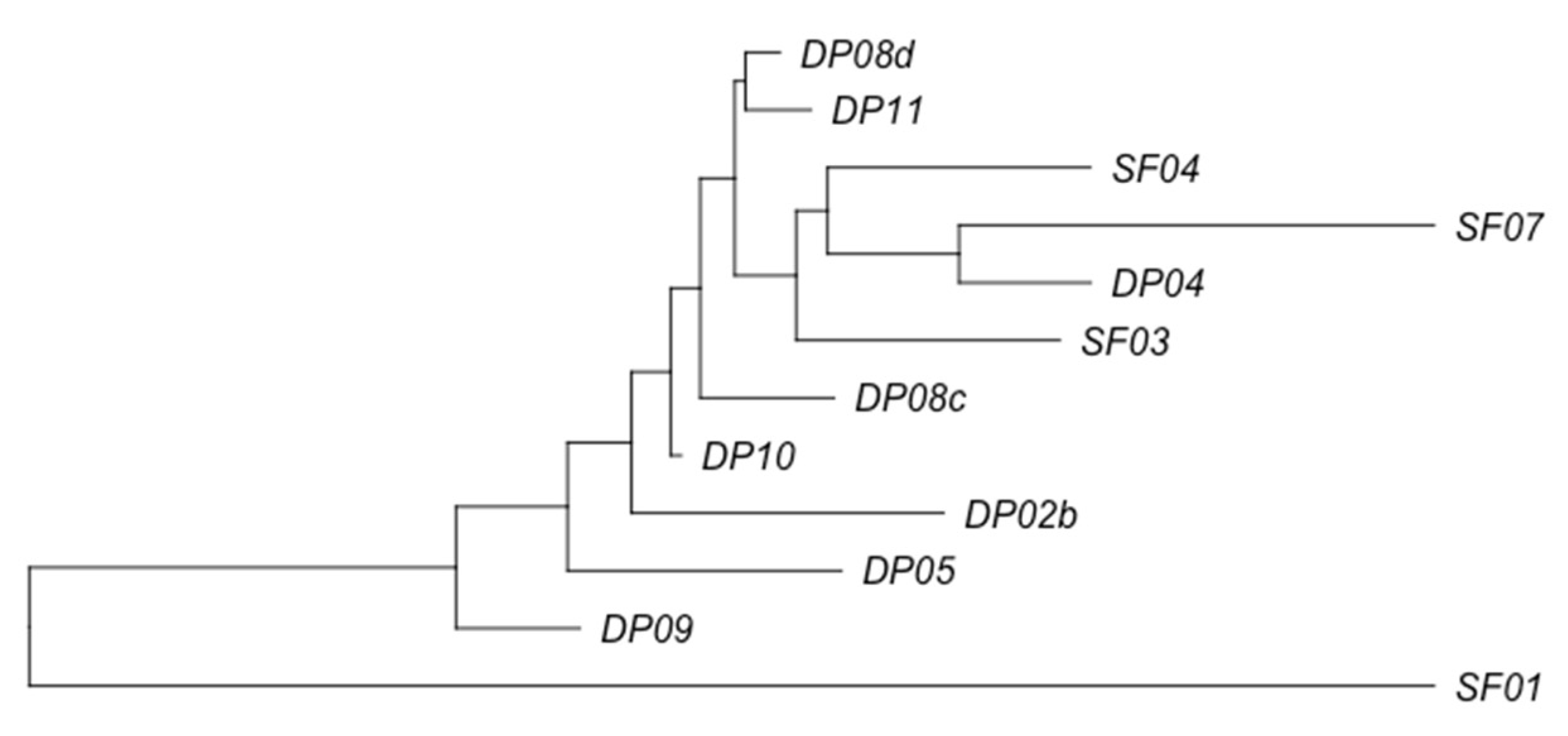
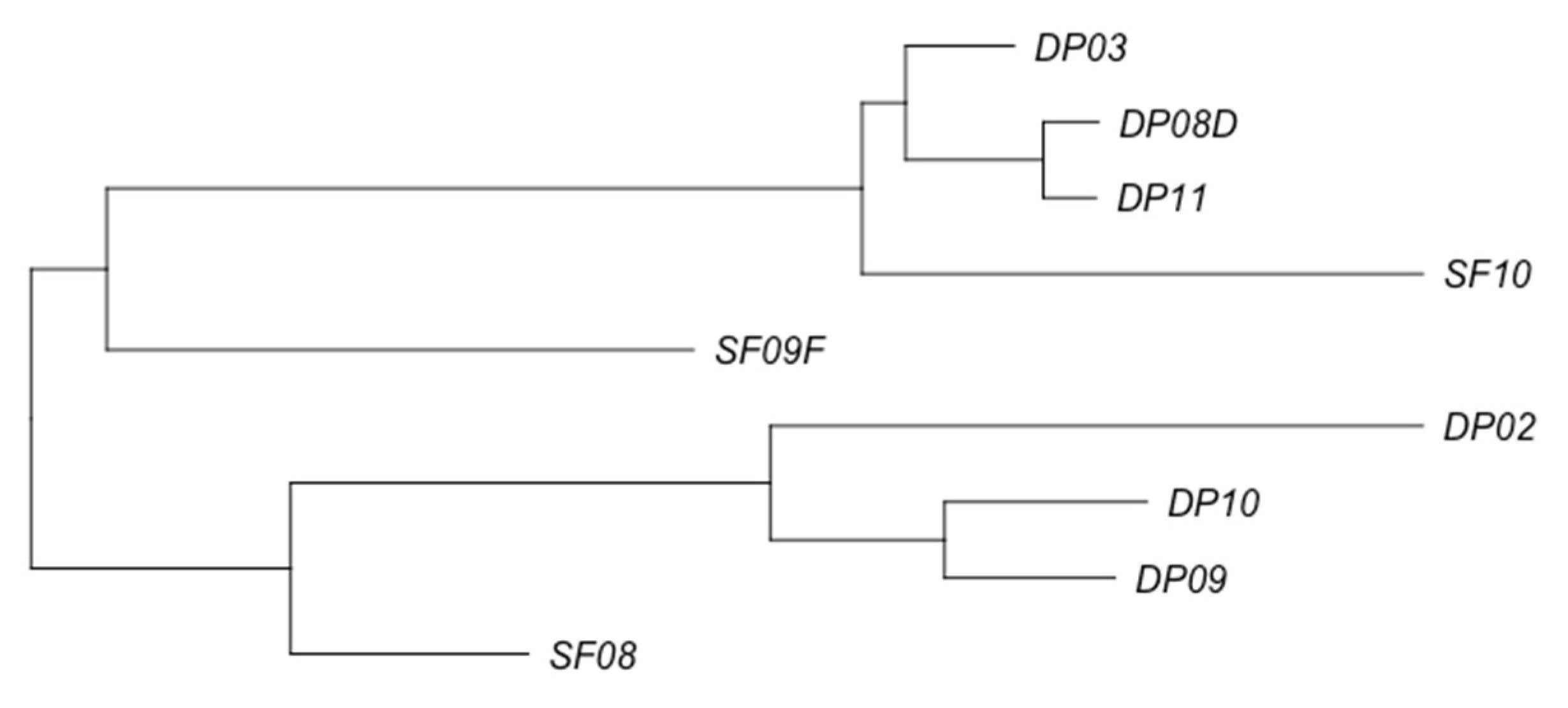
3. Results
3.1. Phage Survey
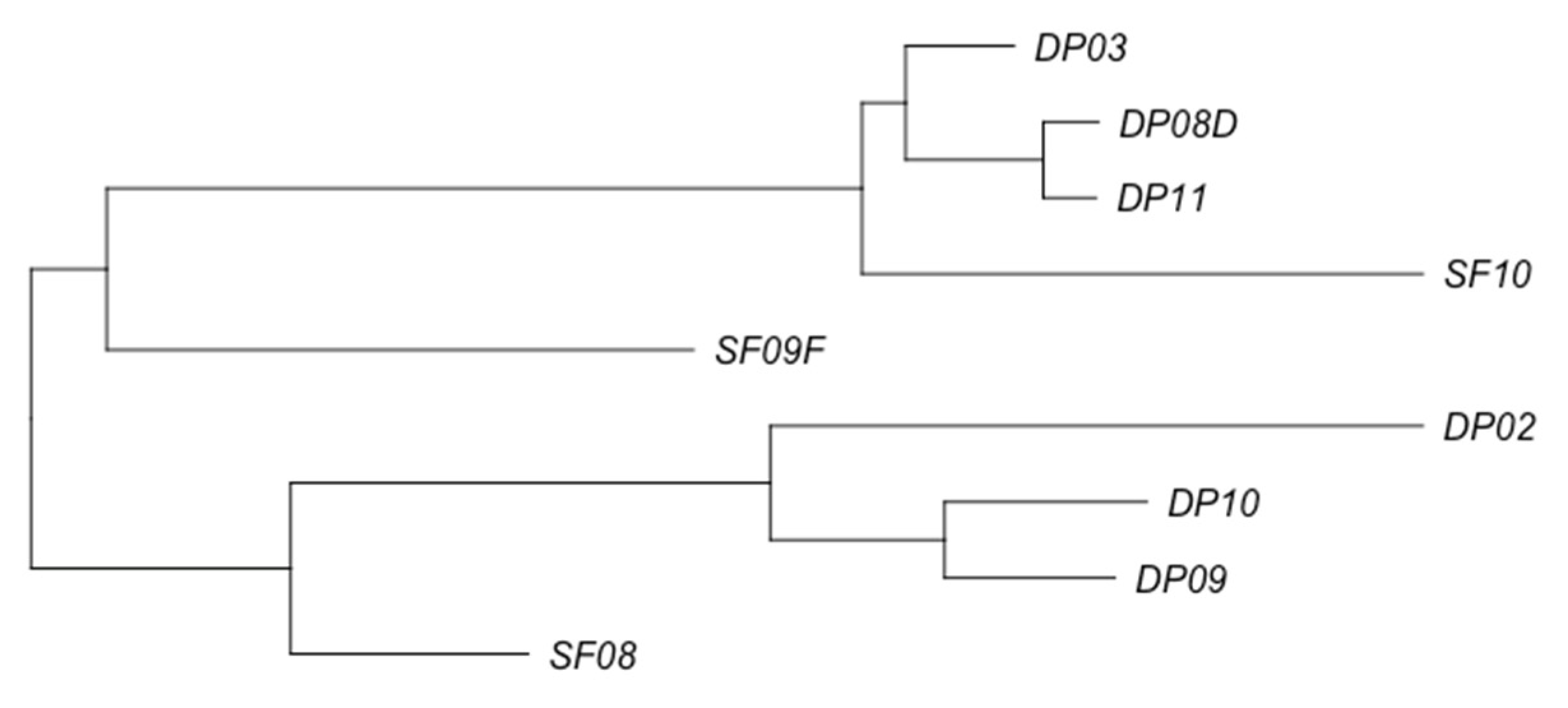
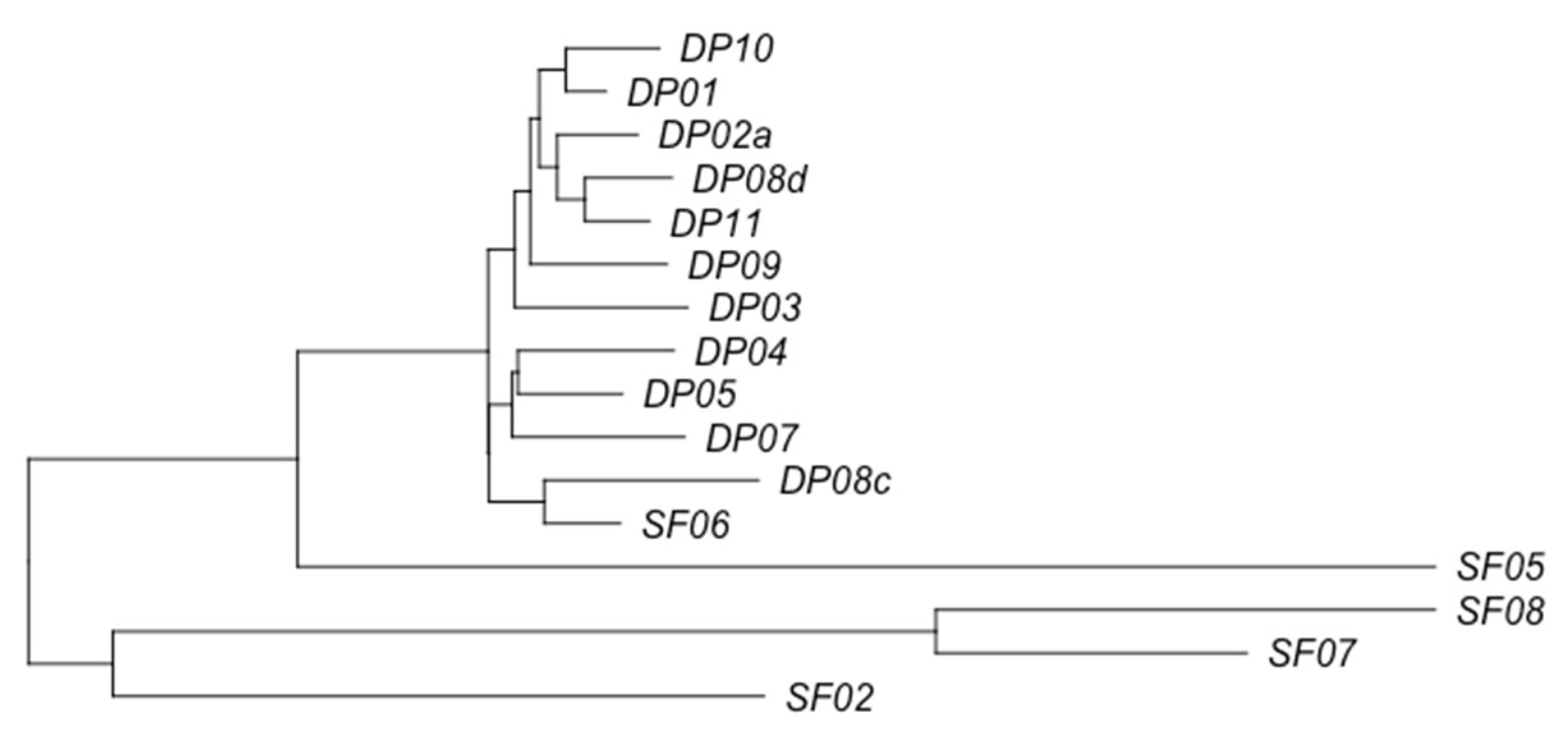
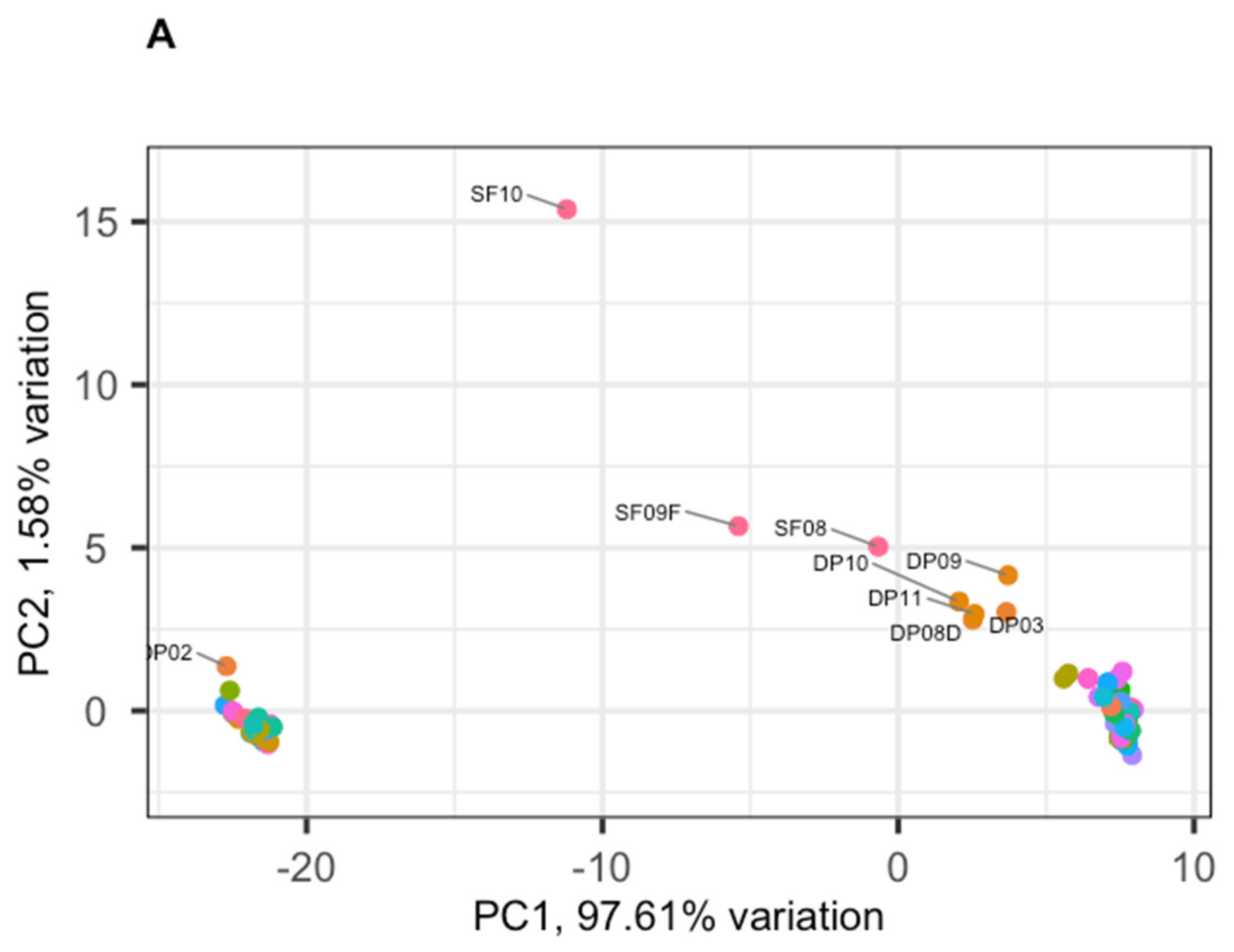
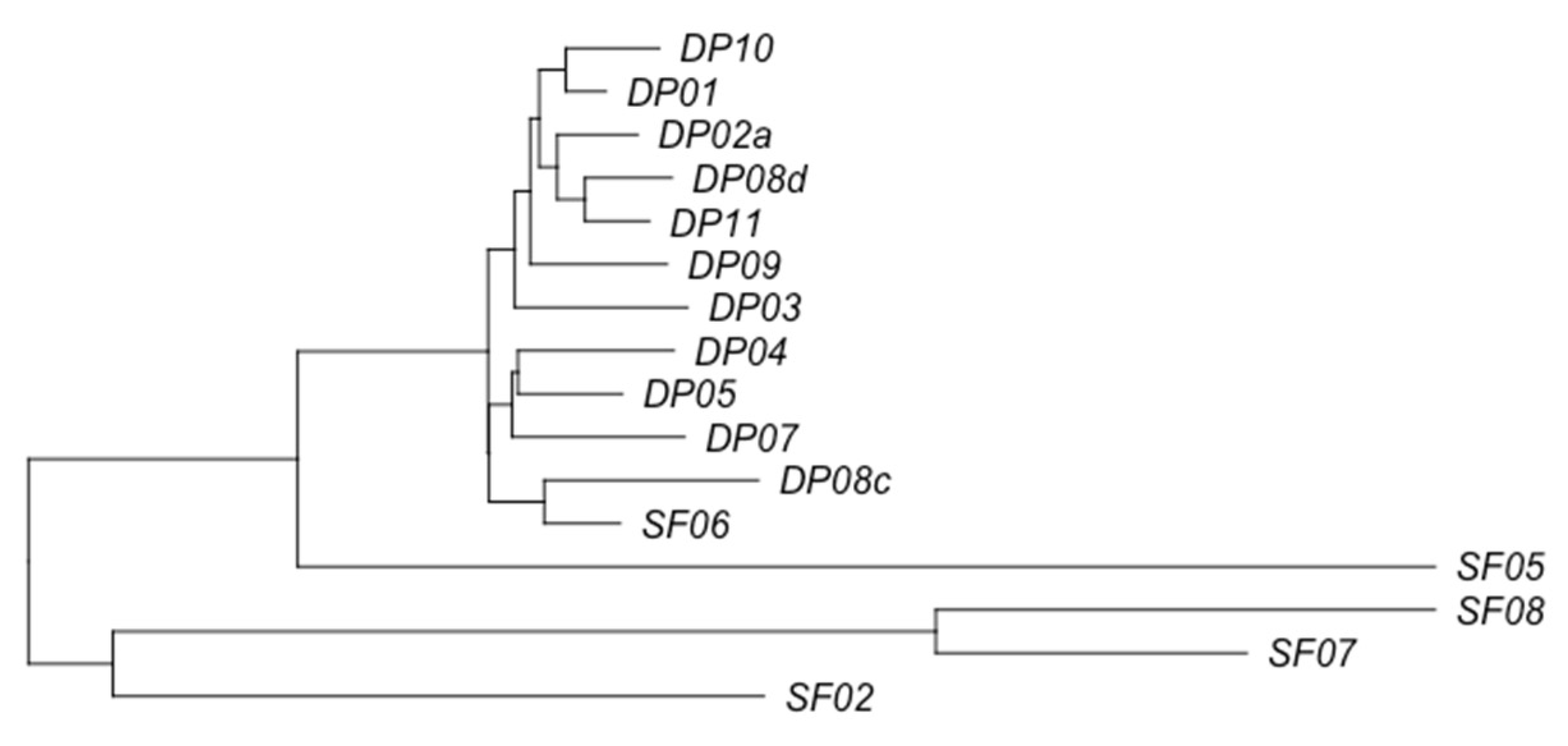
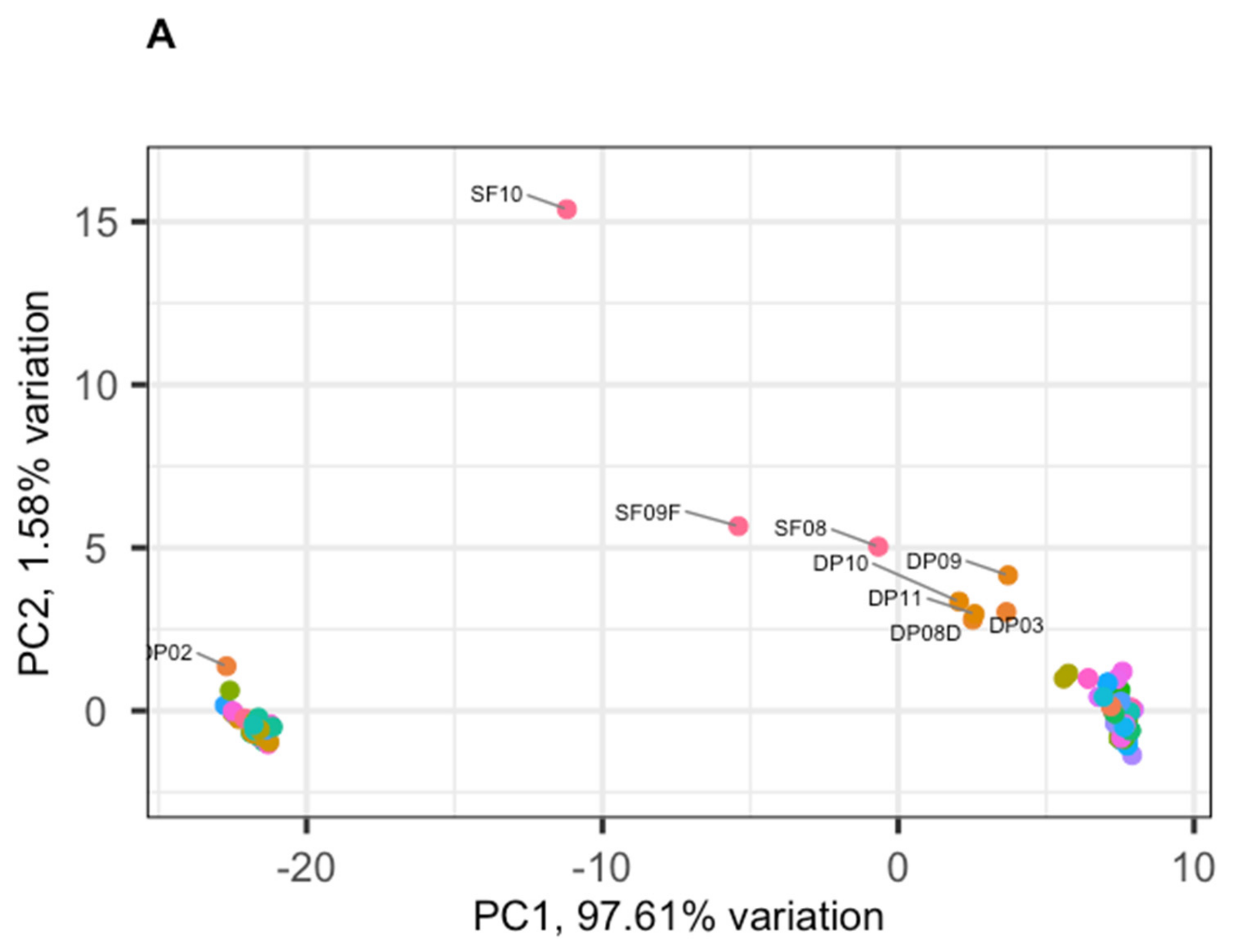
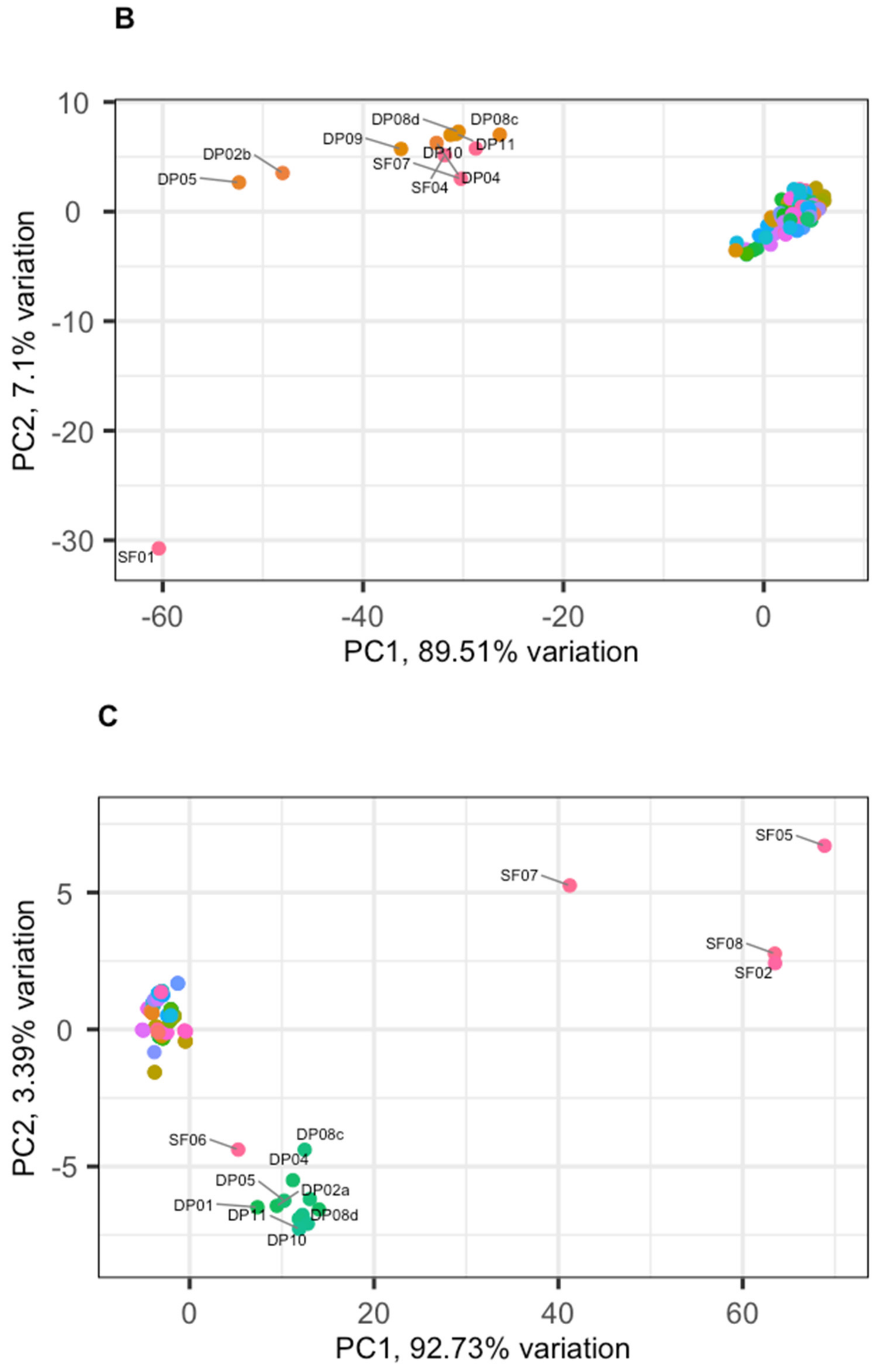
3.2. Phage Diversity Analysis between Samples
3.3. Global Phage Diversity Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marcó, M.B.; Moineau, S.; Quiberoni, A. Bacteriophages and dairy fermentations. Bacteriophage 2012, 2, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Kleppen, H.P.; Bang, T.; Nes, I.F.; Holo, H. Bacteriophages in milk fermentations: Diversity fluctuations of normal and failed fermentations. Int. Dairy J. 2011, 21, 592–600. [Google Scholar] [CrossRef]
- Schmidt, M.T.; Olejnik-Schmidt, A.K.; Zareba, A.; Pezacki, M.; Wojewoda, I.; Grajek, W. Induction of loci mutation during lactococcus lactis spontaneous conversion to bacteriophage-insensitive phenotype. Food Biotechnol. 2010, 24, 332–348. [Google Scholar] [CrossRef]
- Mahony, J.; van Sinderen, D. Current taxonomy of phages infecting lactic acid bacteria. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Deveau, H.; Labrie, S.J.; Chopin, M.C.; Moineau, S. Biodiversity and classification of lactococcal phages. Appl. Environ. Microbiol. 2006, 72, 4338–4346. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, J.; Mahony, J.; Hanemaaijer, L.; Kouwen, T.R.H.M.; van Sinderen, D. Biodiversity of bacteriophages infecting Lactococcus lactis starter cultures. J. Dairy Sci. 2018, 101, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.; Escobedo, S.; Gutiérrez, D.; Portilla, S.; Martínez, B.; García, P.; Rodríguez, A. Bacteriophages in the dairy environment: From enemies to allies. Antibiotics 2017, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Del Rio, B.; Martín, M.C.; Ladero, V.; Martnez, N.M.D.; Fernndez, M.A.M. Bacteriophages in Dairy Industry: PCR Methods as Valuable Tools. In Bacteriophages; InTech: London, UK, 2012. [Google Scholar]
- Cattonaro, F.; Spadotto, A.; Radovic, S.; Marroni, F. Do you cov me? Effect of coverage reduction on metagenome shotgun sequencing studies. F1000Research 2020, 7. [Google Scholar] [CrossRef]
- Pacwa-Płociniczak, M.; Biniecka, P.; Bondarczuk, K.; Piotrowska-Seget, Z. Metagenomic Functional Profiling Reveals Differences in Bacterial Composition and Function During Bioaugmentation of Aged Petroleum-Contaminated Soil. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Zotta, T.; Ricciardi, A.; Condelli, N.; Parente, E. Metataxonomic and metagenomic approaches for the study of undefined strain starters for cheese manufacture. Crit. Rev. Food Sci. Nutr. 2021. [Google Scholar] [CrossRef]
- Frantzen, C.A.; Holo, H. Unprecedented diversity of lactococcal group 936 bacteriophages revealed by amplicon sequencing of the portal protein gene. Viruses 2019, 11, 443. [Google Scholar] [CrossRef] [Green Version]
- Labrie, S.; Moineau, S. Multiplex PCR for detection and identification of lactococcal bacteriophages. Appl. Environ. Microbiol. 2000, 66, 987–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eric, S.D.; Nicholas, T.K.D.D.; Theophilus, K.A. Bioinformatics with basic local alignment search tool (BLAST) and fast alignment (FASTA). J. Bioinforma. Seq. Anal. 2014, 6, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Ihaka, R.; Gentleman, R. R: A Language for Data Analysis and Graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar] [CrossRef]
- Wright, E.S. Using DECIPHER v2.0 to analyze big biological sequence data in R. R J. 2016, 8, 352–359. [Google Scholar] [CrossRef] [Green Version]
- PCAtools GitHub—kevinblighe/PCAtools: PCAtools: Everything Principal Components Analysis. Available online: https://github.com/kevinblighe/PCAtools?s=03 (accessed on 8 June 2021).
- Som, A. Theoretical foundation to estimate the relative efficiencies of the Jukes-Cantor+gamma model and the Jukes-Cantor model in obtaining the correct phylogenetic tree. Gene 2006, 385, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Szczepańska, A.K.; Hejnowicz, M.S.; Kołakowski, P.; Bardowski, J. Biodiversity of Lactococcus lactis bacteriophages in Polish dairy environment. Acta Biochim. Pol. 2007, 54, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Mahony, J.; Moscarelli, A.; Kelleher, P.; Lugli, G.A.; Ventura, M.; Settanni, L.; van Sinderen, D. Phage biodiversity in artisanal cheese wheys reflects the complexity of the fermentation process. Viruses 2017, 9, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavelle, K.; Martinez, I.; Neve, H.; Lugli, G.A.; Franz, C.M.A.P.; Ventura, M.; Dal Bello, F.; van Sinderen, D.; Mahony, J. Biodiversity of streptococcus thermophilus phages in global dairy fermentations. Viruses 2018, 10, 577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moisan, M.; Moineau, S. Multilocus sequence typing scheme for the characterization of 936- like phages infecting Lactococcus lactis. Appl. Environ. Microbiol. 2012, 78, 4646–4653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olejnik-Schmidt, A.; Pietrzak, B.; Kawacka, I.; Malak, K.; Wawrzyniak, W.; Schmidt, M. A Simple Method for Assessing Diversity and Dynamics of Microbial Community: Comparison of Dairy Phages from Industrial and Spontaneous Fermentation. Appl. Sci. 2021, 11, 8915. https://doi.org/10.3390/app11198915
Olejnik-Schmidt A, Pietrzak B, Kawacka I, Malak K, Wawrzyniak W, Schmidt M. A Simple Method for Assessing Diversity and Dynamics of Microbial Community: Comparison of Dairy Phages from Industrial and Spontaneous Fermentation. Applied Sciences. 2021; 11(19):8915. https://doi.org/10.3390/app11198915
Chicago/Turabian StyleOlejnik-Schmidt, Agnieszka, Bernadeta Pietrzak, Iwona Kawacka, Klaudia Malak, Weronika Wawrzyniak, and Marcin Schmidt. 2021. "A Simple Method for Assessing Diversity and Dynamics of Microbial Community: Comparison of Dairy Phages from Industrial and Spontaneous Fermentation" Applied Sciences 11, no. 19: 8915. https://doi.org/10.3390/app11198915
APA StyleOlejnik-Schmidt, A., Pietrzak, B., Kawacka, I., Malak, K., Wawrzyniak, W., & Schmidt, M. (2021). A Simple Method for Assessing Diversity and Dynamics of Microbial Community: Comparison of Dairy Phages from Industrial and Spontaneous Fermentation. Applied Sciences, 11(19), 8915. https://doi.org/10.3390/app11198915